

The most widely used pharmacological treatments for depression are reuptake inhibitors, which inhibit the reuptake of serotonin (5-hydroxytryptamine, 5-HT) or noradrenaline from the synapse back into the presynaptic terminal. Drugs that specifically inhibit the reuptake of 5-HT (selective serotonin reuptake inhibitors, SSRIs) now predominate in the pharmacological management of depression. However, noradrenaline reuptake inhibitors (NARIs) have broadly similar efficacy, although a network meta-analysis has suggested that reboxetine could be less effective. Reference Freemantle, Anderson and Young1,Reference Cipriani, Furukawa, Salanti, Geddes, Higgins and Churchill2 Antidepressants are widely prescribed. In 2007–8 in England, 34 million prescriptions for antidepressants were written, 3 and in the USA the number of prescriptions rose from 154 million in 2002 to 170 million in 2005. 4 The pattern of response to SSRIs is variable and so, as in other areas of medicine, it would be advantageous if one could predict response and individually tailor treatments. From the viewpoint of SSRI treatment, there has been much interest in the gene for the serotonin transporter (SLC6A4), and in particular in a 44 base pair insertion/deletion polymorphism within a repetitive unit in the promoter region (5-HTTLPR). In vitro evidence suggests the long (insertion) form of 5-HTTLPR is functionally more active than the short (deletion) form. Reference Lesch, Bengel, Heils, Sabol, Greenberg and Petri5,Reference Laux, Konig, Lesch and Stein6 Studies in humans are less consistent, with some evidence of enhanced serotonin transmission in those homozygous for the insertion allele, Reference Whale, Quested, Laver, Harrison and Cowen7 but no consistent support for an increase in transporter binding sites. Reference Murthy, Selvaraj, Cowen, Bhagwagar, Riedel and Peers8 There is also inconsistent evidence for the proposal that SSRIs are more effective in individuals with the long/long (l/l) genotype. A meta-analysis has provided support (odds ratio 2.0, 95% CI 1.4–2.9), Reference Serretti, Kato, De and Kinoshita9 but the large Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study (n = 1272) provided first a ‘negative’ finding, Reference Hu, Rush, Charney, Wilson, Sorant and Papanicolaou10 and then a ‘positive’ finding in a smaller subsample restricted to non-Hispanic White participants. Reference Mrazek, Rush, Biernacka, O'Kane, Cunningham and Wieben11 A more recent meta-analysis suggested no association but used the larger STAR*D sample in their calculations. Reference Taylor, Sen and Bhagwagar12 Of more importance for attempts to target treatments on individuals is whether variation at 5-HTTLPR has some specific benefit for treatment with SSRIs rather than a more general influence on outcome irrespective of whether the patient is treated with an SSRI or an NARI.
Studies that have compared SSRIs with NARIs have not provided consistent findings. The Genome Based Therapeutic Drugs for Depression (GENDEP) study reported that only men with the l/l genotype had a better response to escitalopram than to nortriptyline. Reference Huezo-Diaz, Uher, Smith, Rietschel, Henigsberg and Marusic13 Neither Joyce in a smaller study (n = 169) nor Kim et al reported a differential effect, Reference Joyce, Mulder, Luty, McKenzie, Miller and Rogers14,Reference Kim, Lim, Kim, Kim, Chang and Carroll15 and those in Kim's South Korean cohort with l/l had a worse outcome for both SSRI and NARI (nortriptyline) treatment.
When comparing the effects of interventions in randomised controlled trials, CONSORT guidelines are clear that the primary outcome, the principal comparison and method of analysis should be specified in advance. Reference Moher, Schulz and Altman16 For hypotheses such as that posed here, where the primary analyses are subgroup analyses, the need for advance specification and adequate sample size planning is especially important. Reference Patsopoulos, Tatsioni and Ioannidis17 We therefore conducted a randomised controlled trial, Genetic Predictors of Outcome in Depression (GENPOD), comparing an SSRI and an NARI, in which we specified the hypothesis that those with depression and the l/l genotype of 5-HTTLPR would show a better response to the SSRI citalopram than to the NARI reboxetine.
Method
The trial methods have been reported in detail elsewhere, Reference Thomas, Mulligan, Mason, Tallon, Wiles and Cowen18 and are summarised in brief below. The study was a multicentre randomised controlled trial in which patients with depression, recruited in primary care, were randomly allocated to receive either an NARI (reboxetine 4 mg twice daily) or an SSRI (citalopram 20 mg daily). The study included those aged 18–74 years who had already agreed with their general practitioner (GP) that antidepressants should be prescribed. The study was conducted in three centres in the UK: Bristol, Birmingham and Newcastle. Ethical approval was obtained from the South West Ethics Committee (MREC 02/6/076) as well as research governance approval from Bristol, Birmingham and Newcastle primary care National Health Service (NHS) trusts. The trial was registered with the International Standard Randomised Controlled Trials Number registry (ISRCTN31345163) and the European Union Drug Regulating Authorities Clinical Trials (EudraCT) number is 2004-001434-16.
