


Acute agitation, represented by a state of motor restlessness and accompanying mental tension, is a serious medical problem that can present in a number of psychiatric disorders, including schizophrenia Reference Osser and Sigadel1 and bipolar mania. Reference Alderfer and Allen2 Characterised by symptoms that include pacing, hand wringing, fist clenching, pressured speech, yelling and threatening other people, Reference Battaglia3 agitation may escalate and necessitate physical restraint or seclusion to protect the individual, care providers and others in the immediate environment. Rapid, effective, and safe intervention that does not produce excessive sedation is important in returning the agitated person to a less aroused and less potentially dangerous state, thereby facilitating further assessment of the individual and their treatment options.
Antipsychotic drugs administered with or without supplemental benzodiazepines are the current standard of care in the acute treatment of agitation. Reference Currier and Trenton4–Reference Zimbroff10 The Expert Consensus Guidelines for Treatment of Behavioral Emergencies cite speed of onset as one of the most important factors in choosing a route of medication administration. Reference Allen, Currier, Hughes, Reyes-Harde and Docherty11 Although intravenous administration of antipsychotic drugs affords a rapid onset of action, it is often impractical unless intravenous access is already established. Reference Allen, Currier, Hughes, Reyes-Harde and Docherty11 As a result, oral and intramuscular administrations are more commonly used, but these routes can entail a notably delayed onset of action. For example, controlled studies of intramuscular antipsychotics demonstrate a statistically significant difference from placebo in agitation from 15 to 60 min. Reference Breier, Meehan, Birkett, David, Ferchland and Sutton12–Reference Battaglia, Lindborg, Alaka, Meehan and Wright15 During such a delay, symptoms can escalate. In addition, intramuscular administration is often resisted by individuals, further increasing the risk of escalating symptoms. Therefore, there is a clear need for novel anti-agitation treatments that are rapid in onset, well tolerated, easy to administer and accepted by individuals and staff.
People with schizophrenia are highly vulnerable to acute episodes of agitation, especially during exacerbation of the disease. Loxapine, which was introduced more than 25 years ago in the USA, Canada and Europe, has a well-established efficacy and safety profile in the treatment of schizophrenia, Reference Heel, Brogden, Speight and Avery16–Reference Chakrabarti, Bagnall, Chue, Fenton, Palaniswamy and Wong17 and an intramuscular injection formulation has been shown to be effective in the treatment of agitation. Reference Paprocki and Versiani18–Reference Gaussares, Gerard and Bosc21 In France, intramuscular loxapine is frequently used in the emergency department setting for the acute treatment of agitation. Reference Bourdinard and Pochard22 The antipsychotic effects of loxapine are similar to those of other antipsychotic drugs such as haloperidol, and are likely attributable to its action at dopamine D2 receptors. Reference Heel, Brogden, Speight and Avery16 There is limited evidence that loxapine shares some of its clinical effects with atypical antipsychotic drugs, such as clozapine and olanzapine, Reference Glazer23 including action at 5-hydroxytryptamine (5-HT2A) receptors although the immediate impact on agitation at these receptors is not known.
In the present study in people with schizophrenia and agitation, loxapine was delivered by inhalation using the Staccato system (product also known as AZ–004), a proprietary, breath-actuated delivery system that delivers loxapine with intravenous-like pharmacokinetics. Reference Spyker, Munzar and Cassella24 A previous phase I study in healthy volunteers (0.62–10 mg doses Reference Spyker, Munzar and Cassella24 ) and a phase II study in participants with schizophrenia or schizoaffective disorder (5 and 10 mg doses Reference Cassella, Fishman and Spyker25 ) demonstrated that inhaled loxapine was well-tolerated and, in participants with schizophrenia, had dose-related anti-agitation effects without evidence of excessive sedation. This first phase III study, conducted in an in-patient setting, evaluated the efficacy and safety of inhaled loxapine in treating agitation in individuals with schizophrenia (ClinicalTrials.gov number NCT00628589). Based on the results of the phase II study, dose levels of 5 and 10 mg were evaluated in this phase III study.
Method
Participants
The study enrolled agitated people with schizophrenia (according to DSM–IV criteria). 26 The DSM–IV criteria were applied by research trained psychiatrists on the basis of clinical presentation, psychiatric examination, known previously documented diagnosis when available and by history provided by a second source when available. Males and females, 18 to 65 years old, were drawn from the following settings: individuals admitted to a hospital setting or a research unit in order to be enrolled; people already hospitalised for treatment of schizophrenia who had agitation; and individuals who were treated at a psychiatric emergency department that could allow extended patient stays in a secluded observation room for the period of the study.
At screening, agitation was evaluated by the Positive and Negative Syndrome Scale–Excited Component (PANSS–EC). Reference Kay, Fiszbein and Opler27,Reference Kay28 Individuals were eligible if they had a total score of ≥14 (out of 35) and a score ≥4 (out of 7) on at least one of the five items. In addition, individuals were confirmed to be in good general health as assessed by medical history, physical examination, 12-lead electrocardiogram, and standard serum chemistry, haematology and urinalysis tests. (An investigator could choose to enrol the person while laboratory results were pending if the individual appeared to be in good health based on the other assessments.) Females were non-pregnant and non-lactating.
