


Introduction
Regulations are necessary to ensure the safety and effectiveness of healthcare products such as drugs and medical devices. The need became pressing with the rapid expansion of options for effective pharmacotherapy and with the development of many diagnostic technologies and implantable medical devices. Both trends have accelerated from the middle of the twentieth century.
European-wide directives concerning medical devices were enacted in the European Economic Community (EEC) from 1990. Within 20 years it became apparent that the directives would need to be adapted, as the rules concerning clinical evidence requirements were insufficient (European Commission 2012a). The process of regulatory revision was initiated in 2008, and new European Union (EU) laws were passed in 2017 that required higher levels of evidence and more thorough oversight. Their full implementation was postponed first during the COVID-19 pandemic, and again until 2027. By then, European regulatory reform for medical devices will have taken 20 years, with the possibility that the system will remain suboptimal.
There will be another review, of the current EU regulations for medical devices and in vitro diagnostic devices (European Parliament 2017a), as required by Article 121 of the Medical Device Regulation 745/2017 (MDR) (European Parliament 2017b). It is being conducted by the Medical Devices Unit in the European Commission (Unit D3 in DG SANTE), which has launched a public consultation on the MDR framework.
As a foundation for logical reconsideration of the regulatory framework, it is important to understand the principles and motives that guided its initial development. We compare the origins of systems for evaluating devices with the history of regulations for pharmaceutical products, first in the USA and later in Europe. We identify common themes as well as differences that provide context for the development of an alternative and improved approach.
Methods
We performed a narrative review of the history of regulation of pharmaceutical products and medical devices, first in the USA, and then in Europe. The scope included initial regulation for food safety, since that led to the foundation of the Food and Drug Administration (FDA) in the USA, but the main focus was on high-risk medical devices and particularly in the European Economic Community (EEC, until 1993) and the European Union (EU). We did not review the history of regulation of in vitro diagnostic medical devices, which in Europe followed some years after the directives for medical devices and adopted a similar model, perhaps inappropriately; that would merit a separate analysis.
The timespan for our review was the twentieth century, and developments are described in chronological order. We aimed to consult and cite primary sources, including accounts published by individuals who were personally involved in the events that they described. We recorded personal recollections during interviews with some individuals who were directly involved in the development of the first European legislation for medical devices.
The Medical Devices Unit of the European Commission has no records of the background, procedures and negotiations leading to the first European directives in 1990 (European Council 1990) and (European Council 1993). The archives of the Council are limited to minutes of some Council working group meetings, which we have reviewed, but they provide no insights into the reasons for decisions that were made. The Historical Archives of the EU, which we searched using the term ‘Medical Devices’, hold a copy of the proposal from the Commission in 1991 for the first EU directive concerning general medical devices, including an explanatory memorandum. The History Office of the US FDA provided some original documents and references. We consulted with trade associations MedTech Europe (founded as EucoMed in 1979), and COCIR (the European Coordination Committee of the Radiological, Electromedical and Healthcare IT Industry; founded in 1959), to identify any records which they retain relating to this period.
Origins of Regulations in the USA
Food Safety
In 1883 the US Department of Agriculture appointed Dr Harvey Washington Wiley as its chief chemist. Concerns had been raised for some decades (US FDA 2023) because potentially toxic additives were being added to food products, such as formaldehyde to preserve (or ‘embalm’) milk and copper sulphate to enhance the green colour of French beans. This came to prominence during the Spanish-American War of 1898 when canned meat containing borax intended as a preservative was putrid, causing dysentery on top of other acquired diseases, such as infections that together killed more troops than the conflict (Keuchel Reference Keuchel1974; O’Reilly Reference O’Reilly2019).
In order to understand the effects of adulterated food and chemicals on health, Wiley organized tests on healthy volunteers, who were dubbed by journalists as the ‘poison squad’. His advocacy is credited with providing the principal motivation for the US Food and Drugs Act of 1906, which prevented marketing of adulterated and misbranded food and drugs. Wiley directed the Bureau of Chemistry in the US Department of Agriculture from 1907 to 1912; it became the US FDA in 1930.
Pharmaceutical Products
By the 1930s, it was recognized that the 1906 Act was obsolete but there was no consensus on what should replace it (Ballentine Reference Ballentine1981).
Then, in 1937, more than 100 people including many children died after taking sulfanilamide, within a few weeks after a new preparation had been distributed by its manufacturer, the S.E. Massengill company (Ballentine Reference Ballentine1981). Sales staff had transmitted requests for the drug to be made available as a syrup, and the company had identified diethylene glycol as a suitable solvent. The new formulation was released without any investigations of its safety; the only studies that had been performed had identified raspberry as the preferred flavouring. In fact, diethylene glycol causes acute renal failure and severe neurological problems (Schep et al. Reference Schep, Slaughter and Temple2009). The deaths from this syrup led to the passage of the 1938 Food, Drug, and Cosmetic Act, which increased the FDA’s authority to regulate drugs (Ballentine Reference Ballentine1981; Wax Reference Wax1995; Paine Reference Paine2017). It was made necessary to submit an application to the FDA before marketing a drug, and to provide evidence that it was safe.
In 1960, the FDA appointed Dr Frances Kelsey, who was a clinical pharmacologist, as an assessor of new drug applications (Vanderbes Reference Vanderbes2023; Tobbell Reference Tobbell2012). One of her first assignments was to review an application for approval in the USA of thalidomide. She recommended that it should not be supported, because of lack of evidence of safety, and she maintained her expert opinion despite considerable external lobbying and legal threats (Vanderbes Reference Vanderbes2023). The tragedy of thalidomide was largely, but not completely, avoided in the USA because some physicians who had been given free samples from the company for ‘research’ projects gave them to their patients. This experience provided influential support for passage of the Kefauver–Harris amendment to the US Federal Food, Drug, and Cosmetic Act, in 1962. Known as the ‘Drug Efficacy Amendment’, it required manufacturers thereafter to provide proof of the efficacy as well as the safety of their drugs, from adequate and well-controlled clinical trials, and to disclose accurate information about side-effects. Sadly, issues remain with use of diethylene glycol and thalidomide (Box 1) (British Broadcasting Corporation 2022a; British Broadcasting Corporation 2022b; Diggle Reference Diggle2001; Schuler-Faccini et al. Reference Schuler-Faccini, Soares and de Sousa2007; Rehman et al. Reference Rehman, Arfons and Lazarus2011).