Patients who had taken antidepressant medication within the 2 weeks prior to the baseline assessment and those who could not complete self-administered scales were excluded. General practitioners also excluded those with medical contraindications, psychosis, bipolar affective disorder, major substance or alcohol misuse, and others whose participation was deemed inappropriate. However, we do not know how many were excluded or refused further information at this stage, before the GP referred the potential participant to the trial team. At the baseline assessment only patients with a current diagnosis of ICD–10 depressive episode F32 from the computerised Clinical Interview Schedule – Revised (CIS–R), 19–Reference Lewis21 and a Beck Depression Inventory (BDI) score above 14, Reference Beck, Ward, Mendelsohn, Mock and Erbaugh22 were eligible to continue in the study.
Randomisation procedure
Following the baseline assessment the participant, if eligible, was asked to consent to randomisation. Randomisation was conducted using a computer-generated code, administered centrally and communicated by telephone and thereby concealed in advance from the researcher. Allocation was stratified by severity of symptoms (CIS–R total score ≥28 or <28) and by centre, using variable block sizes to maximise concealment. The researcher gave the randomised medication to the participant. Those randomly allocated to reboxetine were advised to begin with a dose of 2 mg twice daily and increase it to 4 mg twice daily after about 4 days. The patients were advised that they could approach their GP if they wished to increase the dose of antidepressant, whether citalopram or reboxetine.
Sampling of DNA and genotyping
Blood samples were taken from eligible patients in order to investigate the potential links with the 5-HT transporter (SLC6A4) gene. Their DNA was extracted using routine methods and banked at the Cardiff Medical Research Council (MRC) Centre for Neuropsychiatric Genetics and Genomics, a group with extensive experience of banking DNA under MRC guidelines. Stock DNA was stored at –20°C in a linked anonymised format. All molecular genetic personnel were masked to all clinical data, including drug response.
Polymerase chain reaction (PCR) amplification was carried out with the primers 5′-CGCTCTGAATGCCAGCACCTAACC-3′ and 5′-GGGATTCTGGTGCCACCTAGACGC-3′, in a total volume of 12 l solution containing 20 ng genomic DNA, 0.2 mol/l of each primer, 400 mol/l each of deoxynucleotide triphosphate (dNTP), 4% dimethyl sulfoxide, 1.5 mmol/l MgCl2,10×PCR buffer (Qiagen; www.qiagen.com) and 0.3 U HotStarTaq (Qiagen). Initial denaturation at 95°C for 15 min was followed by 45 cycles of denaturation at 95°C for 20 s, primer annealing at 60°C for 30 s and primer extension at 72°C for 60 s, and a final extension reaction was performed at 72°C for 3 min. The DNA fragments were resolved by electrophoresis in a 3% agarose gel (2.5% Metaphor Agarose, Lonza, www.lonza.com, and 0.5% Hi-Standard Agarose, AGTC Bioproducts Ltd, www.agtcbioproducts.com) stained with ethidium bromide.
Genotypes were called by two experienced researchers working independently and masked to each other's results. For additional quality control, 166 samples were assayed again masked to the prior assigned genotypes. Concordance rates were 100%. The overall call rate was 99%. The di-allelic system was in Hardy–Weinberg equilibrium (P = 0.74).
Outcome measures
Clinical outcome data were recorded 6 weeks and 12 weeks after randomisation. The primary outcome was the total BDI score at 6 weeks. There were a number of secondary outcomes, Reference Thomas, Mulligan, Mason, Tallon, Wiles and Cowen18 and here we also report the proportion ‘in remission’ (defined as a total BDI score <10) at 6 weeks, Short Form Health Survey (SF–12) mental and physical subscale scores and Hospital Anxiety and Depression Scale (HADS) total scores. Reference Jenkinson and Layte23,Reference Zigmond and Snaith24 Adherence to pharmacotherapy was assessed at 6 weeks by asking the patient and counting returned tablets. The participants and research assistants were aware of the randomised allocation and all outcomes were self-administered. We considered using clinician-administered measures such as the Hamilton Rating Scale for Depression, but decided that these would be susceptible to bias in a non-masked trial. The BDI score has been widely used in depression trials.