Key exclusion criteria were agitation primarily because of acute intoxication; a urine drug screen positive for psychostimulants; a history of drug or alcohol dependence in the previous 2 months; a serious risk of suicide; use of benzodiazepines or other hypnotics or oral or short-acting intramuscular antipsychotic drugs in the 4 h before study treatment; use of injectable depot antipsychotics within a one-dose interval before study treatment; use of an investigational drug in the 30 days before screening; clinically significant acute or chronic pulmonary disease; or clinically significant hepatic, renal, gastroenterological, cardiovascular, endocrinological, neurological or haematological disease.

Fig. 1 Staccato drug-device combination product.
Inhalation initiates controlled rapid heating of excipient-free loxapine to form a thermally generated, highly pure drug vapour that condenses into aerosol particles with a size distribution appropriate for efficient delivery to the deep lung.
At baseline, in the 30 min before study treatment, agitation was reconfirmed using the PANSS–EC scale (i.e. a total score of ≥14 and a score ≥4 on at least one item). Individuals not meeting this criterion at baseline were not treated but could be re-evaluated for eligibility in the subsequent 24 h.
Study drug
Inhaled loxapine was delivered by the Staccato system, a single-dose, single-use, hand-held drug-device combination product (Fig. 1). Reference Spyker, Munzar and Cassella24,Reference Rabinowitz, Lloyd, Munzar, Myers, Cross and Damani29,Reference Rabinowitz, Wensley, Lloyd, Myers, Shen and Lu30 For each dose, trained study centre staff instructed the participant to take a deep breath through the product, followed by a short breath hold. Oral inhalation through the product initiates the controlled rapid heating of a thin film of excipient-free loxapine on the sealed stainless steel heat package to form a thermally generated, highly pure drug vapour. The vapour condenses into aerosol particles with a particle size distribution appropriate for efficient delivery to the deep lung. The rapid absorption of the drug provides peak plasma levels in the systemic circulation with a median T max (25, 75 percentiles) of 2 (1, 3) min after administration. Reference Spyker, Munzar and Cassella24
Study design
This was a multicentre, randomised, double-blind, repeat-dose (as required), placebo-controlled, parallel-group study conducted at 24 psychiatric research facilities in the USA between February and June 2008. The study objectives were to confirm the efficacy and safety of the 5 and 10 mg dose levels of inhaled loxapine in the treatment of acute agitation in people with schizophrenia and to confirm the tolerability of up to 3 doses administered in a 24 h period.
Baseline assessments, which were conducted in the 30 min before study treatment, were the PANSS–EC scale, the Clinical Global Impression–Severity scale Reference Guy31 (CGI–S, a pre-treatment assessment of agitation), the Agitation–Calmness Evaluation Scale Reference Meehan, Wang, David, Nisivoccia, Jones and Beasley32 (ACES, a scale developed Eli Lilly and Company) and vital signs measurements. Eligible participants were then randomised to inhaled loxapine 5 mg, inhaled loxapine 10 mg or inhaled placebo. Randomisation was 1:1:1, with a block size of three. Participants, investigators and the sponsor of the study remained masked to treatment assignment. Bilcare Inc (Phoenixville, Philadelphia) produced the computer-generated randomisation sequence and applied masked labels to the study drug; the label displayed the patient number, but the treatment assignment was obscured by a scratch-off coating. Patient numbers were assigned sequentially by the investigator from the allotment of study drug provided to the centre. After study completion, labels were returned to the sponsor and inspected to confirm that treatment assignment remained masked.
After randomisation, dose one was administered and the 24 h evaluation period began. If necessary, a maximum of three doses of the study drug were allowed during that 24 h period: if agitation did not subside sufficiently after dose one or it recurred, dose two could be given >2 h after dose one (after completion of the 2 h assessments); if necessary, dose three could be given ≥4 h after dose two. Unless medically required, rescue medication (intramuscular lorazepam) was not allowed until after the 2 h assessments had been completed, dose two had been given, and at least 20 min had elapsed after the last dose of the study drug. Participants who received lorazepam rescue medication were not eligible to receive additional doses of the study drug.
Throughout the 24 h evaluation period, participants were not to receive any other psychotropic drug that, in the opinion of the investigator, would confound study efficacy or safety end-points. Previously prescribed drugs for extrapyramidal symptoms were prohibited, as was prophylaxis for extrapyramidal symptoms. Participants developing extrapyramidal symptoms were to be treated with anti-Parkinsonian or antihistaminic agents as clinically indicated.
After the 24 h evaluation period ended, all study assessments had been completed, and at least 12 h had elapsed after the last dose of the study drug, participants were discharged or stayed in the hospital depending on their clinical status and the judgement of the investigator.
The study design was reviewed and approved by independent institutional review boards (Western Institutional Review Board, Olympia, Washington; University of California, San Diego Human Research Protections Program, La Jolla, California; Schulman Associates Institutional Review Board, Cincinnati, Ohio; Albert Einstein Healthcare Network, Philadelphia, Philadelphia). The study was conducted in compliance with institutional review board requirements, informed consent regulations and the International Conference on Harmonisation Good Clinical Practice Guidelines. 33 All participants provided written informed consent.