Late sequels of adverse effects from sulfanilamide and thalidomide
Neither story ends with the initial regulatory responses.
Incredibly, children died in 2022 in the Gambia in West Africa after taking a cough syrup (British Broadcasting Corporation News 2022a). Gambia did not have any national capacity to assess the quality or purity of drugs, but tests performed with support from the World Health Organization showed that the syrups contained traces of diethylene glycol and ethylene glycol. The products were marketed by a generic drug manufacturer from India, Maiden Pharmaceuticals, despite the company having failed tests for some of its products in India (British Broadcasting Corporation News 2022b).
Thalidomide was found to be effective for cutaneous reactions in leprosy, and intermittent cases of birth defects have been reported since 1965 in Brazil (Diggle Reference Diggle2001; Schuler-Faccini et al. Reference Schuler-Faccini, Soares and de Sousa2007). Thalidomide was approved by the FDA in 1998, for very specific indications and with stringent precautions (Rehman et al. Reference Rehman, Arfons and Lazarus2011). A derivative of thalidomide, lenalidomide, is now used routinely to treat myeloma.
The 1962 act required the FDA to review, within two years, about 3400 drugs that had been approved since 1938, and to classify them as effective, probably effective, possibly effective, or ineffective (Chhabra et al. Reference Chhabra, Kremzner and Kiliany2005). The FDA was inadequately funded and staffed for this (Schwartz Reference Schwartz2016), however, so ‘the first thing the agency did after the new drug laws were passed in 1962 was nothing’ (Crout Reference Crout1997). The situation changed in 1966 when James Goddard became Commissioner of the FDA, since he set delivery of the retrospective review as one of his priorities (Langer Reference Langer1966). He agreed to a partnership with the National Academy of Science and National Research Council to bring together scientists and clinicians to provide ‘authoritative opinions’. The project became known as the Drug Efficacy Study Implementation or ‘DESI’ study (Chhabra et al. Reference Chhabra, Kremzner and Kiliany2005). The opinions were non-binding on the FDA but generally followed: drugs that were effective were listed in the ‘Orange Book’ which to this day provides a list that can be used for decisions about reimbursement and to identify generic drugs that can be substituted when a patent expires (Hickey Reference Hickey2024). In this way, the FDA was able to use both external expertise and market incentives to improve the evidence-base for drug products (Meadows Reference Meadows2006).
The Academy appointed 30 committees (Institute of Medicine Reference Rettig, Earley and Merrill1992). The FDA had no role in their selection and the composition of each panel was not made public until its work had been completed. There was no assessment of conflicts of interest but members would recuse themselves when appropriate (Langer 1996). The success of the DESI study led to formalization of standing advisory committees by the FDA, for drugs and later for biologics and medical devices (Schwartz Reference Schwartz2016; Institute of Medicine Reference Rettig, Earley and Merrill1992).
Medical Devices
As early as 1872, the US Post Office had authority to assess any device for which a medical claim had been made, while it was in transit; if the Postmaster General considered the device worthless it was returned to sender, marked ‘Fraudulent’ (Junod Reference Junod2017). Despite this evidence of concerns, the Pure Food and Drug Act of 1906 did not refer to medical devices, and in 1917 the Bureau of Chemistry, in its annual report to the US Congress, stated that the 1906 act ‘has its serious limitations […] which render it difficult to control […] fraudulent mechanical devices used for therapeutic purposes’ (Rados Reference Rados2006).
It was not until the Food, Drug and Cosmetic Act of 1938 (1938 Act) that the FDA was given some authority with respect to medical devices. This was limited to cases in which a medical device could be shown to be adulterated or misbranded, and the product had to enter interstate commerce before the FDA could take action. As a result, early enforcement activity was directed towards obviously ‘quack’ devices (Bartlett Foote Reference Bartlett Foote1992). The FDA had no authority to determine if a device was safe and effective, through pre-market testing, review, or approval. In 1962, when proposing the Drug Amendments, President John F. Kennedy also proposed legislation for medical devices (Institute of Medicine Reference Rettig, Earley and Merrill1992) but Congressional hearings about whether or not devices should be evaluated similarly to, but separately from, drugs were sidelined by the need to respond to the thalidomide scandal.
Ultimately, the plan of Congress to enact medical device legislation would take another 14 years. Lobbying for better regulation of medical devices continued throughout the 1960s (Link Reference Link1972), and the need was mentioned by President Lyndon Johnson in three consumer messages (Hutt Reference Hutt and Estrin1990). The FDA resorted in some cases to treating a device as a ‘drug’. Examples included contact lenses, injectable silicone, pregnancy-test kits, and bone cement (Institute of Medicine Reference Rettig, Earley and Merrill1992). In 1969, the Supreme Court ruled that an antibiotic sensitivity disc used as a laboratory screening test could be considered as a drug in order to apply pre-market clearance requirements (US Supreme Court 1969). The FDA feared that an increase in litigation could delay enforcement of its actions and limit its ability to protect the public (Cooper Reference Cooper1971). The judgement meant either that the FDA could designate whatever devices it deemed necessary, as drugs, or else that Congress needed to approve new device legislation.
In his October 1969 consumer message, President Richard Nixon stated that ‘certain minimum standards should be established’ for medical devices and ‘the government should be given additional authority to require premarket clearance in certain cases’ (Nixon Reference Nixon1969). In December 1969, a Study Group on Medical Devices was convened, with the remit to recommend procedures for the establishment of standards for certain medical devices, and for the review of other devices, prior to marketing (Study Group on Medical Devices 1970). It was led by Dr Theodore Cooper, who was then Director of the National Heart and Lung Institute of the National Institutes of Health.
