


Stimulants have been used for many centuries but only latterly have there been reports of associated psychosis (Reference Guttmann and SargantGuttmann & Sargant, 1937), culminating in Connell's monograph (Reference ConnellConnell, 1958), which reviewed cases of ‘stimulant’ psychosis that resolved rapidly. In Japan, where there was an epidemic of injected amphetamine use, the duration of psychosis appeared to be prolonged and chronic (Reference Koyama, Muraki and NakayamaKoyama et al, 1991). The theory was proposed that repeated low doses of a stimulant lead to changes in the central nervous system (CNS) (Reference Ellingwood and KilbeyEllingwood & Kilbey, 1980), a form of ‘kindling’, which produces a psychotic illness similar to schizophrenia. Animal experiments seem to support such an effect (Reference Post and KopandaPost & Kopanda, 1976). Others dispute this theory of sensitisation (e.g. Reference Brabbins and PooleBrabbins & Poole, 1996). If sensitisation is occurring, then early treatment and retention of stimulant users in mental health care services would appear to be desirable to prevent chronic psychoses developing. There is a lack of good-quality evidence as to the effectiveness of this: a recent Cochrane review found no relevant trials (Reference Srisurapanont, Kittiratanapaiboon and JarusuraisinSrisurapanont et al, 2004).
The purpose of this study is to examine evidence for the theory of sensitisation. The hypothesis is that stimulant psychoses can be divided into a ‘toxic’ type of response and a chronic persisting response resulting from longer-term use of stimulants.
METHOD
We searched for experimental and observational studies in humans taking stimulants that investigated or described the development of psychotic symptoms. We did not include case series or cross-sectional studies, as these give little information as to the direction of effect or changes over time.
We performed electronic searches on Medline, PsycLIT and EMBASE psychiatry from the earliest dates available to 2001, using the search terms COCAINE, CRACK, AMPHETAMINE, METHYLAMPHETAMINE, METHAMPHETAMINE, METHAMFETAMINE, d-AMPHETAMINE, DEXAMPHETAMINE, METHYLPHENIDATE, PSYCHOACTIVE DRUGS, CNS STIMULANT DRUGS and DRUG-INDUCED PSYCHOSIS (for stimulants) and PSYCHOSIS, PSYCHOSES, SCHIZOPHRENIA and SCHIZO-AFFECTIVE (for psychoses). Where Medical Subject Headings (MeSH) terms were available, they were exploded and combined. Papers were checked for references to other relevant studies.
Identifying and evaluating the studies
Following the initial searches by C.C., all experimental case–control and longitudinal studies were independently appraised by C.C. and N.B. Any disagreements on whether a study should be included were resolved by reference to the criteria. Three methodologically distinct types of studies were identified, which were reviewed separately. Studies were included if they met the following criteria.
Experimental studies
Studies were included if:
-
(a) participants were given stimulants (cocaine, amphetamines or methylphenidate); and
-
(b) participants were monitored for possible psychotic reactions; and
-
(c) circumstances of administration were controlled for dose, route and timings (if variable doses were given, this was related to dose per kilogram or dose according to physiological response or blood level); and
-
(d) psychosis or changes in psychosis were measured in a standardised fashion.
Longitudinal studies
Studies were included if:
-
(a) a cohort of substance users with or without psychosis, defined by operational criteria, was followed up for a defined period; and
-
(b) stimulant users were identified and differentiated from other substance users in the report.
Case-control studies
Studies were included if:
-
(a) individuals using stimulants with psychosis were compared with those using stimulants with no psychosis; or
-
(b) individuals with psychosis using stimulants were compared with control individuals with psychosis but with no history of drug use; or
-
(c) individuals using stimulants were compared with individuals using non-stimulant substances; and
-
(d) Stimulant users are identified and differentiated from other substance users in the report.
RESULTS
A total of 84 experimental or observational studies were identified by the search and cross-referencing strategies. Initial agreement on studies meeting the criteria in the review was present for 89% of the experimental studies, 82% of the longitudinal studies and 75% of the case–control studies. After discussion between the raters, it was agreed that 43 studies met the criteria and were thus included in the review.
Experimental studies
A total of 32 experimental studies were included (Table 1). Twenty-eight of these involved single doses of oral or intravenous (i.v.) dexamfetamine or methylphenidate given to individuals with schizophrenia, and 9 of these 28 studies included a control group. One of the remaining 4 studies included a heterogeneous group of individuals with psychosis and controls given two doses of dexamfetamine orally 48 h apart (Reference Strakowski, Sax and SettersStrakowski et al, 1997). Two studies involved substance users (Reference Cami, Farre and MasCami et al, 2000; Reference Farren, Hameedi and RosenFarren et al, 2000). The final study (Reference Casey, Hollister and KlettCasey et al, 1961) was a randomised controlled trial of 520 individuals with schizophrenia in which one group received dexamfetamine orally for 20 weeks. All studies used some form of standardised rating scale – most commonly the Brief Psychiatric Rating Scale (BPRS) – to measure changes resulting from stimulant use. A ‘response’ was considered to have occurred when changes were measured in the psychosis component of the various scales. The response to a single dose of stimulant, when present, was brief, seldom lasting more than a few hours.