Efficacy measures
During the 24 h evaluation period, two scales were used to assess agitation: the PANSS–EC scale and the CGI–Improvement (CGI–I) scale. Reference Guy31 The PANSS–EC scale measures the following five symptoms associated with agitation: poor impulse control, tension, hostility, uncooperativeness and excitement. Each symptom is rated on a scale of one (absent) to seven (extreme) and scores are summed. Therefore, total scores can range from 5 (all symptoms absent) to 35 (all symptoms extreme). Participants were evaluated with the PANSS–EC scale at 10, 20, 30 and 45 min and 1, 1.5, 2, 4 and 24 h after dose one. The CGI–I scale was used to assess the change from baseline agitation. Scores range from one (very much improved) to seven (very much worse). Participants were evaluated using the CGI–I scale at 2 h after dose one. Raters were trained and certified in the use of the PANSS–EC and CGI scales by one of the authors (M.H.A.) and i3 Research (Basking Ridge, New Jersey).
The primary end-point was the change from baseline in the PANSS–EC score 2 h after dose one of inhaled loxapine compared with the change from baseline after inhaled placebo. The key secondary efficacy end-point was the absolute CGI–I score 2 h after dose one of inhaled loxapine compared with inhaled placebo. Additional efficacy end-points included the changes from baseline in the PANSS–EC scores at each assessment time from 10 min through to 24 h after dose one (both 5 and 10 mg v. placebo, with the 5 mg/placebo comparison done as a post hoc analysis); an analysis of CGI–I ‘responders’ at 2 h (i.e. the number of participants with scores of one (very much improved) or two (much improved) (both 5 and 10 mg v. placebo); and the time to dose two (both 5 and 10 mg v. placebo).
Safety and tolerability measures
Safety was assessed by treatment-emergent adverse events, periodic assessments of vital signs (systolic and diastolic blood pressure, heart rate and respiratory rate), and pre- and post-treatment physical examinations and laboratory tests (serum chemistry, haematology and urinalysis). Adverse events were recorded when identified by the study staff or reported by the participant, as were medications used to treat adverse events. The severity of each adverse event and its relationship to the study drug were evaluated by the investigator. Furthermore, any adverse events reported in the 30 days after discharge from the study were to be recorded and followed, although no such adverse events were reported. Clinically significant changes from baseline in blood chemistry, haematology and urinalysis parameters were assessed before discharge from the study.
Sedation was assessed using the ACES, which rates the participant on an agitated–calm–sleeping continuum. Scores range from one (marked agitation) to nine (unarousable), with a score of four indicating ‘normal’. Participants were evaluated using the ACES at 2 h after dose one. Raters were trained in use of the ACES by i3 Research (Basking Ridge, New Jersey).
Statistical methods
Power calculations were based on the results of the previous phase II study of inhaled loxapine. For the primary efficacy end-point, 100 participants per group (300 participants total) would provide 99% statistical power for the 10 mg/placebo pair-wise comparison and 79% statistical power for the 5 mg/placebo pair-wise comparison (family-wise α = 0.05 for the two active/placebo comparisons).
The efficacy population was the intent-to-treat (ITT) population, which included all participants who received any study drug and had both baseline and at least 1 post-dose efficacy assessment or used rescue medication before 2 h after dosing. In this population, missing values were replaced using the last-observation-carried-forward (LOCF), except in the survival analysis, which used the standard right-censoring approach. Observations recorded after the use of rescue medications were considered missing and subject to the LOCF algorithm. The safety population included all participants who were treated.
An α-level of 0.05 was the criterion for all statistical comparisons. Furthermore, by the following multiple-comparison statistical methods, a family-wise α-level of 0.05 was maintained for the primary efficacy end-point (5 and 10 mg v. placebo), the key secondary efficacy end-point (5 and 10 mg v. placebo) and the change in the PANSS–EC score from baseline to 45, 30, 20 and 10 min after dose one (10 mg v. placebo only). Statistical testing of the primary efficacy end-point – change in PANSS–EC score from baseline to 2 h after dose one – was performed via analysis of covariance (ANCOVA) with terms for baseline PANSS–EC score, treatment and centre, using a global F-test and pair-wise Dunnett's t-tests for the two follow-up loxapine/placebo pair-wise comparisons. If the primary efficacy end-point was statistically significant on the global F-test, the key secondary end-point – CGI–I score at 2 h after dose one – was tested via analysis of variance (ANOVA) with terms for treatment and centre, using a global F-test and Dunnett's t-tests for the two follow-up pair-wise comparisons. If the primary and key secondary end-points were both statistically significant for the 10 mg/placebo pair-wise comparisons, then the 10 mg/placebo PANSS–EC scores at 45, 30, 20, and 10 min after dose one were tested using a downward step-wise testing rule via ANCOVA with the same structure as used for the primary end-point.
Although not protected as part of the family-wise α-level, changes in PANSS–EC score from baseline to 1, 1.5, 4, and 24 h after dose one were also analysed with ANCOVA using the same structure as for the primary end-point. Active/placebo pair-wise comparisons of the frequency distributions of CGI–I responders/non-responders (i.e. CGI–I responder analysis; responders were participants with a CGI–I score of one or two) were made using Fisher's exact tests. The time to use of an as-required dose two during the 24 h after dose one was analysed using Kaplan–Meier survival curves and log rank tests. The ACES scores at 2 h after dose one were summarised with descriptive statistics.

Fig. 2 Study flow chart.