Other measures
Life events in the previous 6 months were assessed with the following questions: ‘Has someone close to you died?’ ‘Have you separated/divorced from your spouse/partner?’ ‘Has a serious illness or injury occurred to yourself or someone close to you?’ ‘Has a mugging, burglary or other serious assault happened to you or to someone close to you?’ ‘Have you or someone close to you had problems with the police involving a court appearance?’ ‘Have you had a serious dispute with a close friend/relative or neighbour?’ ‘Have you been made redundant or sacked from your job?’ Responses were scored 1 for ‘yes’ and 0 for ‘no’. Total scores could range from 0 to 7.
Statistical analysis
An analysis plan was agreed with the trial steering committee before the results were analysed. The study was designed to test two hypotheses. As well as that stated we wished to investigate whether those with more severe depression would respond better to the NARI than to the SSRI, but the results of that analysis are not presented here because this is a distinct hypothesis with a separate literature that requires a different and extensive discussion. The primary analyses for this trial were therefore two pre-specified subgroup analyses for the primary outcome of BDI score at 6 weeks, with analysis performed on an intention-to-treat basis. Both of these included an interaction term between treatment and the relevant predictor variable (genotype or severity) in a multiple regression model for the primary outcome, adjusting for baseline BDI score, centre and the stratification variable of CIS–R total score (<28 or ≥28). The genotype variable was a linear term for the three-level variable: long/long (l/l), long/short (l/s) and short/short (s/s). For the power calculation the genetic hypothesis was formulated in terms of a binary variable comparing those with the l/l genotype v. the remainder, so we also performed analyses to illustrate the findings using this categorisation. A further secondary analysis was performed including only participants who reported taking their allocated medication for at least 4 weeks. All differences were calculated by subtracting BDI scores for the citalopram group from those in the reboxetine group (so a positive value indicates a worse outcome for reboxetine). The reported odds ratios are the ratio of the odds of recovery for those on reboxetine to citalopram, so an odds ratio of more than 1 indicates a better response with reboxetine. We used a repeated-measures linear regression analysis in order to incorporate data from both the 6-week and 12-week assessments, including time in weeks as a variable in the model along with the other adjustments mentioned in the primary analysis. The main effect of genotype was studied in models without an interaction term, but retaining the other variables. We also adjusted for the time between randomisation and follow-up by incorporating it as a continuous variable in the model.
We investigated the association of baseline characteristics with missing outcome data at 6 weeks by comparing percentages of participants with missing data. The impact of missing values on our results was investigated by adjusting for factors in the regression model that were associated with missing outcome data at 6 weeks (www.missingdata.lshtm.ac.uk). This method should address any bias resulting from attrition, assuming the missing at random assumption is correct. All analyses used Stata version 10 for Windows.
Justification of the sample size
The protocol paper gives further detail on the sample size calculations and the impact that our final recruitment had on the trial. The least statistically powerful analysis was for the genotype analysis so we will summarise the power calculation for this analysis. We used the binary outcome for the BDI, remission, for the sample size calculation as this made it easier to formulate. The transporter allele was also represented as a binary variable. Both these decisions would be expected to lead to an underestimate of statistical power. Studies in the UK have observed 25% of those studied to be homozygous for the insertion allele and 50% heterozygous. Reference Kirov, Rees, Jones, MacCandless, Owen and Craddock25,Reference Ogilvie, Battersby, Bubb, Fink, Harmar and Goodwim26 The original power calculation, based on the methods of Lubin et al, Reference Lubin and Gail27,Reference Garcia-Closas and Lubin28 assumed that the proportions in remission at 6 weeks were as follows: for those in the SSRI group 80% of those homozygous for the insertion allele (l/l) would be in remission compared with 60% for the other genotypes (l/s and s/s). For those in the NARI group we assumed 65% in remission irrespective of genotype. For 90% power it was calculated this would require 754 participants in total, increased to 887 to allow for 15% attrition at 6 weeks. This corresponds to an interaction odds ratio (θ) of 0.375 (with reboxetine coded as 1 and citalopram 0; l/l coded as 1 and l/s plus s/s coded as 0).
Recruitment into the trial was less than planned and the final number randomised was 601, with a target of 570 for (primary) analysis. This target was specified in our published protocol paper. Reference Thomas, Mulligan, Mason, Tallon, Wiles and Cowen18 The actual remission rates in the trial were about 25%, substantially less than the 65% expected, although similar to the results of the STAR*D trial, which also attempted to recruit a broadly representative sample of people with depression in the USA. Reference Trivedi, Rush, Wisniewski, Nierenberg, Warden and Ritz29 We estimated that an interaction odds ratio of 0.33 could be detected with 80% power at the 5% level, based on similar assumptions to those described earlier. This is equivalent to remission rates of 25% for the NARI group, and 36.8% and 19.1% respectively for the homozygous and remainder subgroups in the SSRI group.