Table 1 Experimental studies
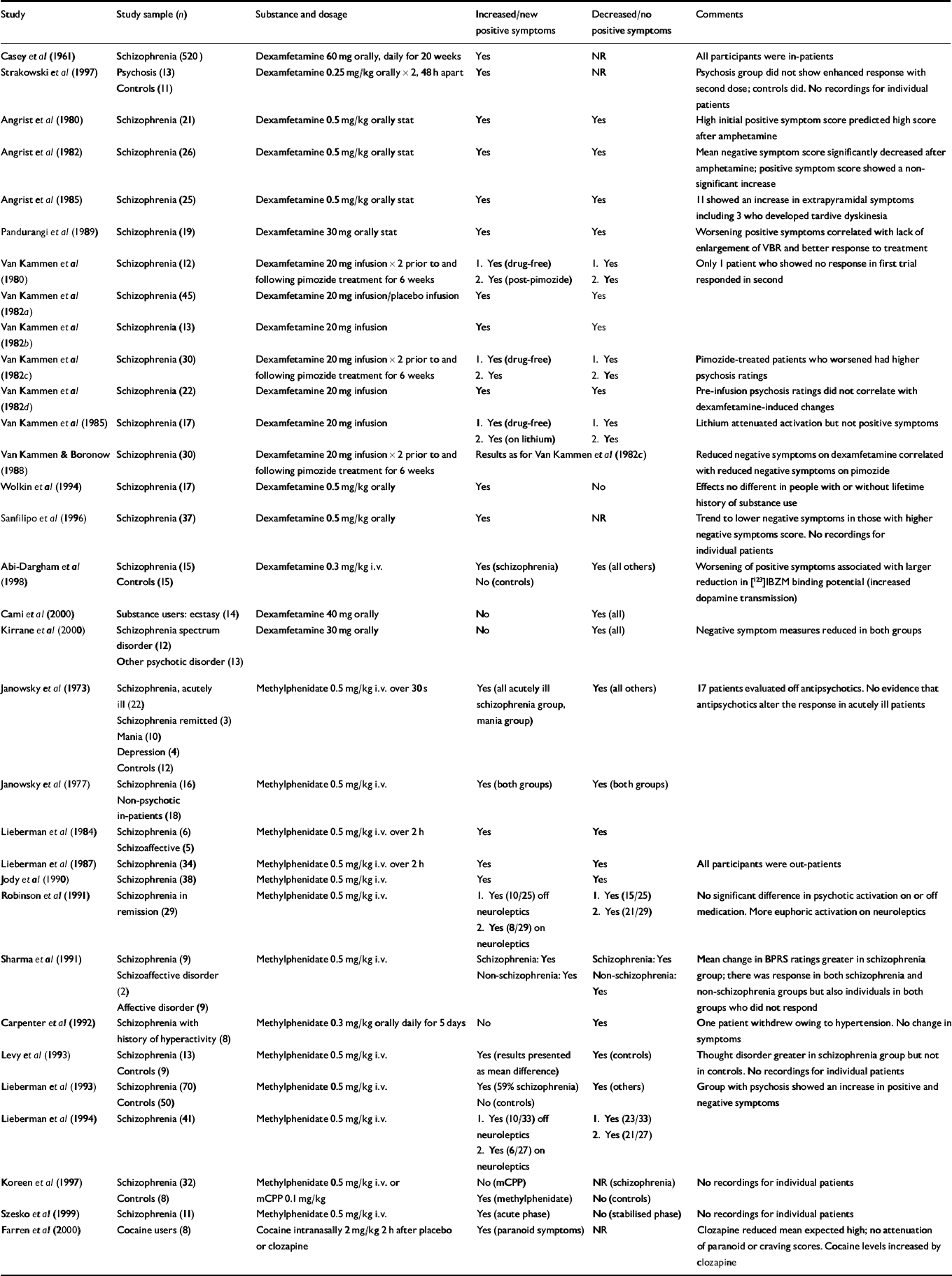
| Study | Study sample (n) | Substance and dosage | Increased/new positive symptoms | Decreased/no positive symptoms | Comments |
|---|---|---|---|---|---|
| Casey et al (Reference Casey, Hollister and Klett1961) | Schizophrenia (520) | Dexamfetamine 60 mg orally, daily for 20 weeks | Yes | NR | All participants were in-patients |
| Strakowski et al (Reference Strakowski, Sax and Setters1997) | Psychosis (13) | Dexamfetamine 0.25 mg/kg orally × 2, 48 h apart | Yes | NR | Psychosis group did not show enhanced response with second dose; controls did. No recordings for individual patients |
| Controls (11) | |||||
| Angrist et al (Reference Angrist, Rosen and Gershon1980) | Schizophrenia (21) | Dexamfetamine 0.5 mg/kg orally stat | Yes | Yes | High initial positive symptom score predicted high score after amphetamine |
| Angrist et al (Reference Angrist, Peselow and Rubinstein1982) | Schizophrenia (26) | Dexamfetamine 0.5 mg/kg orally stat | Yes | Yes | Mean negative symptom score significantly decreased after amphetamine; positive symptom score showed a non-significant increase |
| Angrist et al (Reference Angrist, Peselow and Rubinstein1985) | Schizophrenia (25) | Dexamfetamine 0.5 mg/kg orally stat | Yes | Yes | 11 showed an increase in extrapyramidal symptoms including 3 who developed tardive dyskinesia |
| Pandurangi et al (Reference Pandurangi, Goldberg and Brink1989) | Schizophrenia (19) | Dexamfetamine 30 mg orally stat | Yes | Yes | Worsening positive symptoms correlated with lack of enlargement of VBR and better response to treatment |
| Van Kammen et al (Reference Van Kammen, Docherty and Marder1980) | Schizophrenia (12) | Dexamfetamine 20 mg infusion × 2 prior to and following pimozide treatment for 6 weeks | 1. Yes (drug-free) | 1. Yes | Only 1 patient who showed no response in first trial responded in second |
| 2. Yes (post-pimozide) | 2. Yes | ||||
| Van Kammen et al (Reference Van Kammen, Bunney and Docherty1982a ) | Schizophrenia (45) | Dexamfetamine 20 mg infusion/placebo infusion | Yes | Yes | |
| Van Kammen et al (Reference Van Kammen, Docherty and Bunney1982b ) | Schizophrenia (13) | Dexamfetamine 20 mg infusion | Yes | Yes | |
| Van Kammen et al (Reference Van Kammen, Docherty and Marder1982c ) | Schizophrenia (30) | Dexamfetamine 20 mg infusion × 2 prior to and following pimozide treatment for 6 weeks | 1. Yes (drug-free) | 1. Yes | Pimozide-treated patients who worsened had higher psychosis ratings |
| 2. Yes | 2. Yes | ||||
| Van Kammen et al (Reference Van Kammen, Docherty and Marder1982d ) | Schizophrenia (22) | Dexamfetamine 20 mg infusion | Yes | Yes | Pre-infusion psychosis ratings did not correlate with dexamfetamine-induced changes |
| Van Kammen et al (Reference Van Kammen, Docherty and Marder1985) | Schizophrenia (17) | Dexamfetamine 20 mg infusion | 1. Yes (drug-free) | 1. Yes | Lithium attenuated activation but not positive symptoms |
| 2. Yes (on lithium) | 2. Yes | ||||
| Van Kammen & Boronow (Reference Van Kammen and Boronow1988) | Schizophrenia (30) | Dexamfetamine 20 mg infusion × 2 prior to and following pimozide treatment for 6 weeks | Results as for Van Kammen et al (Reference Van Kammen, Docherty and Marder1982c ) | Reduced negative symptoms on dexamfetamine correlated with reduced negative symptoms on pimozide | |