Testing of the PANSS–EC 5 mg/placebo comparisons at time points other than 2 h was not included in the statistical analysis plan, but post hoc tests were carried out at all other time points with ANCOVA using the same structure as for the primary end-point.
Results
Study population
In total 344 participants were randomised, all received at least one dose of the study drug (loxapine or placebo) and 338 completed the study. Participant disposition and the numbers of participants included in the efficacy analysis populations are summarised inFig. 2 (all 344 participants were included in the safety analysis). Participant characteristics and baseline disease severity are summarised inTable 1. Most participants had had schizophrenia for many years and had multiple previous hospitalisations. At baseline, the mean PANSS–EC scores were similar in the three groups (placebo group: 17.4; 5 mg group: 17.8; 10 mg group, 17.6) (Fig. 3). Mean CGI–S scores were also similar in the three groups (placebo group: 3.9; 5 mg group: 4.0; 10 mg group: 4.1), corresponding to moderate agitation. Adherence to the treatment regimen was high: once randomised, no participants dropped out because of failure to follow the inhalation instructions or an inability (or refusal) to take a dose of the study drug. Notable protocol deviations were infrequent, affecting a similar number of participants in each treatment group (placebo group: 4/115; 5 mg group: 4/116; 10 mg group: 3/113). No deviation was judged to have affected the findings of the study.
Table 1 Baseline characteristics and disease severity (safety population)
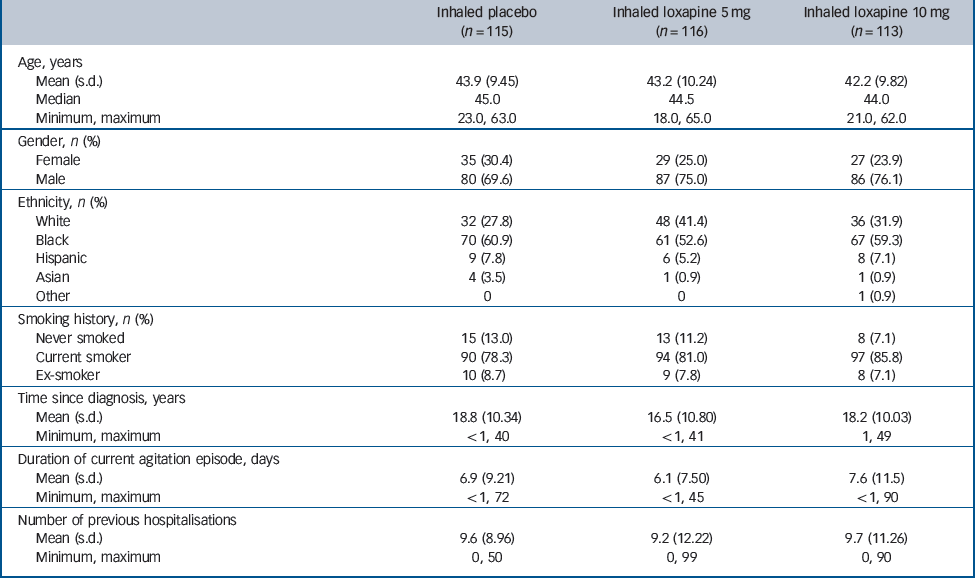
| Inhaled placebo (n = 115) | Inhaled loxapine 5 mg (n = 116) | Inhaled loxapine 10 mg (n = 113) | |
|---|---|---|---|
| Age, years | |||
| Mean (s.d.) | 43.9 (9.45) | 43.2 (10.24) | 42.2 (9.82) |
| Median | 45.0 | 44.5 | 44.0 |
| Minimum, maximum | 23.0, 63.0 | 18.0, 65.0 | 21.0, 62.0 |
| Gender, n (%) | |||
| Female | 35 (30.4) | 29 (25.0) | 27 (23.9) |
| Male | 80 (69.6) | 87 (75.0) | 86 (76.1) |
| Ethnicity, n (%) | |||
| White | 32 (27.8) | 48 (41.4) | 36 (31.9) |
| Black | 70 (60.9) | 61 (52.6) | 67 (59.3) |
| Hispanic | 9 (7.8) | 6 (5.2) | 8 (7.1) |
| Asian | 4 (3.5) | 1 (0.9) | 1 (0.9) |
| Other | 0 | 0 | 1 (0.9) |
| Smoking history, n (%) | |||
| Never smoked | 15 (13.0) | 13 (11.2) | 8 (7.1) |
| Current smoker | 90 (78.3) | 94 (81.0) | 97 (85.8) |
| Ex-smoker | 10 (8.7) | 9 (7.8) | 8 (7.1) |
| Time since diagnosis, years | |||
| Mean (s.d.) | 18.8 (10.34) | 16.5 (10.80) | 18.2 (10.03) |
| Minimum, maximum | <1, 40 | <1, 41 | 1, 49 |
| Duration of current agitation episode, days | |||
| Mean (s.d.) | 6.9 (9.21) | 6.1 (7.50) | 7.6 (11.5) |
| Minimum, maximum | <1, 72 | <1, 45 | <1, 90 |
| Number of previous hospitalisations | |||
| Mean (s.d.) | 9.6 (8.96) | 9.2 (12.22) | 9.7 (11.26) |
| Minimum, maximum | 0, 50 | 0, 99 | 0, 90 |
Efficacy
The primary efficacy end-point – the change in the PANSS–EC score from baseline to 2 h after dose one – demonstrated the efficacy of both doses of inhaled loxapine compared with inhaled placebo (Fig. 3). The overall treatment effect was highly statistically significant (P<0.0001), and both the 5 and 10 mg doses resulted in significantly larger decreases in the PANSS–EC score from baseline to 2 h, relative to inhaled placebo (5 mg group/placebo group, P = 0.0004; 10 mg group/placebo group, P<0.0001). Furthermore, inhaled loxapine produced a rapid onset of effect as assessed by the PANSS–EC score (Fig. 4), with highly statistically significant differences between inhaled loxapine and inhaled placebo at 10 min after dose one, the earliest assessment time (10 mg group/placebo group planned comparison, P<0.0001; 5 mg group/placebo group post hoc comparison, P = 0.0003). A continued treatment effect was evident at all subsequent assessments throughout the 24 h period after dose one (10 mg group/placebo group, P<0.0001; 5 mg group/placebo group, P<0.05).