Results
General practitioners from 72 practices referred 842 patients to the GENPOD study. Of these, 90 (11%) declined to participate and 23 (3%) were excluded prior to baseline assessment (Fig. 1). There were 126 participants ineligible at baseline and 110 did not receive a diagnosis of ICD–10 depression. Of the 729 who completed a baseline assessment, 601 were randomised between October 2005 and February 2008 to receive either citalopram (n = 298) or reboxetine (n = 303). The baseline characteristics of these groups were similar (Table 1, details shown in online Table DS1). Most of those randomised were from Bristol (n = 521), with the remainder from Birmingham (n = 72) and Newcastle (n = 8).
Table 1 Baseline characteristics of randomised participants

| Citalopram group (n = 298) | Reboxetine group (n = 303) | |
|---|---|---|
| Gender, n (%) | ||
| Male | 98 (33) | 95 (31) |
| Female | 200 (67) | 208 (69) |
| Age, years: mean (s.d.) | 38.6 (12.1) | 39.1 (12.6) |
| Age group, n (%) | ||
| 18–34 years | 122 (41) | 124 (41) |
| 35–54 years | 146 (49) | 136 (45) |
| 55–74 years | 30 (10) | 43 (14) |
| Previous treatment, n (%) | 165 (56) | 160 (53) |
| CIS–R score, mean (s.d.) | 31.0 (7.5) | 30.7 (8.5) |
| Depression category, n (%) | ||
| Moderate (CIS–R <28) | 102 (34) | 105 (35) |
| Severe (CIS–R ≥28) | 196 (66) | 198 (65) |
| BDI score, mean (s.d.) | 33.9 (9.3) | 33.4 (10.0) |
| Suicidal thoughts,a n (%) | 38 (13) | 43 (14) |
| Life events, mean (s.d.) | 1.7 (1.3) | 1.6 (1.4) |
| Social support, mean (s.d.) | 11.9 (3.9) | 12.1 (3.7) |
| Employment status, n (%) | ||
| Employed full-time | 114 (38) | 129 (43) |
| Employed part-time | 55 (18) | 59 (19) |
| Student | 12 (4) | 11 (4) |
| Retired | 6 (2) | 15 (5) |
| Housekeeper | 45 (15) | 32 (11) |
| Unemployed job seeker | 22 (7) | 19 (6) |
| Unemployed owing to ill health | 44 (15) | 38 (13) |
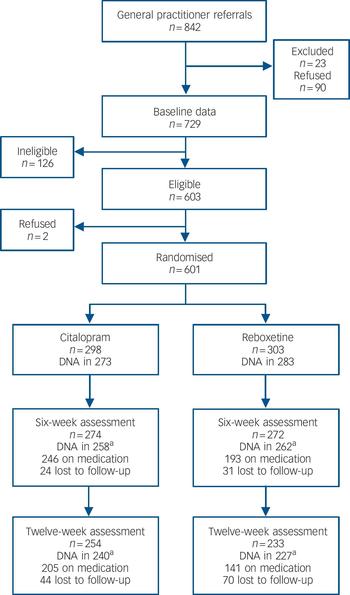
Fig. 1 Participant flow through the trial.
a. Available for analysis.
Baseline comparability of genotype
A blood sample was taken from 561 participants but the genotype of 5 patients could not be determined from their sample. Genotype data were available for 556 (93%) randomised patients: 189 (34%) of these were categorised as l/l, 267 (48%) as l/s and 100 (18%) as s/s. Although different from our assumed distribution of genotypes in the power calculation, these frequencies are in good agreement with recent estimates in a large White non-Hispanic sample from the USA, Reference Mrazek, Rush, Biernacka, O'Kane, Cunningham and Wieben11 and with a large multicentre study sample recruited from Europe. Reference Huezo-Diaz, Uher, Smith, Rietschel, Henigsberg and Marusic13 Table 2 compares the characteristics of these patients according to their 5-HTTLPR genotype. Overall, the characteristics of patients were similar regardless of genotype.