| Wolkin et al (Reference Wolkin, Sanfilipo and Angrist1994) | Schizophrenia (17) | Dexamfetamine 0.5 mg/kg orally | Yes | No | Effects no different in people with or without lifetime history of substance use |
| Sanfilipo et al (Reference Sanfilipo, Wolkin and Angrist1996) | Schizophrenia (37) | Dexamfetamine 0.5 mg/kg orally | Yes | NR | Trend to lower negative symptoms in those with higher negative symptoms score. No recordings for individual patients |
| Abi-Dargham et al (Reference Abi-Dargham, Gil and Krystal1998) | Schizophrenia (15) | Dexamfetamine 0.3 mg/kg i.v. | Yes (schizophrenia) | Yes (all others) | Worsening of positive symptoms associated with larger reduction in [123]IBZM binding potential (increased dopamine transmission) |
| Controls (15) | No (controls) | ||||
| Cami et al (Reference Cami, Farre and Mas2000) | Substance users: ecstasy (14) | Dexamfetamine 40 mg orally | No | Yes (all) | |
| Kirrane et al (Reference Kirrane, Mitropoulou and Nunn2000) | Schizophrenia spectrum disorder (12) | Dexamfetamine 30 mg orally | No | Yes (all) | Negative symptom measures reduced in both groups |
| Other psychotic disorder (13) | |||||
| Janowsky et al (Reference Janowsky, El-Yousef and Davis1973) | Schizophrenia, acutely ill (22) | Methylphenidate 0.5 mg/kg i.v. over 30 s | Yes (all acutely ill schizophrenia group, mania group) | Yes (all others) | 17 patients evaluated off antipsychotics. No evidence that antipsychotics alter the response in acutely ill patients |
| Schizophrenia remitted (3) | |||||
| Mania (10) | |||||
| Depression (4) | |||||
| Controls (12) | |||||
| Janowsky et al (Reference Janowsky, Huey and Storms1977) | Schizophrenia (16) | Methylphenidate 0.5 mg/kg i.v. | Yes (both groups) | Yes (both groups) | |
| Non-psychotic in-patients (18) | |||||
| Lieberman et al (Reference Lieberman, Kane and Gadaleta1984) | Schizophrenia (6) | Methylphenidate 0.5 mg/kg i.v. over 2 h | Yes | Yes | |
| Schizoaffective (5) | |||||
| Lieberman et al (Reference Lieberman, Kane and Sarantakos1987) | Schizophrenia (34) | Methylphenidate 0.5 mg/kg i.v. over 2 h | Yes | Yes | All participants were out-patients |
| Jody et al (Reference Jody, Lieberman and Geisler1990) | Schizophrenia (38) | Methylphenidate 0.5 mg/kg i.v. | Yes | Yes | |
| Robinson et al (Reference Robinson, Mayerhoff and Alvir1991) | Schizophrenia in remission (29) | Methylphenidate 0.5 mg/kg i.v. | 1. Yes (10/25) off neuroleptics | 1. Yes (15/25) | No significant difference in psychotic activation on or off medication. More euphoric activation on neuroleptics |
| 2. Yes (8/29) on neuroleptics | 2. Yes (21/29) | ||||
| Sharma et al (Reference Sharma, Javaid and Pandey1991) | Schizophrenia (9) | Methylphenidate 0.5 mg/kg i.v. | Schizophrenia: Yes | Schizophrenia: Yes | Mean change in BPRS ratings greater in schizophrenia group; there was response in both schizophrenia and non-schizophrenia groups but also individuals in both groups who did not respond |
| Schizoaffective disorder (2) | Non-schizophrenia: Yes | Non-schizophrenia: Yes | |||
| Affective disorder (9) | |||||