The Study Group reported their findings in September 1970, in what became known as the ‘Cooper Report’, noting that they were:
distressed by the lack of data in many areas related to the interaction of medical devices with the human body, and by the seeming unquestioning acceptance of claims for medical device safety and performance unsubstantiated or inadequately supported by scientific fact. (Study Group on Medical Devices 1970)
Giving testimony to the US House of Representatives in 1975, Cooper further emphasized that ‘we believe […] that the public is now being exposed to an undesirable level of risk’ (Cooper Reference Cooper1975). Delineating the nature of this risk, the Study Group noted that ‘while its exact magnitude could not be quantified, there were convincing “indicators” of its dimension’ (Cooper Reference Cooper1971). Examples of this included 512 deaths from heart valves, 89 deaths from pacemakers, and also a report that 40% of the devices tested in one New York hospital had been found to be defective (Cooper Reference Cooper1971; Bartlett Foote 1978). Other examples of hazards associated with medical devices, which were documented in Congressional hearings in 1973, included a cardiac defibrillator with faulty electrical circuitry, and faulty valves on emergency oxygen respirators (Gluck Reference Gluck1985). The influential case of the Dalkon shield is summarized in Box 2 (Hutt Reference Hutt and Estrin1990; US FDA 1975; Mumford and Kessel Reference Mumford and Kessel1992; Pisac and Wilson Reference Pisac and Wilson2021).
The Dalkon Shield
The Dalkon Shield, an intra-uterine contraceptive device, was marketed from 1971 without having undergone any prior regulatory scrutiny. Sales in the USA were suspended by the FDA in 1974 after reports that it was associated with an increased risk of sepsis (US FDA 1975). There were public hearings on the Dalkon shield, in the US House of Representatives and Senate, in 1973 and 1975 (Hutt Reference Hutt and Estrin1990).
Doubt was cast later when no significant safety signals were observed in controlled clinical trials, unlike the earlier case-control studies; instead, complications were attributed to other factors such as operative technique and experience (Mumford and Kessel Reference Mumford and Kessel1992). Nonetheless, most investigators concluded that the design of the Dalkon Shield was associated with a small increase in risk compared with other intrauterine devices. A review published by the FDA concluded that ‘patient harm has been a driver of change’ (Pisac and Wilson Reference Pisac and Wilson2021).
The Cooper Report recommended the creation of three classes of medical devices, as the basis for regulation according to their potential risks and benefits (Study Group on Medical Devices 1970). In 1976, the Federal Food, Drug, and Cosmetic Act established Class I devices, which were exempt from standards or pre-market clearance and subject only to general controls applied by the manufacturer; class II devices which would require controls through the use of standards; and class III devices which would be subject to a pre-market review (Library of Congress 1976). The 1976 amendments also mandated the FDA to review the safety and effectiveness of new devices before marketing, and to require manufacturers to develop and adhere to good manufacturing practices (Comptroller General 1983).
A further requirement, for the FDA to review all devices on the market, was challenging because of limited capacity, as with implementing the Kefauver-Harris amendments for drugs. An inventory identified about 8000 devices manufactured by about 1000 manufacturers (Link Reference Link1972). The FDA then set up two advisory panels as a pilot, to classify class III orthopaedic and cardiovascular devices, while the whole classification project took decades to complete (US FDA Center for Devices and Radiological Health 2017; Mooghali et al. Reference Mooghali, Rathi and Kadakia2023). For class II devices, it was intended that performance specifications and standards would be created, but fewer standards were produced than expected. In 1990, the law was changed to allow manufacturers to market a device based upon a demonstration of ‘substantial equivalence’ to a previously cleared device. This 510(k) pathway is now used for the majority of medical devices in the USA (Darrow et al. Reference Darrow, Avorn and Kesselheim2021).
Origins of Regulations in Europe
The Treaty of Rome (on the founding of the EEC) in 1957 contained two provisions that could be considered to impact directly on health services, namely the free mobility of workers, and the harmonization of social security and health insurance systems. Member states have responsibility for their own healthcare systems but the Treaties of Maastricht in 1992 and Lisbon in 2007, which established the EU, broadened its supra-national responsibilities by adding a requirement for joint actions by the member states when necessary to protect public health.
Food Safety
Regulations for food have their origins in antiquity, possibly from the prohibition of colouring and flavouring of wine in ancient Greece and Rome (van Waarden Reference van Waarden, Ansell and Vogel2006). National food laws existed in member states long before the European project, and initial EU food law from the 1960s was directed principally towards establishment of the single market. The first related EU directive in 1962 concerned colourings in foodstuffs (Higson Reference Higson2001).
The landmark ‘Cassis de Dijon’ case established the principle of mutual recognition, when the European Court of Justice ruled in 1979 that a product that has been produced and marketed lawfully in one member state cannot be prohibited in another member state on the grounds of non-compliance with national rules, in the absence of any urgent need to intervene (European Court of Justice 1979).
The single-market approach deriving from this judgement was challenged profoundly in the 1990s by a series of food safety crises, the greatest of them arising from bovine spongiform encephalopathy. Elsewhere, late deaths had resulted from the toxic oil syndrome when rapeseed oil had been adulterated with aniline (James Reference James1994); in total, the fraudulent use of commercial oil rather than oil for consumption was associated with perhaps 20,000 cases and 1663 deaths between 1981 and 1994 (Van der Meulen Reference Van der Meulen2013).
It was noted subsequently that the ‘heyday of market-oriented food law based on mutual recognition ended in tears’ (van Waarden Reference van Waarden, Ansell and Vogel2006). A major change in EU policy occurred with publication of a Green Paper which laid out new general principles of food law (European Commission 1997) and led to passage of EU Regulation 178/2002 (European Council and Parliament 2002). It established the European Food Safety Authority, which has been based in Parma since 2002.
Pharmaceutical Products
Thalidomide was developed in 1953 by chemists working for the German company Chemie Grünenthal, which had been formed after the Second World War to take part in the boom for antibiotics (Knightley et al. Reference Knightley, Evans, Potter and Wallace1979). Chemists who had been looking for simpler methods of making peptides, tested a substance that appeared to have a sedative effect in laboratory animals. Subsequently, and despite reports of peripheral neuropathy, thalidomide was marketed as a powerful antiemetic agent for pregnant women, which led to an estimated 2000 deaths and 10,000 children being born with birth defects, principally in Europe, Australasia, and Canada. In the United Kingdom alone, 2000 ‘thalidomide babies’ were born with phocomelia and/or other abnormalities during the three years after the drug was licensed for use in 1958. The first alert of a possible association between these effects and exposure to thalidomide, which came in a short letter to The Lancet in 1961 (McBride Reference McBride1961) and in reports from Germany, prompted the company to withdraw the drug. It is now known that thalidomide has an anti-angiogenic effect, especially during the critical period of early embryogenesis (Vargesson Reference Vargesson2015).