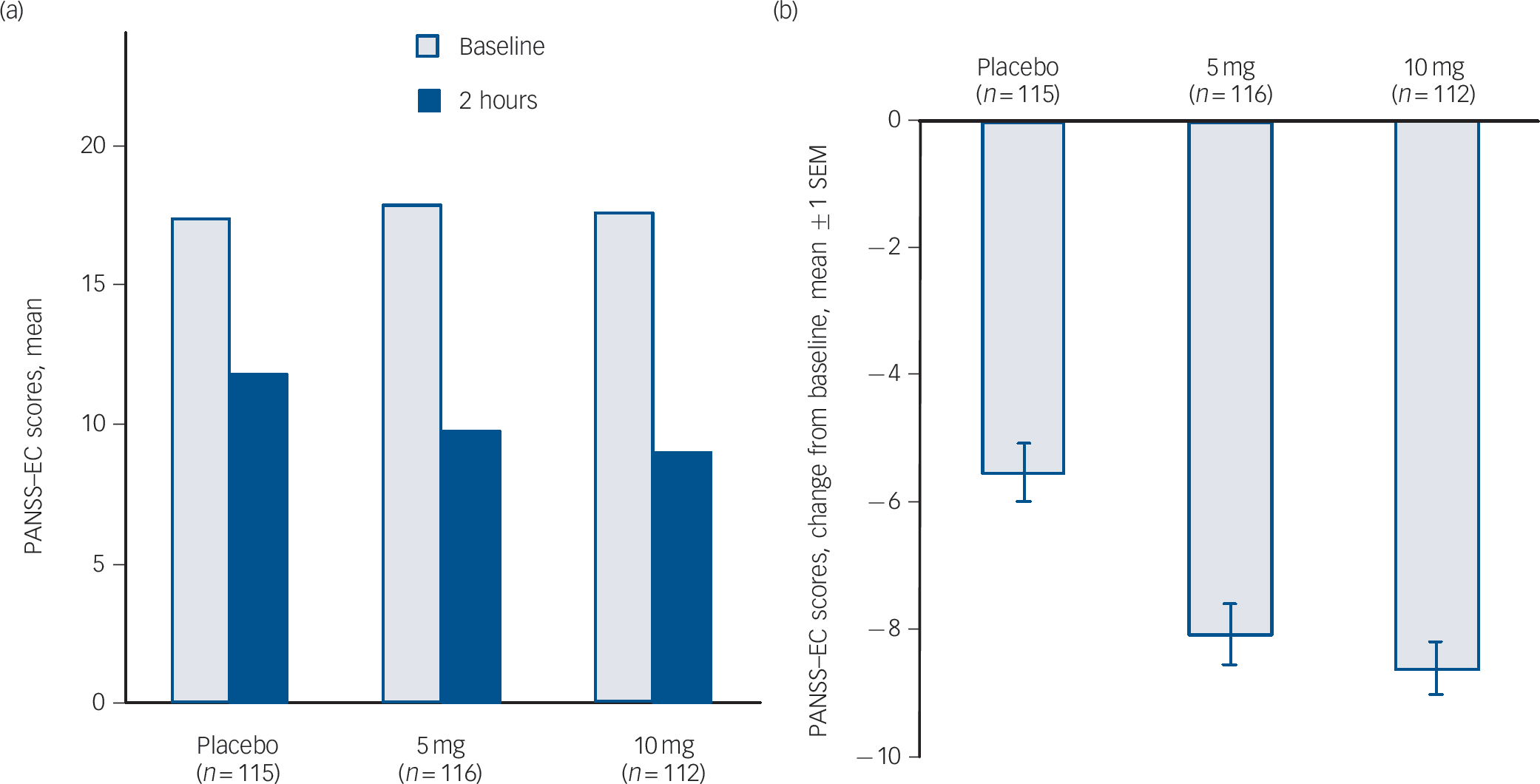
Fig. 3 Positive and Negative Syndrome Scale–Excited Component (PANSS–EC) scores in intention-to-treat population.
(a) PANSS–EC at baseline and 2 h assessment. (b) Primary end-point – change in PANSS–EC from baseline to 2 h assessment: highly statistically significant decreases in PANSS–EC score in 5 and 10 mg groups compared with placebo. SEM, standard error of the mean.