| Carpenter et al (Reference Carpenter, Winsberg and Camus1992) | Schizophrenia with history of hyperactivity (8) | Methylphenidate 0.3 mg/kg orally daily for 5 days | No | Yes | One patient withdrew owing to hypertension. No change in symptoms |
| Levy et al (Reference Levy, Smith and Robinson1993) | Schizophrenia (13) | Methylphenidate 0.5 mg/kg i.v. | Yes (results presented as mean difference) | Yes (controls) | Thought disorder greater in schizophrenia group but not in controls. No recordings for individual patients |
| Controls (9) | |||||
| Lieberman et al (Reference Lieberman, Jody and Alvir1993) | Schizophrenia (70) | Methylphenidate 0.5 mg/kg i.v. | Yes (59% schizophrenia) | Yes (others) | Group with psychosis showed an increase in positive and negative symptoms |
| Controls (50) | No (controls) | ||||
| Lieberman et al (Reference Lieberman, Alvir and Geisler1994) | Schizophrenia (41) | Methylphenidate 0.5 mg/kg i.v. | 1. Yes (10/33) off neuroleptics | 1. Yes (23/33) | |
| 2. Yes (6/27) on neuroleptics | 2. Yes (21/27) | ||||
| Koreen et al (Reference Koreen, Lieberman and Alvir1997) | Schizophrenia (32) | Methylphenidate 0.5 mg/kg i.v. or mCPP 0.1 mg/kg | No (mCPP) | NR (schizophrenia) | No recordings for individual patients |
| Controls (8) | Yes (methylphenidate) | No (controls) | |||
| Szesko et al (Reference Szesko, Bilder and Dunlop1999) | Schizophrenia (11) | Methylphenidate 0.5 mg/kg i.v. | Yes (acute phase) | No (stabilised phase) | No recordings for individual patients |
| Farren et al (Reference Farren, Hameedi and Rosen2000) | Cocaine users (8) | Cocaine intranasally 2 mg/kg 2 h after placebo or clozapine | Yes (paranoid symptoms) | NR | Clozapine reduced mean expected high; no attenuation of paranoid or craving scores. Cocaine levels increased by clozapine |
The Strakowski et al (Reference Strakowski, Sax and Setters1997) study looked for a response to repeated doses of stimulants. In the control group there was a greater response to the second dose of dexamfetamine than to the first. Participants with pre-existing psychosis showed no such enhanced response to a second dose.
The study by Casey et al (Reference Casey, Hollister and Klett1961) examined additional drug therapy in patients with schizophrenia, all of whom were taking antipsychotic medication regularly and had not responded to 200–600 mg of chlorpromazine taken daily for at least 2 months. One arm of the study examined the addition of dexamfetamine as an adjunctive treatment for schizophrenia. There was no benefit from the addition of dexamfetamine 60 mg daily compared with placebo, with worsening of ‘hostile belligerency, paranoid belligerency and thinking disturbance’.
For 26 studies it was possible to perform a statistical analysis of differences in psychotic response between controls, those with schizophrenia in remission and those with positive symptoms, using the definitions provided by the studies to determine the presence or absence of positive symptoms. There was a methodological difference between participants given i.v. dexamfetamine and those given oral dexamfetamine or i.v. methamphetamine (see Table 6): the doses of dexamfetamine used intravenously were lower and fixed, as opposed to being variedaccording to body weight (dexamfetamine 20 mg as opposed to 0.5 mg/kg methylphenidate).