The thalidomide scandal was a major catalyst for the development of regulations to control drugs. It had been possible for it to be marketed in Germany (where it was even sold over the counter) and in other European countries without proper studies of safety and without appropriate regulatory oversight. For example, in the UK, ‘Before thalidomide, the only regulation of medicines […] was control of the quality of therapeutic substances through their manufacture and supply; there was no requirement for provision of evidence to support claims for either clinical safety or efficacy’ (Emanuel et al. Reference Emanuel, Rawlins and Duff2012).
That tragedy became a key driver of the evolution of evidence standards for drug development and licensing. It also reinforced the notion that a knowledgeable and trusted third party – the drug regulator – is needed to protect patients’ interests and to decide for them which drugs would be available to them. (Eichler et al. Reference Eichler, Abadie and Baker2012)
The first EEC legislation governing pharmaceutical products was Directive 65/65/EEC on the approximation of provisions laid down by Law, Regulation or Administrative Action relating to proprietary medicinal products (European Council 1965). The European Medicines Evaluation Agency (EMEA) was established in 1995. It introduced a centralized procedure for the assessment of innovative medicines, and a decentralized procedure for the mutual recognition of licences granted by individual medicines authorities (Jones and Jefferys Reference Jones and Jefferys1994). Over time, centralized assessments became predominant and mandatory for certain medicines (Kohler Reference Kohler2011). The agency was renamed the European Medicines Agency (EMA) in 2004.
Medical devices
Early attempts to develop implants failed, such as the first joint replacement by Themistocles Glück in Germany in 1890 using an ivory device. Development and wide clinical use followed only much later, after technological advances. The first successful implantation of an intra-ocular lens made from Perspex was in 1949. The first fully implantable cardiac pacemaker was inserted by Åke Senning in Sweden in 1958, and the first heart valve replacement with a caged-ball prosthesis in the aortic position was pioneered by Dwight Harken in the USA in 1960. John Charnley in England did his first total hip replacement in 1962. Arguably, systematic regulation was not needed until the second half of the twentieth century.
EU Member State Regulations before 1990
Standards were developed in Europe early in the twentieth century to limit exposure to X-rays (Rolleston Reference Rolleston1927; Spear Reference Spear1953), once the dangers had been recognized, but otherwise national systems evolved slowly. Italy (in 1927) was the only country to have specific legislation before the mid-point of the century. The frameworks that applied in the 12 member countries of the EEC in 1990, at the time when the first European medical device directive was adopted, were described by Gordon Higson, a physicist who directed the Scientific and Technical Branch of the Procurement Directorate of the Department of Health in the UK (Higson Reference Higson2001). They are summarized in Table 1.
Summary of national regulatory frameworks for medical devices, existing in EU member states before the first EU legislation in 1990

Note: Arranged by date of accession, and in alphabetical order.
Source: Adapted, expanded, and summarized after Higson (Reference Higson2001).
In the absence of specific medical device laws, some countries had adapted their legislation for drugs so that it also applied to certain devices – by the expedient manoeuvre of including devices in their definitions of drugs (France, Germany, Italy, Spain) (Higson Reference Higson2001). Germany distinguished between ‘true drugs’ and ‘fictitious drugs’. France had definitions of device types, and an approval procedure called homologation, which included inspections. France, Germany and Italy delegated the examination of certain devices to testing houses to check if they conformed to national or international standards. In 1985, directors of medical device testing organizations from several EEC member states (France, Germany, Italy, the Netherlands, and the United Kingdom) met to determine areas of common interest (Ilsey and Runciman Reference Ilsley and Runciman1988). There were provisions concerning clinical trials in France, Germany, Italy and Spain. The UK introduced procedures for registering manufacturers of medical devices, auditing their quality assurance systems, and investigating reports of defects. Assessments of devices and advice on evaluation and selection were published in a series of technical guides (JT, personal communication; Higson Reference Higson2001).
Between 1988 and 1990 the Commission was notified of 25 sets of draft national regulations, most of them relating to specific products (European Commission 1991). It concluded that:
In view of the differences in approach, which by their nature are incompatible with a single Community market in medical devices, it is essential to set up a Community system of laws based on Article 100a of the EEC Treaty to abolish and prevent technical barriers to trade. (European Commission 1991)
Involvement by European Pacemaker Companies
Issues with cardiac pacemakers had been noted since the 1960s (Jeffrey and Parsonnet Reference Jeffrey and Parsonnet1998). Technical challenges such as the reliability of batteries and the durability of pacemaker leads prompted suggestions for collective actions to develop standards and/or regulation, and calls for much greater transparency. Seymour Furman, a clinical pioneer of cardiac pacing in the USA, wrote in 1972 that ‘the manufacturers have inherited both prosperity, and an obligation to the patient. […] Not all live up to that standard’ (Furman Reference Furman1972). He was referring to the risks that a new manufacturer entering the field might not meet the performance of the best existing devices.
In December 1975 and August 1976, models of the Medtronic Xytron pacemaker were recalled by the FDA because of moisture intrusion and battery depletion, leading to a rate of premature failure estimated at 1% per month (MacGregor et al. Reference MacGregor, Noble and Morrow1977). In 1976, when Alan Howard joined Medtronic as its European legal director, he was asked to consider the implications of these actions for their business in Europe. He arranged to meet ministries of health in several countries but found that most were unfamiliar with the concept of device recalls and uncertain how to respond (AJH, personal communication). There was very little relevant legislation. Ultimately, recalls of Xytron pacemakers affected more than 40,000 units, with up to 15% risk of sudden death for some models (Tyers Reference Tyers1989).
Within Medtronic, Alan Howard collaborated with an engineer, Joseph (Joe) Cordonnier. They developed ideas about product liability and how to ensure quality, and they debated the possibility of a European regulatory approval system, with other manufacturers. The companies founded the International Association of Pacemaker Manufacturers (IAPM) in 1978 as a trade association, so that they could speak with an authoritative single voice; Joe Cordonnier became its first Secretary General. The goals of IAPM included collaboration with all parties interested in establishing international norms and standards, and cultivation of regular contacts with governmental authorities (Willingmyre Reference Willingmyre1990).