Table 2 Characteristics of participants according to 5-HTTLPR genotype
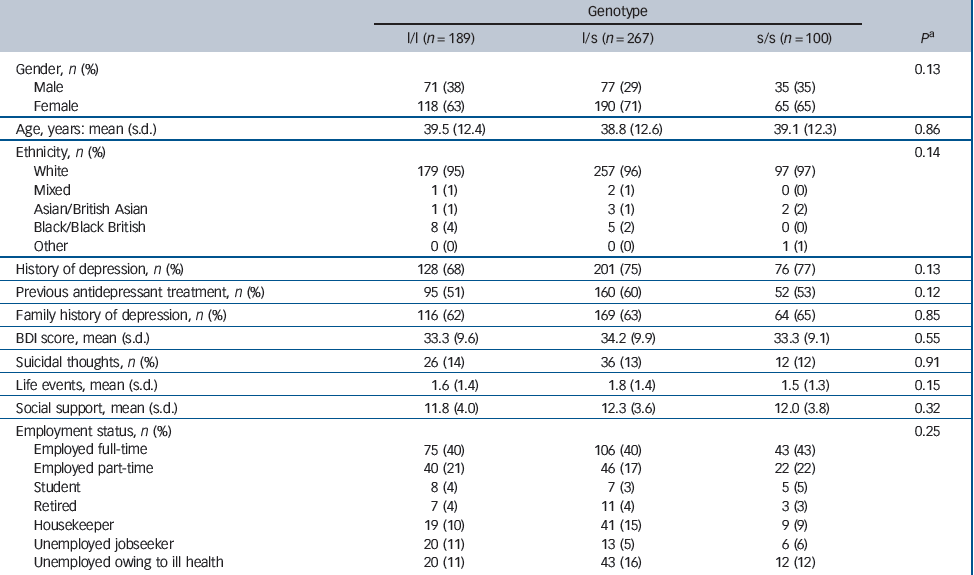
| Genotype | ||||
|---|---|---|---|---|
| l/l (n = 189) | l/s (n = 267) | s/s (n = 100) | P a | |
| Gender, n (%) | 0.13 | |||
| Male | 71 (38) | 77 (29) | 35 (35) | |
| Female | 118 (63) | 190 (71) | 65 (65) | |
| Age, years: mean (s.d.) | 39.5 (12.4) | 38.8 (12.6) | 39.1 (12.3) | 0.86 |
| Ethnicity, n (%) | 0.14 | |||
| White | 179 (95) | 257 (96) | 97 (97) | |
| Mixed | 1 (1) | 2 (1) | 0 (0) | |
| Asian/British Asian | 1 (1) | 3 (1) | 2 (2) | |
| Black/Black British | 8 (4) | 5 (2) | 0 (0) | |
| Other | 0 (0) | 0 (0) | 1 (1) | |
| History of depression, n (%) | 128 (68) | 201 (75) | 76 (77) | 0.13 |
| Previous antidepressant treatment, n (%) | 95 (51) | 160 (60) | 52 (53) | 0.12 |
| Family history of depression, n (%) | 116 (62) | 169 (63) | 64 (65) | 0.85 |
| BDI score, mean (s.d.) | 33.3 (9.6) | 34.2 (9.9) | 33.3 (9.1) | 0.55 |
| Suicidal thoughts, n (%) | 26 (14) | 36 (13) | 12 (12) | 0.91 |
| Life events, mean (s.d.) | 1.6 (1.4) | 1.8 (1.4) | 1.5 (1.3) | 0.15 |
| Social support, mean (s.d.) | 11.8 (4.0) | 12.3 (3.6) | 12.0 (3.8) | 0.32 |
| Employment status, n (%) | 0.25 | |||
| Employed full-time | 75 (40) | 106 (40) | 43 (43) | |
| Employed part-time | 40 (21) | 46 (17) | 22 (22) | |
| Student | 8 (4) | 7 (3) | 5 (5) | |
| Retired | 7 (4) | 11 (4) | 3 (3) | |
| Housekeeper | 19 (10) | 41 (15) | 9 (9) | |
| Unemployed jobseeker | 20 (11) | 13 (5) | 6 (6) | |
| Unemployed owing to ill health | 20 (11) | 43 (16) | 12 (12) | |
Follow-up
The number of questionnaires returned at 6 weeks and at 12 weeks were 546 (91%) and 487 (81%) respectively. The mean time of completion for the 6-week follow-up was 43.2 (s.d. = 6.4) days in the citalopram group and 44.9 (s.d. = 10) days for the reboxetine group (P = 0.02). Nine-tenths (91%) of the 6-week follow-up questionnaires were received between 37 and 50 days after randomisation. Of those followed up at 6 weeks, 520 also had genotype data. At 6 weeks, 24 (8%) of those allocated to citalopram were lost to follow-up, compared with 31 (10%) in the reboxetine group. There were 520 participants with both 6-week outcome data and genotype data (87% of the randomised sample).
Adherence
Reboxetine appeared to be less well tolerated than citalopram. At 6 weeks, 36% of patients randomised to reboxetine had stopped taking the allocated medication compared with 17% for citalopram. For those with outcome data at 6 weeks, 91.2% (250 of 274) had taken citalopram for at least 4 weeks and 82.4% (224 of 272) had taken reboxetine for at least 4 weeks. Some of the participants also received additional quantities of their randomised medication from their GP. For those prescribed citalopram, 11 patients had their daily dosage increased to 30 mg, 33 patients had it increased to 40 mg and 11 patients to 60 mg (1 further participant had an increase in prescribed dose of an unknown amount). For reboxetine, 3 patients had an increase in daily dosage to 10 mg, 9 patients had an increase to 12 mg and 1patient's dosage wasincreased to 16mg.