Across the 26 studies, 51.4% of those with schizophrenia who had positive symptoms (n=149), 28.3% of those with schizophrenia in remission (n=69) and 10.2% of controls (n=9) had a temporary increase in positive symptoms, usually lasting for only a matter of hours. An analysis of the effects of the presence of positive symptoms v. absence of positive symptoms in participants with schizophrenia found a significant difference (χ2=46.3, d.f.=1, P<0.0001). We also examined modulating effects of antipsychotic drugs on the psychotic response. We did not detect a significant effect of antipsychotic medication in the response of participants with schizophrenia to a single dose of stimulant (χ2=0.06, d.f.=1, P=0.80); this was true whether the participants were defined as having positive symptoms or as being in remission (χ2=0.16, d.f.=1, P=0.68 for those with positive symptoms; χ2=0.36, d.f.=1, P=0.55 for those in remission).
Longitudinal studies
Seven longitudinal studies met the inclusion criteria (Table 2). Studies of this type were most commonly excluded because of the difficulty of separating stimulants from the other substances used. Two studies examined individuals prescribed stimulants: adults with narcolepsy (Reference Pawluck, Hurwitz and SchluterPawluck et al, 1995) and children with attention-deficit hyperactivity disorder (Reference Cherland and FitzpatrickCherland & Fitzpatrick, 1999). Two of the 11 adults in the first study developed acute psychotic symptoms, as did 9 of the 192 children in the latter study. Two follow-up studies of cocaine users (Reference Gawin and KleberGawin & Kleber, 1986; Reference Carroll, Power and BryantCarroll et al, 1993) reported no case of chronic psychosis. Sato et al (Reference Sato, Chen and Akiyama1983) studied amphetamine amphetamine users who had previously had long-lasting psychotic episodes who reused a stimulant after long periods of abstinence. These individuals were found to relapse after using a lower dose of amphetamine than they had used before first becoming psychotic. In one case the person's relapse seemingly was due to stress, without drug use. The researchers also conducted a small, uncontrolled trial of haloperidol 3 mg daily in eight of these individuals, none of whom then relapsed following subsequent amphetamine use. Iwanami et al (Reference Iwanami, Sugiyama and Kuroki1994) studied individuals who presented with a psychotic illness in the presence of amphetamine use; they identified a small group whose psychotic symptoms persisted for several months after ceasing amphetamine use who were being prescribed antipsychotic treatment. This group did not meet criteria for DSM–III schizophrenia (American Psychiatric Association, 1980) but had definite psychotic symptoms.
Table 2 Longitudinal studies

| Study | Study sample | Follow-up | Findings | Comments |
|---|---|---|---|---|
| Pawluck et al (Reference Pawluck, Hurwitz and Schluter1995) | Adults with narcolepsy on methylphenidate (> 100 mg/day) | 5 years | 2/11 psychotic symptoms | Both premorbid difficulties, former had paranoid ideas, latter family history of psychosis and head injury |
| 1 hallucinations and persecutory delusions | ||||
| 1 hypnogogic hallucinations with no insight | ||||
| Cherland & Fitzpatrick (Reference Cherland and Fitzpatrick1999) | Children with ADHD on methylphenidate, pemoline or dextroamfetamine | 5 years | 9/192 developed mood-incongruent psychotic symptoms | Notes three symptom clusters: |
| 11/192 developed mood-congruent psychotic symptoms | MPH toxic hallucinations (first doses) slower-developing paranoia mood-congruent psychotic symptoms | |||
| Gawin & Kleber (Reference Gawin and Kleber1986) | Cocaine users in treatment programme | 4—6 weeks | Screened with DIS, no reported case of psychosis | Looking for withdrawal symptoms |
| Carroll et al (Reference Carroll, Power and Bryant1993) | Treatment-seeking cocaine users | 1 year | No evidence of any chronic psychotic disorder | Most abstinent or markedly decreased use |
| Sato et al (Reference Sato, Chen and Akiyama1983) | Methamphetamine users with chronic psychosis | > 1 month (variable within group) | 16 patients reused MAP after long-term abstinence (up to 5 years) and relapsed with less MAP than previously, 4 with only one injection, 1 with none | 8 patients treated with haloperidol 3 mg daily did not relapse with MAP use after abstinence |
| Iwanami et al (Reference Iwanami, Sugiyama and Kuroki1994) | Methamphetamine users with psychosis | > 1 month (variable within group) | Two groups, symptoms lasting for: | Excluded if met DSM—III criteria for schizophrenia |
| 1 week after abstinence (transient group, n=54) | All given antipsychotics | |||
| 3 months after abstinence (persistent group, n=17) | Abstinence ensured | |||
| Persistent group more likely to have non-auditory non-visual hallucinations | ||||
| Kwapil (Reference Kwapil1996) | High scores on Chapman | 10 years | Psychosis-prone group used more stimulants than controls | Of 8000 screened, 193 were ‘psychosisprone’; 182 followed up: |
| Questionnaire (‘psychosis-prone) using substances | Substance use disorder at initial interview not predictive of later psychosis | DSM—III—R cocaine use disorder 12 | ||
| DSM—III—R amphetamine use disorder II power therefore small to detect link between psychosis and stimulants (controls n=153) |
Kwapil (Reference Kwapil1996) reported a 10-year follow-up study of substance-using individuals and controls who scored highly on the Chapman Questionnaire ‘psychosis proneness’ section. This self-report questionnaire is designed to measure symptoms and traits reported to be characteristic of proneness to schizophrenia or psychosis. The study showed that psychosis was not predicted by earlier substance use, but the small number of stimulant users meant that the power of the study was insufficient for a meaningful analysis of any link between psychosis and stimulants.