The first European directive concerning medical technology was passed in 1984, with specific relevance to electro-medical equipment (European Council 1984b). The European Commission issued this directive in order to promote adoption of the International Electrotechnical Commission (IEC) standard 60101 from 1977 that referred to general requirements for safety and performance (for updated version, see International Electrotechnical Commission 1988) – but it did not concern implantable devices, adherence was not monitored, and its impact was limited. The first EU legislation for general product liability followed in 1985 (Directive 85/374 EEC). After IAPM had expanded its charter to include the formulation of EEC directives, and also the coordination of industry representation in the European standards organizations the Comité Européen de Normalisation (CEN), and the Comité Européen de Normalisation Électrotechnique (CENELEC) (Willingmyre Reference Willingmyre1990), it provided input that was described as instrumental for a European standard on ‘Safety of implantable cardiac pacemakers’ (CENELEC 1988). In 1986, the scope of IAPM was broadened to include heart valve manufacturers, and it was redesignated as the International Association of Medical Prosthesis Manufacturers. Later it amalgamated with Eucomed, now MedTech Europe.
Engagement with the European Commission
In the mid-1980s, and independently, both IAPM and COCIR approached Directorate General III of the European Commission (DG III), which was responsible for the Internal Market. Alan Howard recalls that in an early meeting, he and Cordonnier on behalf of IAPM found themselves explaining the scope of the 1976 Medical Device Amendments in the USA, including categories of pre-market approvals and recalls (AJH, personal communication). They summarized the medical device regulatory situations in key European countries that they had overviewed, which led to discussions about what to do in Europe for medical devices. An approach similar to the one being applied for pharmaceutical products was deemed inappropriate.
The European Commission invited COCIR to nominate an expert who could join a new sub-unit that was to be formed in DG III. At that time, ‘there was no medical device expertise or experience of any kind within the Commission’ (Higson Reference Higson2001). COCIR signed an agreement with the Commission in 1987. Its presidency was held then by the Italian association ANIE (Federazione Nazionale Imprese delle Elettrotecniche ed Elettroniche), and it proposed Dario Pirovano, an engineer employed by Saccab SpA in Milan, who represented the Italian standards body (Comitato Elettrotecnico Italiano, CEI) on Technical Committee 121 of the International Organization for Standardization (ISO) which prepared guidance for anaesthetic and respiratory equipment. After an interview with Ernesto Previdi, who was responsible for ‘Technical harmonization and standardization’ in DG III, he was offered the position and went to Brussels to work on contract as a technical expert at the European Commission from June 1987 until 1991. His first duty was to establish a panel or working group of about 10–15 experts to advise about medical devices (DP, personal communication).
It was decided that the first directive would be for active implantable devices, for two reasons: it would be relevant to pacemakers, and it was the preference of Previdi (and the head of the section, Tom Garvey) to start with a focused approach to an identifiable and limited group of products. The experts who were appointed included people from national ministries of health (including Higson), people from industry with experience of standardization, and clinicians who had contributed to ISO standards (including an anaesthetist with an interest in technology, Peter Thompson) (Thompson Reference Thompson1987). The main objective for the panel was to assist the Commission in drafting the text of the first directive.
It was written in 1990 that ‘IAPM has succeeded in establishing itself in the eyes of the Commission of the European Communities as a sufficiently important and responsible body to propose a draft directive for active implantable electromedical devices, the first of its kind in Europe’ (Hutt Reference Hutt and Estrin1990). IAPM did submit suggestions for the directive on active implantable devices, but so too did COCIR. Eucomed (now MedTech Europe) formed a committee and prepared a report for the European Commission describing ‘appropriate features’ for a directive on non-active medical devices (Higson Reference Higson2001). Thus, there were interactions between all the trade associations and the European Commission, with each association seeking to deliver legislation relevant to its members’ interests. Some working documents were produced for three legislative proposals, but later they were reduced to two (active implantable, and general medical devices).
Decision to Adopt the ‘New Approach’
Article 30 of the Treaty of Rome had prohibited ‘quantitative restrictions on imports and all measures having equivalent effect’ between member states, but in the 1980s the consensus view was that in many sectors there was still not a properly functioning common European market. Having to comply with different conditions across various member states was judged to impact negatively on the competitiveness of European industry. A 1985 White Paper set a target date of 1992 for completing the internal market (European Commission 1985).
The ‘New Approach’ was established by a resolution from the Council in 1985 which stated: ‘National bodies authorized to issue marks or certificates of conformity shall be notified by each Member State to the Commission and to the other Member States’ (European Council 1985). Thus were born the European notified bodies, at a time when economic priorities and the functioning of the internal market dominated the agenda of the Delors presidency of the European Commission (Petrini Reference Petrini2016). Their role was to introduce more fair competition, by ensuring that all approved products met the prescribed standards. Once a certificate of conformity had been awarded, then its manufacturer could affix a CE-mark (for Conformité Européenne) to its product and sell it anywhere in the EEC.
The White Paper (at Annex page 14) proposed that in 1988 the Commission should draft a directive for electro-medical equipment (European Commission 1985). The unit in the European Commission that was given the job of managing medical device legislation, also had responsibility for New Approach legislation. It designed and structured the first directive ‘as regards technical requirements, in the light of the Council Resolution of 7 May 1985 on a new approach to technical harmonization and standardization’ (European Commission 1991: 436). The earlier directive 84/539 (European Council 1984b) was considered to have failed because it lacked such a structure.
Higson recalled the expert group as a ‘Working Group on the Reduction of Technical Barriers to Trade in Medical Equipment’, drawn from trade associations, standards bodies (CEN and CENELEC), member states, the European Free Trade Association (EFTA), and ‘occasionally, members of the medical profession’ (Higson Reference Higson2001). The Commission issued working papers that were discussed at working group meetings. An agreed Commission text was proposed formally to the Council at the end of 1988, and the working group was then dissolved (Higson Reference Higson2001).