Analysis for the genotype hypothesis
Table 3 reports the mean BDI scores at 6 weeks and the differences in response between citalopram and reboxetine according to genotype. At 6 weeks the difference between citalopram and reboxetine was greater for those with the s/s genotype. The primary analysis was to test for an interaction between genotype and allocated treatment, with 6-week BDI scores as the outcome, adjusting for baseline BDI score, severity stratum and centre. This analysis provided no evidence for a differential effect (interaction term 0.50, 95% CI –2.04 to 3.03, P = 0.70). Restricting this regression to the 451 participants with 6-week follow-up data who reported taking their allocated medication for at least 4 weeks led to results well within the confidence intervals of the primary analysis (interaction term –0.72, 95% CI –3.40 to 1.95, P = 0.60). Adjusting the primary analysis for the time interval between randomisation and 6-week follow-up led to little difference (interaction term 0.49, 95% CI –2.05 to 3.02, P = 0.71), nor did restricting the analysis to patients of White ethnic origin (interaction term 0.39, 95% CI –0.22 to 2.99, P = 0.77; n = 499). We also investigated a main effect of genotype on outcome, irrespective of treatment, after adjustment for the random allocation and the other adjustments listed. We found no evidence for any association between genotype and outcome at 6 weeks (mean BDI score difference 0.49, 95% CI –0.78 to 1.76, P = 0.58) in the whole sample, nor when we restricted the analysis to those receiving citalopram (mean BDI score difference 0.25, 95% CI – 1.64 to 2.15). We studied the influence of genotype on outcome in a repeated-measures analysis in which both 6-week and 12-week data were used. The results for the interaction term were similar (mean BDI score difference 0.41, 95% CI –1.96 to 2.78, P = 0.73), even when the analysis was restricted to those of White ethnic origin (interaction term 0.38, 95% CI – 2.04 to 2.81) in order to address potential population stratification.
Table 3 Beck Depression Inventory (BDI) mean scores and adjusted differences at 6 weeks according to 5-HTTLPR genotype

| Citalopram group | Reboxetine group | Difference (reboxetine minus citalopram) Adjusted differencea (95% CI) | |||
|---|---|---|---|---|---|
| Genotype | n | BDI score, mean (s.d.) | n | BDI score, mean (s.d.) | |
| l/l | 79 | 18.1 (10.1) | 99 | 18.5 (10.8) | 1.03 (–1.90 to 3.96) |
| l/s | 130 | 19.6 (11.6) | 120 | 20.3 (11.2) | 0.94 (–1.69 to 3.57) |
| s/s | 49 | 18.0 (10.9) | 43 | 19.7 (11.8) | 1.90 (–2.47 to 6.27) |
Secondary outcomes
We also examined our secondary outcomes, the HADS score and the SF–12 standardised mental and physical subscales, in relation to the study hypothesis (online Tables DS2–4). The interaction terms for these outcomes were as follows: HADS score –0.09 (95% CI –2.04 to 1.85); SF–12 mental subscale score –0.13 (95% CI –3.06 to 2.79); SF–12 physical subscale score –0.57 (95% CI –2.51 to 1.36). Again, there was no evidence to support an interaction between genotype and treatment allocation. We also carried out repeated-measures analyses on these secondary outcomes, using the same approach as for the primary outcome. The interaction terms were as follows: HADS score 0.20 (95% CI –1.63 to 2.03), SF–12 mental subscale score –0.80 (95% CI –3.38 to 1.78), and SF–12 physical subscale score 0.17 (95% CI –1.58 to 1.91).
Size of possible interaction effects
To further illustrate the results in relation to the power calculation, we carried out an analysis in which both the genotype and outcome were dichotomous, the interaction odds ratio was 1.07 (95% CI 0.58 to 2.12) compared with the target interaction odds ratio in the sample size calculation of 0.33. In addition,Table 4 shows the percentages in remission at 6 weeks according to the dichotomous genotype exposures. The original hypothesis argued for a larger difference between the genotypes in the citalopram group, and no difference in the reboxetine group. The results clearly do not support this hypothesis.