Case–control studies
Most case–control studies identified by the search strategy were excluded because it was impossible to separate stimulant use from other drug use known to be associated with psychotic states, such as cannabis.
Four studies compared cocaine users with psychosis with users with no psychosis (Table 3). Heavier cocaine use was shown among participants with psychosis compared with controls in three studies (Reference Manschreck, Laughery and WeissteinManschreck et al, 1988; Reference Brady, Lydiard and MalcolmBrady et al, 1991; Reference Bartlett, Hallin and ChapmanBartlett et al, 1997). In two studies it was reported that the psychotic episodes worsened over time (Reference Brady, Lydiard and MalcolmBrady et al, 1991; Reference Bartlett, Hallin and ChapmanBartlett et al, 1997). Five studies compared individuals with schizophrenia or another psychotic illness who had been using stimulants with matched groups who had not been using stimulants (Table 4). These studies showed a lower age of onset of psychosis in the stimulant-user group, fewer negative symptoms and more paranoid themes. First-rank symptoms were noted to be fewer and hallucinatory experiences more common. Seibyl et al (Reference Seibyl, Satel and Anthony1993) showed that most of the people misusing drugs in their study had begun their cocaine use after psychosis had developed.
Table 3 Case—control studies of stimulant users: with v. without psychosis

| Study | Cases (n) | Controls (n) | Significant differences (cases v. controls) | Comments |
|---|---|---|---|---|
| Brady et al (Reference Brady, Lydiard and Malcolm1991) | Cocaine users with psychosis (29) | Cocaine users, no psychosis (26) | Greater duration and amount of use prior to admission in psychosis group; greater proportion of males in psychosis group | 72% reported psychosis occurring with increased frequency, greater speed of onset and with smaller amounts of cocaine over time |
| Satel & Edell (Reference Satel and Edell1991) | Cocaine users with paranoia (10) | Cocaine users without paranoia (10) | ‘Psychosis proneness’ score on the Perceptual Aberration Scale and Magic Ideation Scale positively correlated with paranoia | Unable to determine direction or causality of relationship |
| Bartlett et al (Reference Bartlett, Hallin and Chapman1997) | Cocaine users with paranoia (22) | Non-paranoid users (18) | Greater duration of cocaine use in sensitised group | Sensitisation linked to other psychotic features of cocaine |
| Sensitised users1 (11) | Non-sensitised users (7) | Reduced dose escalation in sensitised group Increased referentiality and unease in sensitised group | ||
| Manschreck et al (Reference Manschreck, Laughery and Weisstein1988) | Psychosis > 24 h, cocaine users (31) | Cocaine users, non-psychotic (28) | Past psychiatric history, violence and total drug use all greater in cases | Freebase cocaine used; psychosis present in 29% of cocaine-using patients hospitalised in 1 year |
Table 4 Case-control studies of people with psychosis: stimulant users v. non-users

| Study | Cases (n) | Controls (n) | Significant differences (cases v. controls) | Comments |
|---|---|---|---|---|
| Seibyl et al (Reference Seibyl, Satel and Anthony1993) | Schizophrenia, cocaine users (16) | Schizophrenia, non-users (20) | Age at onset of schizophrenia lower in cocaine users | In the cases group, 5 used cocaine prior to disease onset and 8 after onset (3 undefined) |
| Lysaker et al (Reference Lysaker, Bell and Beam-Goulet1994) | Schizophrenia, cocaine users (25) | Schizophrenia, non-users (18) | Negative symptoms reduced and age at first admission lower in cocaine users | Cocaine users more likely to be paranoid |
| Rosse et al (Reference Rosse, Collins and Fas McCarthy1994) | Cocaine users with psychosis (29) | Schizophrenia, non-users (16) | Number and intensity of first-rank symptoms less in cases, but paranoid themes more common | No formication reported |
| Dermatis et al (Reference Dermatis, Galanter and Egelko1998) | Schizophrenia, cocaine users (43) | Schizophrenia, non-users (27) | Lower educational level and more prior hospitalisation in cocaine users | |
| Serper et al (Reference Serper, Alpert and Richardson1995) | Schizophrenia, cocaine users (32) | Schizophrenia, non-users (54) | Hallucinatory experiences more common in cocaine users with schizophrenia than in the other two groups | Cocaine users with schizophrenia similar to users without psychosis on negative symptoms and moods, and similar to non-users with schizophrenia on most positive symptoms |
| Cocaine users, no psychosis (30) | Negative symptoms in schizophrenia groups less among cocaine users |
Two studies compared people misusing stimulants with those misusing other drugs (Table 5). Graf et al (Reference Graf, Baer and Comstock1977) showed an increase in the psychotic profile on the Minnesota Multiphasic Personality Inventory at discharge in people using stimulants rather than other drugs, and Dalmau et al (Reference Dalmau, Bergman and Brismar1999) showed a significant difference in the rates of psychosis between patients formerly using amphetamines and those using opiates in a study of residents of a drug rehabilitation unit.