There were some disagreements during the preparation of the draft legislation. There were reservations from France, and the UK argued strongly for confidentiality (DP, personal communication). Some countries wanted to retain their existing testing houses. When the proposals were considered by the Council, there were some suggestions to use the same system as for pharmaceutical products. ‘Germany (G Schorn) spoke of the need for evaluation to show therapeutic or diagnostic effectiveness and to make observations of side-effects being needed for devices through clinical trials’ (Higson Reference Higson2001). That proposal was not adopted, since it was considered that the economic burden of establishing an agency to implement these requirements was not justified, and because member states had too few people with sufficient knowledge of devices to be able to staff a new agency (DP, personal communication).
The legislative proposal was reviewed by the Economic and Social Committee (ECOSOC, an advisory body of civil society in the EU) and by the European Parliament during 1989. It was also scrutinized by a Council Working Group consisting of Member State officials with responsibility for the safety of medical devices, in 1989 and 1990. A Common Position, reflecting agreement between the Commission, the Parliament and the Council Working Group, was agreed early in 1990 (Higson Reference Higson2001), and the Directive on the approximation of the laws of the Member States relating to active implantable medical devices (90/385/EEC) was adopted on 20 June 1990 (European Council 1990).
In November 1993 Norbert Anselmann, who was then Principal Administrator in the new unit in DG III, with Jos Putzeys as head, acknowledged tacitly that the approach was not designed from a medical perspective, when he wrote that:
The Medical Device Directives follow the ‘New Approach’, a methodology to be applied to the harmonization of industrial products. The EC legislation is confined to the setting up of Essential Requirements which are drafted in rather abstract terms. The technical expression of these requirements is ensured by European standards, the application of which is at the discretion of manufacturers. (Anselmann Reference Anselmann and Higson1993)
Clinical Problems Accelerating Development of Directives
The development of European directives for medical devices may have been triggered by manufacturers, but the debates were also influenced by clinical problems with devices (Personal communications AJH, DP, JT). Foremost amongst these, during the 1980s, were deaths caused by fractures of the outlet strut of the Björk-Shiley convexo-concave mechanical heart valve. In 1978, its manufacturer (Shiley) increased the angle at which the strut was welded to the valve ring, from 60° to 70°; the rate of strut fracture increased from 0.6% for the 60° model, to 3.9% for the 70° model. There had been insufficient bench testing, but valve failure was described as an anomaly and the company (Shiley had been bought by Pfizer in 1979) continued to sell valves with the modified design, but only outside the USA. Ultimately it was estimated that acute mechanical complications (such as escape and embolism of the valve occluder, leading to torrential regurgitation) had occurred in about 1% of 86,000 valves implanted in patients, leading to about 800 deaths with a peak incidence between 1983 and 1994 (Hiratzka et al. Reference Hiratzka, Kouchoukos and Grunkemeier1988; Blot et al. Reference Blot, Ibrahim and Ivey2005).
Implementation of the EU Medical Device Directives
The primary goal of the Active Implantable Medical Device Directive 90/385/EEC (AIMD) (European Council 1990) was focused on health, as noted in its first recital: ‘Whereas in each Member State active implantable medical devices must give patients, users and other persons a high level of protection and achieve the intended level of performance when implanted in human beings’. The focus of the second, general directive that was introduced in 1993 (Medical Device Directive 93/42/EC; ‘MDD’) (European Council 1993) was more on trade and the single market. Its preamble opens with this statement: ‘Whereas measures should be adopted in the context of the internal market; whereas the internal market is an area without internal frontiers in which the free movement of goods, persons, services and capital is ensured…’ (European Council 1993).
The MDD stated that clinical data were required ‘in particular’ for implantable and Class III devices, implying that they were not needed or were less relevant for other devices. Those data could be sourced from any published or unpublished report, either for the device in question or for another device if it was shown to be ‘equivalent’. As a result, in the first years of the Directives, some medical devices came to market without any compilation of clinical material, and most medical devices did not need any form of pre-market clinical investigation. For many low-risk devices, of course, a generalized requirement for clinical evidence was unnecessary.
The functioning of the medical device directives was reviewed by the Medical Device Experts Group (MDEG) in 2002. It noted that ‘proper implementation of provisions on clinical data’ had not been undertaken and that there were shortcomings in notified body assessments of clinical data (Medical Device Experts Group 2002; European Commission 2003). The Commission concluded in 2003 that there was a need to adopt specific regulatory measures (European Commission 2003). A requirement for clinical evaluation of all medical devices was introduced into the revision of the directives in 2007, in light of ‘technical innovation’ and ‘the development of initiatives at international level’ (Recital 8) (European Council and Parliament 2007).
The European Notified Bodies
During the nineteenth century, the steam engine underwent continuous technological developments which increased power output by a factor of more than 40, so there was a large increase in boiler explosions. In 1882, the UK parliament passed its first law to regulate this problem (Bartrip Reference Bartrip1980). The world’s first national standards body was formed in the UK in 1901, as the Engineering Standards Committee; it was redesignated as the British Standards Institution (BSI) in 1931. The direct descendent of this organization, BSI Group The Netherlands BV, is a large EU notified body designated for evaluating medical devices.
In Germany, by 1871, there was a Union of Boiler Inspection Associations, with membership exempting the operator of the engine from review by a state inspector. In March 1938, 14 new regional technical inspection associations (Technische Überwachungsvereine, or TÜV) were created (TÜV SÜD 2024). Three of these persist now as independent notified bodies approved for evaluating medical devices, namely TÜV Süd, TÜV Rheinland, and TÜV Nord.
By 1993, the first eight notified bodies designated to review medical devices in the EU included both BSI and TÜV Süd. Another on the list was DEKRA, whose origins were from another sector – Deutscher Kraftfahrzeug Überwachungs-Verein is the German Heavy Goods Vehicle Surveillance Association.
European Standards for Medical Devices
It was the early intention of the European Commission, for the new device directives, that ‘The implementation of the concept of reference to standards will be facilitated […] important work on the establishment of a sectoral system of harmonized standards has already been started by CEN/CENELEC’ (European Commission 1991).
When the Directives were first introduced, there were no harmonized standards for medical devices. In 1993, a request was issued for detailed standards relating to 109 specific medical devices or groups of devices (Higson Reference Higson2001). Very few were issued under the device directives, however, and the MDR has required all standards to be ‘re-harmonized’. As a result, their number has fallen from around 220 under the Directives (the vast majority originating from ISO or IEC), to about 20 (as of January 2025).