The key secondary efficacy end-point – the CGI–I score at 2 h after dose one – provided additional evidence of the efficacy of inhaled loxapine. Both 5 and 10 mg doses resulted in statistically significant decreases in agitation compared with inhaled placebo, as assessed by the mean CGI–I score (placebo group: 2.8; 5 mg group: 2.3; 10 mg group: 2.1 (where 2 is much improved and 3 is minimally improved); overall, P<0.0001; 5 mg group/placebo group P = 0.0015; 10 mg group/placebo group, P<0.0001). In addition, the CGI–I responder analysis, which evaluated the percentage of participants with scores of one (very much improved) or two (much improved) at 2 h after dose one, demonstrated that significantly more individuals in the 5 mg group (66/116) and the 10 mg group (75/112) were judged to be very much improved or much improved compared with participants in the placebo group (41/115) (5 mg group/placebo group: P = 0.0015; 10 mg group/placebo group, P<0.0001).
Figure 5 shows the Kaplan–Meier survival analysis of the time to dose two of the study drug (an as-required dose). The overall difference favouring the groups receiving inhaled loxapine over the placebo group was statistically significant (P = 0.0239). The 10 mg group/placebo group pair-wise comparison was statistically significant (P = 0.0076). The 5 mg group/placebo group pair-wise comparison did not achieve statistical significance (P = 0.1155) but a numerical difference favouring the 5 mg group was evident.
Safety and tolerability
Inhaled loxapine was well tolerated. The percentage of participants who had at least one adverse event was similar in the placebo and loxapine groups (placebo group: 44/115; 5 mg group: 40/116; 10 mg group: 43/113), and most events were judged to be of mild or moderate severity and resolved without intervention.
The most common adverse events in participants receiving inhaled loxapine were sedation, dysgeusia and dizziness (Table 2). Wheezing or bronchospasm was reported in three participants treated with inhaled loxapine: one participant receiving the 10 mg dose had moderate bronchospasm that resolved with use of an inhaled bronchodilator (albuterol, two puffs by metered-dose inhaler) and led to withdrawal from the study; two participants receiving the 5 mg dose had mild wheezing that resolved without treatment. Only one participant reported cough (10 mg group), which was judged to be mild and possibly treatment related and it resolved without intervention.
Table 2 Treatment-emergent adverse events in =2% of participants in any treatment group (safety population)
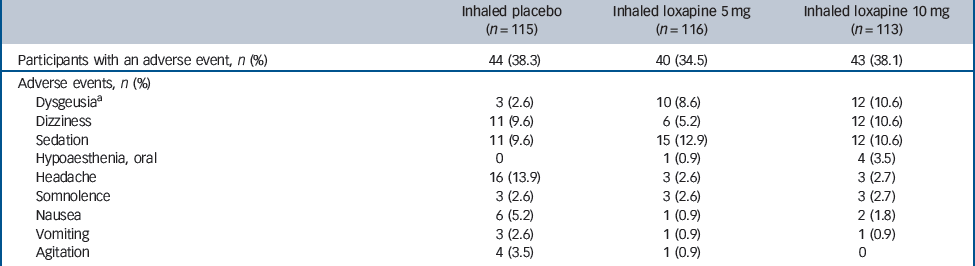
| Inhaled placebo (n = 115) | Inhaled loxapine 5 mg (n = 116) | Inhaled loxapine 10 mg (n = 113) | |
|---|---|---|---|
| Participants with an adverse event, n (%) | 44 (38.3) | 40 (34.5) | 43 (38.1) |
| Adverse events, n (%) | |||
| Dysgeusiaa | 3 (2.6) | 10 (8.6) | 12 (10.6) |
| Dizziness | 11 (9.6) | 6 (5.2) | 12 (10.6) |
| Sedation | 11 (9.6) | 15 (12.9) | 12 (10.6) |
| Hypoaesthenia, oral | 0 | 1 (0.9) | 4 (3.5) |
| Headache | 16 (13.9) | 3 (2.6) | 3 (2.7) |
| Somnolence | 3 (2.6) | 3 (2.6) | 3 (2.7) |
| Nausea | 6 (5.2) | 1 (0.9) | 2 (1.8) |
| Vomiting | 3 (2.6) | 1 (0.9) | 1 (0.9) |
| Agitation | 4 (3.5) | 1 (0.9) | 0 |
Severe adverse events were reported for three people in the 10 mg group and three in the placebo group. In the 10 mg group, one participant experienced concurrent severe adverse events of neck dystonia and oculogyration that were judged probably treatment related, were treated with benztropine and resolved; one person experienced severe sedation (judged probably treatment related and resolved without intervention); and one person experienced severe infectious gastroenteritis (judged unrelated to treatment, involved hospitalisation and resolved), which was the only serious adverse event in a participant receiving inhaled loxapine. In the placebo group, one participant had a severe exacerbation of schizophrenia that resulted in hospitalisation and was judged by the investigator to be unrelated to study treatment (the only other serious adverse event in the study); one participant had severe headache and nausea that resolved with medication; and one participant had severe agitation that resolved with medication.