Table 4 Percentage of patients in remission at 6 weeks according to medication and 5-HTTLPR genotype

| Genotype | Citalopram group % (95% CI) | Reboxetine group % (95% CI) |
|---|---|---|
| l/l | 24.1 (15.1–34.0) | 25.3 (17.1–35.0) |
| l/s, s/s | 22.4 (16.5–29.2) | 20.9 (14.9–27.9) |
Missing data
Although overall levels of attrition were low at 6 weeks (Fig. 1), an exploratory analysis found that participants who were younger, and with more life events and less social support were more likely to have missing data at 6 weeks (online Table DS5). Adjustment for these variables had no effect on the main interaction for genotype, if anything reducing it towards the null (interaction term BDI score 0.25, 95% CI –2.27 to 2.76, P = 0.85).
Discussion
This study was designed to test the hypothesis that 5-HTTLPR leads to a differential response to SSRIs and NARIs in depression. We did not find any evidence to support this suggestion. The main strength of our study was our efforts to reduce the possibility of a chance finding. This was accomplished by prior publication of our protocol and hypotheses, and by preparation of a detailed analysis plan that restricted opportunities for multiple analytical approaches and selective reporting. The main limitation in drawing definitive conclusions concerns the statistical power of the study. Can we conclude that there is no influence of 5-HTTLPR on response to SSRIs? If not, have we at least obtained a result that makes it unlikely that a clinically important interaction is present? Our results exclude the possibility, at least in statistical terms, of an interaction of the size used in our power calculation based on a difference we considered to be clinically important. We have also excluded an interaction of more than 3 points on the BDI (about 0.3 standard deviation). There is no generally accepted definition of a clinically important difference in research on depression but we think that our study was sufficiently large to exclude the possibility of a clinically important interaction between 5-HTTLPR and response to SSRI antidepressants. We have concluded that, given the uncertainties about the assumptions underpinning the power calculations, the shortfall in recruitment had a relatively modest impact on the ability of the trial to answer the primary research questions posed.
For a number of reasons we may have underestimated the influence of 5-HTTLPR on outcome. There was only partial adherence to the medication regimen and this would have made it more difficult to detect any difference. However, restricting the analysis to those who took medication for at least 4 weeks did not alter our findings. Attrition is often a problem in clinical trials, but there was good follow-up response at 6 weeks of 87% once the genotype data had been taken into account. Moreover, adjustment for factors associated with missing data did not influence our conclusions. Finally, depression treated in primary care is often considered to be of mild to moderate severity. In the GENPOD study the mean score on the BDI was about 33 at baseline, indicating that the population included was experiencing depressive illness of at least moderate severity and at a level that one would expect a response to medication. We had a similar response rate to that observed in the STAR*D study. Reference Trivedi, Rush, Wisniewski, Nierenberg, Warden and Ritz29
When considering genetic predictors of antidepressant outcome there is a real danger of reporting type 1 (false positive) errors. Even when analysis is restricted to functional candidate genes, whose function is plausibly related to a target phenotype, there are a large number of polymorphisms which can be analysed using a variety of genetic models. The outcome of depression is usually rated as a continuous score and so the analysis can either use a continuous score or define categories such as response (usually a 50% reduction in score) or remission (below a threshold score). Finally, it is never entirely clear whether genetic hypotheses have been stated beforehand, or whether the statistically significant results have been selected post hoc (especially when two-way interactions are then reported in subgroups such as within-gender). The analysis of subgroups within clinical trials has also received much criticism because of the loss of statistical power and that the many possible effect-modifiers might increase the risk of type 1 errors. Reference Brookes, Whitley, Egger, Davey Smith, Mulheran and Peters30 The impact of such publication and reporting biases are amplified in systematic reviews of the area. Reference Ioannidis and Trikalinos31
Within SLC6A4 there are many variants in and around 5-HTTLPR that could be tested, several of which have been reported to be functional or associated with a range of psychiatric phenotypes, including antidepressant response. Reference Hu, Rush, Charney, Wilson, Sorant and Papanicolaou10,Reference Huezo-Diaz, Uher, Smith, Rietschel, Henigsberg and Marusic13,Reference Kraft, Slager, McGrath and Hamilton32 In particular, a report that haplotypes constructed from polymorphism rs25531 and 5-HTTLPR are functional and associated with obsessive–compulsive disorder has attracted some interest; Reference Hu, Lipsky, Zhu, Akhtar, Taubman and Greenberg33 however, the association between the rs25531/5-HTTLPR haplotypes and the disorder has not been confirmed, Reference Wendland, Kruse, Cromer and Murphy34 and the functionality of this variant is still controversial. Reference Parsey, Hastings, Oquendo, Hu, Goldman and Huang35,Reference Martin, Cleak, Willis-Owen, Flint and Shifman36 Moreover, there is no previous evidence that the haplotype is associated with antidepressant response, Reference Hu, Rush, Charney, Wilson, Sorant and Papanicolaou10,Reference Huezo-Diaz, Uher, Smith, Rietschel, Henigsberg and Marusic13,Reference Kraft, Peters, Slager, Jenkins, Reinalda and McGrath37 except for one report of borderline statistical significance, uncorrected for multiple testing, coming from a small study of 96 participants in which one of the groups (non-responders) contained only 17 individuals. Reference Kraft, Slager, McGrath and Hamilton32 Given the above, we decided that there were insufficient grounds for modifying the predefined design of this study by incorporating this marker at SLC6A4.