Transition to the Medical Device Regulation 745/2017
The long process of recasting the European medical device directives was initiated in 2008. It gathered impetus from clinical problems that were publicized later by the International Consortium of Investigative Journalists (ICIJ). The scandal about Poly Implant Prosthèse (PIP) silicone breast implants was an example of fraud, which went undetected initially because the manufacturer was given notice of pending inspections, which were also infrequent (Oulharj et al. Reference Oulharj, Pauchot and Tropet2014; Greco Reference Greco2015). Local soft-tissue reactions and osteolysis from metallosis due to excessive wear of large-head metal-on-metal hip implants, with loosening of components and loss of stability (van Lingen et al. Reference van Lingen, Zagra and Ettema2016), was detected first by the Australian national joint registry; the arthroplasty prosthesis had been approved without large clinical trials, on the basis of equivalence (Ardaugh et al. Reference Ardaugh, Graves and Redberg2013). Surgical and vaginal meshes had similarly been approved and CE-marked on the basis of equivalence to predicate devices that had been taken off the market or that differed in design (Heneghan et al. Reference Heneghan, Goldacre and Onakpoya2017).
The PIP and hip implant problems prompted urgent regulatory measures in a ‘Joint Action Plan’ (European Commission 2012b) that was announced by the Commissioner for Health, John Dalli, before publication of the texts proposed for the new regulations – for which they had acted as a ‘stress case’ (European Commission 2014). The resultant Medical Device Regulation 2017/745, published in May 2017, combined and greatly expanded the active implantable and general medical device Directives, with rules adapted from existing guidance and standards and other legislation. The same organizations (national competent authorities, notified bodies, and the European Commission) were assigned similar roles, but Expert Panels were introduced as a political compromise to avoid a drug-agency model of regulation. As a response to challenges from implementing the MDR, the European Commission has adopted a variety of delegated acts, and the Medical Device Coordination Group has prepared or updated guidance documents (European Commission 2025). Expert Panels can now provide advice to developers, in addition to opinions on certain high-risk devices (European Medicines Agency 2025). These measures are running in tandem with the consultation on the MDR framework, which allows an opportunity to consider systematic change.
What can be Learned?
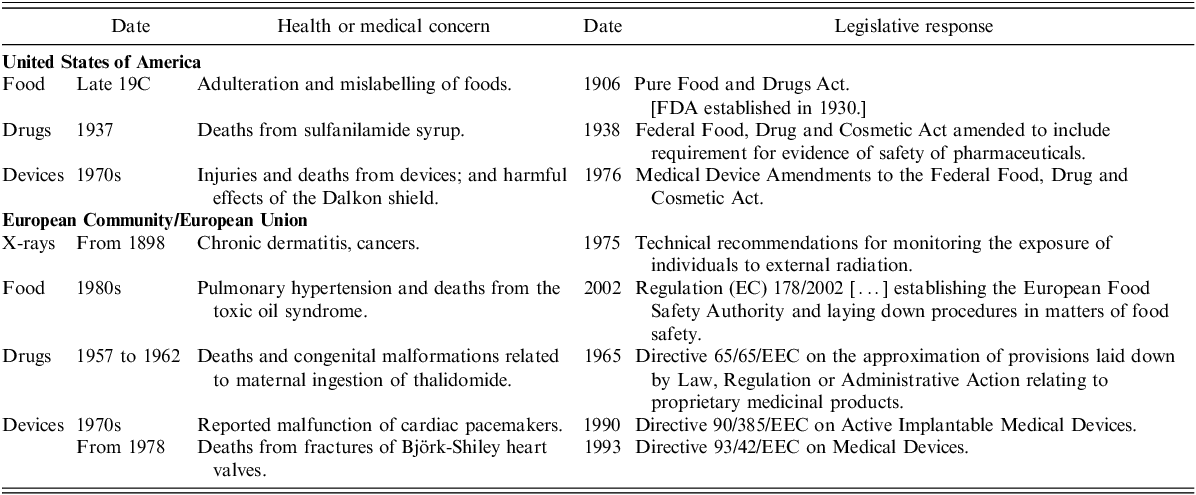
The predominant theme arising from this historical review is that regulation – whether of pharmaceutical products or medical devices and whether in the USA or in Europe – seems almost always to have been a reaction to problems, crises or external pressures, rather than a process planned proactively from first principles (Table 2, and Figure 1). When we consider that European medical device rules have been developed without a thorough evaluation of scientific principles, is it any wonder that the EU regulatory framework for medical devices is not more ‘fit for purpose’?
Summary of temporal relationships (whether causal or coincidental) between clinical events and first US or EU regulations

Notes: 19C – nineteenth century
FDA – Food & Drug Administration
Overview of possible clinical triggers and first US or EU regulations

Principles of Clinical Evidence
One striking difference between the US and EU systems is that the 1976 Medical Device Amendments in the USA were founded on identification of public health hazards and a scientific evaluation of appropriate controls for different levels of risk. They required substantial evidence of safety and efficacy. The focus of EU directives since 1990 has been on safety and performance; no EU legislation for medical devices has been preceded by a scientific evaluation of the effects of policy options on public health.
Having clear and proportionate regulatory pathways is vital in order to provide clarity of expectations for clinical evidence. Drugs and high-risk medical devices in the USA are typically evaluated using experimental study designs, with randomized controlled trials (RCTs) being the norm. Reference to efficacy rather than ‘relative clinical effectiveness’ (as originally intended by Kefauver) led to a focus on RCTs but made evidence for some decisions about reimbursement less clearcut (Greene and Podolsky Reference Greene and Podolsky2012). There is a need to understand when comparative effectiveness is the most informative criterion for decisions about treatment, versus when RCTs should be expected, and the consensus should be reflected in regulations.