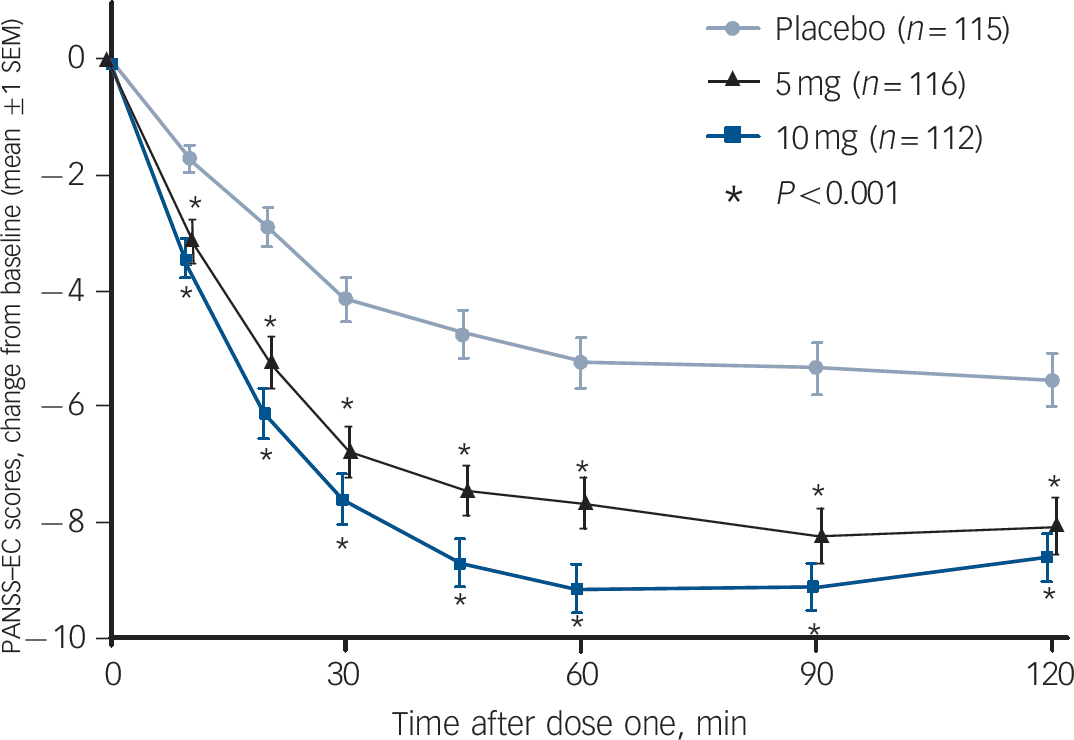
Fig. 4 Positive and Negative Syndrome Scale–Excited Component (PANSS–EC) time-course analysis up to 2 h.
Inhaled loxapine was rapidly effective, with highly statistically significant active–placebo differences 10 min after dose one, the earliest assessment time and significant differences at all subsequent assessments (intention-to-treat population). The testing of the 10 mg dose was planned and the 5 mg dose testing was post hoc.
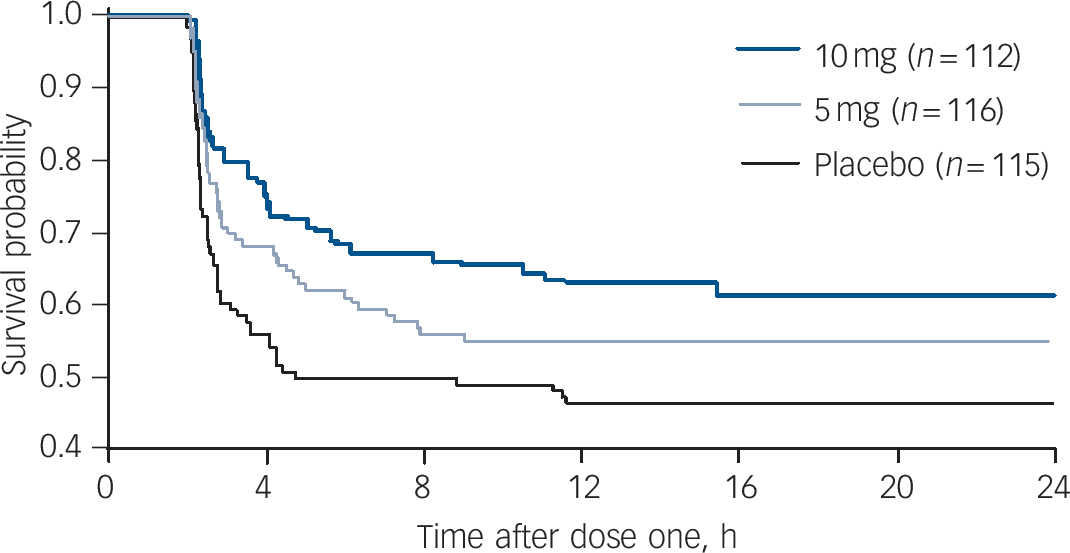
Fig. 5 Survival analysis of time to administration of dose two of study drug (as needed dose).
Participants taking the placebo took dose two significantly sooner than those taking loxapine (Kaplan–Meier overall comparison). In pair-wise comparisons 10 mg/placebo was statistically significant, whereas 5 mg/placebo was not. Lorazepam rescue medication was received by 6, 7 and 18 participants in the 10 mg, 5 mg and placebo groups respectively.