Our results appear to contradict earlier study findings, including the meta-analysis of Serretti et al, Reference Serretti, Kato, De and Kinoshita9 but the latter authors did not investigate or discuss the possibility of a publication bias accounting for their findings. Furthermore, our results are supported by the large STAR*D sample that – at least in the first published analysis – found no association between 5-HTTLPR and outcome. Reference Hu, Rush, Charney, Wilson, Sorant and Papanicolaou10 The subsequent STAR*D analysis on the more ethnically homogeneous subsample was only of borderline significance, Reference Mrazek, Rush, Biernacka, O'Kane, Cunningham and Wieben11 and used a different definition of remission which was not fully justified in the published papers. In the future it would be useful to combine samples and use similar analytical approaches in order to arrive at some consensus about these findings. The generalisability of these results also needs discussion. The presentation and treatment of depression varies between different health systems. Furthermore, it is difficult to obtain a truly representative sample of people, even with treated depression, because of its presentation in primary care. However, it is reassuring that the response rates to antidepressants in our study were similar to those reported in STAR*D, Reference Trivedi, Rush, Wisniewski, Nierenberg, Warden and Ritz29 a study that also attempted to obtain a broadly representative sample of people with depression within the very different US healthcare system. We therefore think that our conclusion that 5-HTTLPR does not influence antidepressant response is at least generalisable to other populations of European origin.
We have considered three possible explanations for our negative result in relation to the 5-HTTLPR gene. First, that the 5-HTTLPR does not alter the availability of 5-HT transporters in the adult human brain. This hypothesis has some support from recent ligand imaging studies that have failed to find convincing differences in 5-HT transporter availability according to 5-HTTLPR. Reference Murthy, Selvaraj, Cowen, Bhagwagar, Riedel and Peers8 Second, even if 5-HT transporter availability is altered, that the influence of 5-HTTLPR on SSRI-induced changes in serotonin neurotransmission is minimal. The SSRIs block over 80% of available 5-HT transporters at minimum therapeutic doses, Reference Meyer, Wilson, Ginovart, Goulding, Hussey and Hood38 so therapeutic response is unlikely to be affected by relatively small differences in transporter availability. Furthermore, even if this polymorphism affects the number of transporter molecules it is possible that compensatory changes in postsynaptic or presynaptic mechanisms might act to maintain steady 5-HT transmission. Neurodevelopmental processes could also have led to brain changes earlier in life in order to minimise any impact that the 5-HTTLPR polymorphism might have on brain function in adults.
The final explanation we have considered is that genetic effects on pharmacological response could be very small. It has become apparent that even if there is a substantial genetic component to a complex disorder this is likely to result from the action of a large number of alleles that individually have a small effect at population level, either because alleles with large effects in individuals are rare in the population, or because common alleles have weak effects on individual risk. Reference Risch39 We cannot rule out a small influence of the 5-HTTLPR variant on outcome, but we think it is unlikely that variation at this locus provides sufficient information to be clinically useful. Of course, it is possible that the predictive value could be improved by combining information from several variants within the gene, or from several genes. Reference Evans, Visscher and Wray40 We think it may be useful to think of recovery from an illness such as depression in the same way as we think of the aetiology of complex diseases. Reference Rothman and Greenland41 Recovery from depression is also a multifaceted process that will be influenced by a large number of biological, psychological and social variables. In order to study genes affecting outcome, it is likely that we will need to adopt a similar approach to that used for studying the genetic aetiology of illness, always remembering the other environmental and treatment factors that might also be important.
Funding
The study was funded by the Medical Research Council and supported by the Mental Health Research Network.
Acknowledgements
We are grateful for the support of the patients who agreed to participate and their general practitioners. We would like to thank the members of the trial steering committee: Ian Anderson (Chair), Tony Johnson, John Geddes and Rodney Elgie, and members of the data monitoring committee: Linda Gask (Chair), Nick Freemantle and Irwin Nazareth. We thank the people who contributed towards the fieldwork, including the following: Helen Lester, Laura Webber, Morag Turnbull, Louise Paterson, Ben Newton, Alex Smith, Nicola Morris, Leigh Franks, Joy Farrimond, Nathan Filer, Caitlin Jarrett and Angela Hill.







eLetters
No eLetters have been published for this article.