Table 5 Case-control studies of stimulant users v. other drug users

| Study | Cases (n) | Controls (n) | Significant differences (cases v. controls) | Comments |
|---|---|---|---|---|
| Graf et al (Reference Graf, Baer and Comstock1977) | Stimulant users (15) | Sedative-hypnotic users (14) | Psychotic profile on MMPI at discharge greater in stimulant user group v. all others | |
| Barbiturate users (17) | ||||
| Multi-drug users (20) | ||||
| Dalmau et al (Reference Dalmau, Bergman and Brismar1999) | Amphetamine users (461) | Opiate users (371) | Psychosis greater in amphetamine and cannabis users v. opiate users (30% v. 6%) | Users recruited from in-patient drugs unit |
| Cannabis users (425) |
Table 6 Change in psychotic ratings per substance used and pre-existing psychosis

| Dexamfetamine | Methylphenidate i.v. n (%) | Total n (%) | ||
|---|---|---|---|---|
| Oral n (%) | i.v. n (%) | |||
| Remission | ||||
| Increased | 13 (27.7) | 5 (27.8) | 51 (28.5) | 69 (28.3) |
| No increase | 34 (72.3) | 13 (72.2) | 128 (71.5) | 175 (71.7) |
| Active psychosis | ||||
| Increased | 28 (73.7) | 79 (39.9) | 42 (77.8) | 149 (51.4) |
| No increase | 10 (26.3) | 119 (60.1) | 12 (22.2) | 141 (48.6) |
| Control | ||||
| Increased | 0 (0.0) | 0 (0.0) | 9 (26.5) | 9 (10.2) |
| No increase | 39 (100.0) | 15 (100.0) | 25 (73.5) | 79 (89.8) |
| Total | ||||
| Increased | 41 (33.1) | 84 (36.4) | 102 (38.2) | 227 (36.5) |
| No increase | 83 (66.9) | 147 (63.6) | 165 (61.8) | 395 (63.5) |
DISCUSSION
The studies reviewed here provide useful evidence about the effect of stimulant use on people with pre-existing psychotic illness, but more limited evidence about the phenomenon of sensitisation.
The expectation that antipsychotic medication might block the action of stimulants and prevent deterioration in psychotic illnesses on exposure is not borne out by these studies. The presence of positive symptoms of schizophrenia (as distinct from being in remission) appears to make an individual more likely to experience a worsening of psychotic symptoms in response to a single dose of a stimulant drug.
There is clear evidence from these studies that, irrespective of the individual's mental state, a large enough dose of a stimulant drug can produce a brief psychotic reaction, usually lasting only hours and being self-limiting in the majority of individuals. The differences between i.v. dexamfetamine, oral dexamfetamine and i.v. methamphetamine in participants with active symptoms are probably due to the lower doses used in the i.v. dexamfetamine condition – usually a maximum of 20 mg. Evidence for sensitisation is found in only two studies. Strakowski et al (Reference Strakowski, Sax and Setters1997) showed that when two doses of a stimulant were given to volunteers free from psychosis, the second dose produced a greater psychotic response as measured by the BPRS – a ‘sensitised’ response. Stimulant users in the study by Brady et al (Reference Brady, Lydiard and Malcolm1991) reported psychotic symptoms occurring with lower doses over time.
The difference between patients who were substance users in the study by Dalmau et al (Reference Dalmau, Bergman and Brismar1999), where psychosis rates were noted to be greater among in-patients who used cannabis or stimulants rather than opiates, is interesting. Sensitisation is a possible contributing factor, but not the only one. The results might have been confounded by differences in rates of admission to the unit. It is possible, for example, that those with opiate problems were admitted more frequently for in-patient detoxification, whereas stimulant users (in whom the withdrawal syndrome is less severe) might have been given out-patient treatment. The proportion presenting with psychosis as in-patients would therefore be greater for those using stimulants rather than opiates.
The difficulties of researching the longer-term effects of stimulants are seen in the two Japanese studies (Reference Sato, Chen and AkiyamaSato et al, 1983; Iwamani et al, 1994). The widespread use of high-dose injected methamphetamine led to hospital admissions of individuals with chronic psychosis that persisted after substance use had ceased. Many patients in these studies could have been given a DSM–IV diagnosis of schizophrenia or other psychotic illness (American Psychiatric Association, 1994) but were classed as having methamphetamine psychosis.
The small open-label trial of haloperidol (Reference Sato, Chen and AkiyamaSato et al, 1983) merits attention, if only because of the paucity of other evidence and the relationship of its results to animal studies. Eight of the cohort of stimulant users with chronic psychoses who had relapsed following stimulant use were prescribed small doses of haloperidol (3 mg daily) following recovery and were observed for further relapse. These participants did not relapse, even if they returned to stimulant use; however, participants who were not given haloperidol relapsed into a psychotic state lasting days to weeks after using stimulants. The results could lead us to postulate that where people are unable to abstain from stimulant use despite repeated psychotic episodes, small doses of regular antipsychotic medication administered once the episode has settled might reduce or prevent sensitisation in the future.