The ACES, which rates individuals on an agitated–calm–sleeping continuum, indicated that the anti-agitation effect was achieved without producing undue sedation: 2 h after dose one, no participants had a score of nine (unarousable) and the mean ratings for the groups receiving inhaled loxapine were in the range of ‘mild calmness’ (5 mg group: 4.7; 10 mg group: 4.9; where 4 is normal and 5 is mild calmness).
Discussion
Main findings
Four main findings emerged from the present study. First, both the 5 and 10 mg doses of inhaled loxapine produced significant improvement compared with placebo in the primary and key secondary efficacy end-points. Additional efficacy assessments (CGI–I responder analysis; additional PANSS–EC time points from 10 min to 24 h; time to use of dose two) provide additional support for the efficacy of inhaled loxapine in reducing agitation in people with schizophrenia. Second, superior anti-agitation effects of inhaled loxapine compared with placebo, as reflected in the PANSS–EC score, were evident at 10 min after dose one, the earliest time point measured. To our knowledge, this 10 min onset demonstrated with inhaled loxapine in this study is the most rapid anti-agitation effect reported in a placebo-controlled trial. Third, inhaled loxapine was well tolerated. The most common adverse events were known effects of loxapine or minor oral effects common with inhaled medications. Finally, no participants refused treatment or were unable to take a dose of the study drug, indicating good ease of use and potential for patient acceptance. Taken together, these results support the conclusion that inhaled loxapine provides a rapid, effective and safe treatment option that may be more acceptable to some individuals with schizophrenia and staff than conventional parenteral administration.
Limitations
There are several potential limitations to the present study. First, the participants received treatment in controlled, in-patient settings. Additional investigation will be needed to evaluate the use of inhaled loxapine in more naturalistic settings. Also, the individuals involved in the study had to be able to provide informed consent and as such may not be completely representative of the population presenting with agitation in the real-world setting. Furthermore, although the PANSS–EC is commonly used to assess agitation in clinical studies, agitation assessment and treatment decisions in the real world are not based on this instrument. Consequently, the real-world clinical significance of changes in the PANSS–EC is unknown, although the significant findings with the CGI–I provide assurance of a clinically relevant effect.
Clinical implications
The rapid onset of anti-agitation effects seen in this study may represent an improvement relative to oral and intramuscular options in reducing the severe distress and injury risks experienced by both agitated individuals and their caregivers. Reference Buckley34 The potential willingness of individuals to self-administer an inhaled medication versus forcible treatment with intramuscular injections may reduce the risk of escalated agitation associated with involuntary treatment. The need for such an intervention is supported by survey data showing that mechanical restraints were used in a mean of 8.5% of visits to psychiatric emergency services settings. Reference Allen and Currier35 The fact that no participant reported difficulty in using the product supports its ease of use. The ACES assessment indicates that efficacy was achieved without producing undue sedation in the majority of individuals. In summary, inhaled loxapine is a well-tolerated and effective novel treatment of agitation in schizophrenia and potentially other disorders.
Funding
The study was sponsored by Alexza Pharmaceuticals.
Acknowledgements
Additional study investigators were Gustavo Alva, MD (ATP Clinical Research, Inc, Costa Mesa, California); Mohammed A. Bari, MD (Synergy Clinical Research Center, National City, California); Prakash Bhatia, MD, PhD (Synergy Escondido, Escondido, California); Ronald Brenner, MD (Neurobehavioral Research, Inc, Cedarhurst, New York); David W. Brown, MD (Community Clinical Research, Inc, Austin, Texas); Eduardo Cifuentes, MD (Carolina Clinical Trials, Inc, Charleston, South Carolina); David Howard Flaherty, DO (Fidelity Clinical Research, Inc, North Miami, Florida); Donald J. Garcia, Jr, MD (Future Search Trials, Austin, Texas); Lev G. Gertsik, MD (California Clinical Trials Medical Group, Glendale, California); Ramanath Gopalan, MD (Comprehensive Neuroscience, Inc, Arlington, Virginia); Richard Louis Jaffe, MD (Belmont Center for Comprehensive Treatment, Philadelphia, Philadelphia); Arifulla Khan, MD (Northwest Clinical Research Center, Bellevue, Washington); Jelena Kunovac, MD (Excell Research, Oceanside, California); Joseph Kwentus, MD (Precise Research Centers, Flowood, Mississippi); Adam Lowy, MD (Comprehensive Neuroscience, Inc, Washington DC); Raymond A Manning, MD (CNRI-Los Angeles, LLC, Pico Rivera, California); Morteza Marandi, MD (Comprehensive Neuroscience, Inc, Cerritos, California); Scott Daniel Segal, MD (Scientific Clinical Research, Inc, North Miami, Florida); Rajinder Shiwach, MD (InSite Clinical Research, DeSoto, Texas); and David P Walling, MD (Collaborative Neuroscience Network, Inc, Garden Grove, California). Each was compensated. Stephen Horohonich, BS (Cognitive Research Corporation, St Petersburg, Florida) was responsible for data collection and management. William Accomando, DSc (Accomando Consulting Enterprises, Mystic, Connecticut) and Julie Jones, PhD (JJo Inc, Breckenridge, Colorado) were responsible for statistical planning and analysis. Each was compensated.








 Open access
Open access
eLetters
No eLetters have been published for this article.