Human experimental studies investigating sensitisation are unlikely because of ethical considerations, but a number of animal experiments have been carried out. Stimulant-induced stereotyped behaviour in small mammals and possible hallucinatory experiences in primates have been used as a model for schizophrenia in humans. In animals, the response to chronic amphetamine use has been divided into two phases. In the ‘initiation’ phase of these experiments animals are ‘sensitised’ by small regular doses of stimulants, insufficient to cause a ‘psychotic’ reaction on their own. The ‘expression’ phase occurs if the animals are either stressed or given a single dose of a stimulant. In the first phase, sensitisation has been shown to be blocked by antipsychotic drugs, whereas the psychotic reaction in the expression phase is not always blocked (Reference Lieberman, Kinon and LoebalLieberman et al, 1990). Castner & Goldman-Rakic (Reference Castner and Goldman-Rakic1999) investigated rhesus monkeys, which were given intermittent, escalating low doses of amphetamine over a 12-week period, followed by an acute challenge with low-dose amphetamine (0.4–0.46 mg/kg). Enhanced responses (hallucinatory-like behaviours, static posturing and motor stereotypies) were noted in response to a low-dose amphetamine challenge 5 days after withdrawal and up to 28 months later. The monkeys also showed an increase in responses ‘independent of stimuli’, possibly indicating hallucinations, in the absence of additional drug challenges. Antipsychotic drugs were not used.
Meng et al (Reference Meng, Feldpaush and Merchant1998) performed a similar experiment on rats, but also pre-treated one group of rats with high-dose haloperidol (0.5 mg/kg) or clozapine (20 mg/kg), withholding the ‘sensitising’ phase of amphetamines. This group showed an enhanced response to amphetamine challenge in a similar way to those sensitised with amphetamines. Rats that had been given low-dose antipsychotic treatment (haloperidol 0.1 mg/kg or clozapine 4 mg/kg) alongside regular amphetamine administration did not show an enhanced effect, suggesting that they were not sensitised, in a similar way to the humans in the study by Sato et al (Reference Sato, Chen and Akiyama1983). The sensitisation following high-dose antipsychotic treatment is presumably related to dopamine receptor upregulation, which occurs in these circumstances, increasing the vulnerability of the brain to stimulants once the antipsychotic treatment is stopped.
Evidence against sensitisation occurring can be found. Seibyl et al (Reference Seibyl, Satel and Anthony1993) noted that for the majority of participants stimulant use began after the onset of psychotic illness, again weakening the case for a causative role for stimulants. We identified only two studies that looked specifically at the therapeutic use of methylphenidate and psychosis (Reference Pawluck, Hurwitz and SchluterPawluck et al, 1995; Reference Cherland and FitzpatrickCherland & Fitzpatrick, 1999), but many studies have established the safety of this agent, although not specifically reporting or examining for psychosis (e.g. Reference Efron, Jarman and BarkerEfron et al, 1997). Illicit use of methylphenidate, however, tends to follow a different pattern, with binges and escalation of dose occurring.
The lack of evidence in this area of psychiatry causes problems for clinicians who must plan management without a solid evidence base for a group of patients whose management is challenging. Using the data from these studies, we can say clearly that use of stimulants leads to a brief psychotic reaction, usually only hours in length, that is more pronounced in people who already have active symptoms of psychosis and is seemingly unaffected by antipsychotic medication. With regard to the hypothesis that stimulant use can produce chronic psychosis, supportive evidence is present in studies of humans but is of lower quality, although supported by experimental animal studies.
In the absence of better evidence, treatment of stimulant-induced psychosis should probably involve efforts to encourage abstinence from stimulants and medication with antipsychotic drugs until the acute symptoms settle. This should be followed by regular low doses of antipsychotics in those who have experienced more than one episode of psychosis. Given that the evidence (however poor) points to sensitisation occurring, it is important that people using stimulants should be assertively managed in an attempt to prevent long-term chronic psychosis.
Clinical Implications and Limitations
Clinical Implications
-
▪ The findings of this review indicate that stimulant use can result in a short-lived psychotic reaction, more pronounced in those with pre-existing psychotic symptoms. This reaction is unaffected by antipsychotic medication.
-
▪ People with schizophrenia who use stimulants will not necessarily be protected from worsening of their clinical condition by compliance with antipsychotic therapy.
-
▪ Longer-term stimulant use may lead to the development of sensitisation and a more chronic psychosis, but low-dose, long-term antipsychotic treatment may prevent the development of this sensitisation.
Limitations
-
▪ There is little evidence for the effects of long-term stimulant use, and because of the methodological difficulties, it is poor in quality or derived from animal experiments.
-
▪ The effects of other psychoactive drugs confound many of the studies of this subject, which were therefore excluded from the review.
-
▪ The only treatment study available is a small open trial.
Acknowledgement
We thank Dr Janice Morgan for her advice.







eLetters
No eLetters have been published for this article.