For devices in the EU, it is necessary to submit ‘clinical data providing sufficient clinical evidence’ (MDR, Article 61(1)) but sufficiency is not defined. Clinical study designs are typically observational rather than experimental, and often there is no comparator. This likely sets a standard that is too low for high-risk permanent implants with a critical function, but perhaps too high for low-risk products. The lack of full transparency of clinical evidence in Europe (Fraser et al. Reference Fraser, Butchart and Szymański2018) – another major difference compared with the USA – was recognized in the Impact Assessment for the recast of the directives (European Commission 2012a), but not satisfactorily resolved in the MDR: the evidence that is submitted to a notified body for high-risk devices will in future be published only in summary (MDR, Article 32).
The basic structure of the regulatory system, which was derived from legislation for industry, can appear to place respect for manufacturers’ confidentiality above the needs and rights of patients. The MDR requires a Summary of Safety and Clinical Performance (SSCP) to be made available for certain high-risk devices (MDR, Article 32), and reports of clinical investigations are required to be made public (MDR, Article 77(7)). Until the implementation of the clinical module of the European Database on Medical Devices (EUDAMED) occurs, perhaps in 2026 or 2027, dissemination of these reports is limited in practice. Partial access to manufacturers’ reports of serious incidents will be available in future when the vigilance module of EUDAMED is active, but the extent of information that will be shared is not yet known. Transparency of clinical evidence and data sharing should be the norm in device regulation.
Regulatory Frameworks
Adoption of the ‘New Approach’ framework for medical devices in the European Union has delivered a single market, which was the primary motivation of the European trade associations when they advocated for legislation in the 1980s, but a consequence has been insufficient priority given to developing standards for clinical evidence and to other initiatives to improve the public health. Assessment and certification of devices was handed to predominantly private sector companies – the notified bodies – when in other jurisdictions those are typically roles for government institutions such as the FDA (Horton Reference Horton1995). Before the directives, they were also the responsibilities of government departments in some of the member states (see Table 1).
The MDR has increased the procedural requirements for notified bodies, for example by introducing unannounced audits as part of the assessment procedure (MDR, Annex VII). Despite these changes, the client–service relationship between notified bodies and manufacturers has the potential to create biases and conflicts of interest; for example, a notified body may be hesitant to withdraw the certification for a manufacturer on safety grounds, as this could lead to the manufacturer moving their files to another notified body. The extent to which increased procedural requirements for notified bodies, and increased oversight by their national designating authorities, can overcome these conflicts and biases remains to be seen. Market dynamics have changed between manufacturers and notified bodies in recent years, as there was a significant shortage of notified body capacity in 2022 that has dissipated as a result of the revised transition timelines.
The EU framework is intended to apply to categories of products that are sufficiently homogeneous to allow their ‘essential requirements’ to be defined. Arguably this was appropriate for the first directive in 1990, on active implantable medical devices including pacemakers, but it is far less suitable for general medical devices. The conceptual challenge of designing the same requirements for all risk classes, including everything from wheelchairs to heart valves, seems to have been given scant regard in the move from EU Directives to the MDR.
Another key difference between the US and EU systems is the clear distinction in the USA between high-risk medical devices in the premarket approval pathway, which require clinical evidence, and medium- and low-risk devices, which require compliance with standards and which can be marketed based on claims of substantial equivalence to another device (by the 510(k) pathway). This was recommended in the Cooper Report and implemented by classifying technologies and linking them to regulatory pathways. The FDA may require some additional clinical evidence to support a medium-risk product, but that can be agreed with the agency in advance. In contrast, the EU system applies the same general requirements for clinical evaluation to medical devices of all risk classes (MDR, Article 61(1)), without defining precisely what clinical evidence will be needed for each type. The MDR requires clinical investigations for high-risk devices (MDR, Article 61(4)), but with exceptions such as when introducing modifications to an approved device; for legacy (i.e. previously marketed) devices; when claiming equivalence to another device; and for implants of low risk (MDR, Article 61(4) – 61(6)). This can lead to overzealous interpretation of the requirements for clinical evaluation by notified bodies in some cases, and an acceptance of limited clinical evidence in others.
Both the US and EU regulatory systems depend on technical standards to provide reliable requirements for approving medium-risk devices. That was the original plan for the class II category introduced by the 1976 US amendments, but it was changed in 1990 to a ‘clearance’ pathway based on a claim of substantial equivalence to a device already on the market. Creating sufficient numbers of recognized (US) or harmonized (EU) device standards has been challenging in both regulatory frameworks. Unintended consequences of too few standards in the EU include the absence of special pathways for orphan products, devices for children, and breakthrough innovations; new legislation is being considered to address this deficiency.
Resources, Capacity and Expertise
One striking similarity across the regulatory systems is that their resources have rarely met the requirements needed to implement significant regulatory changes. This was evident in the USA for the Kefauver-Harris pharmaceutical amendments in 1962 and the medical device amendments in 1976, and in the EU for the medical device directives in 1990 and regulations in 2017. The MDR introduced many new requirements, but the member states and EU institutions did not provide infrastructure and capacity to deliver them properly. The European Commission estimated that an additional 35–50 staff would be required to implement the MDR (European Commission 2012a); the Unit D3 in DG SANTE (as of February 2025) has 18 staff with five more providing administrative support.
The consequences of insufficient political commitment to invest what is needed, were stated well by a UK health minister and cited in the MDEG report in 2002:
It is not enough to produce better guidance, clarify rules and tighten the requirements upon manufacturers. What is needed above all is for national authorities across Europe, and the European Commission itself, to devote sufficient human and other resources to this important area. It is clear that this is not the case now. There is a real risk that unsafe medical devices could be placed on the market. They could remain on the market until they have compromised, perhaps fatally, the health and safety of patient or user. That is unacceptable. (Hunt Reference Hunt2002)
A scarcity of human resources at EU member state level and in EU institutions was identified in 2012, by the European Commission in its Impact Statement (European Commission 2012a).
The regulation and approval of pharmaceutical products and devices in the USA is undertaken by the FDA as a single central agency with about 18,000 employees. Its Center for Devices and Radiological Health (CDRH) had 2230 dedicated staff in 2023 (US FDA, Center for Devices and Radiological Health 2024). The European Medicines Agency employs about 900 people. The EU regulatory system for devices is coordinated by the small unit in the European Commission, with about 10 policy officers, but devolved to national agencies and delivered by the notified bodies. In 2023, 35 notified bodies who are members of their trade association, Team–NB, employed a total of 5267 staff of whom 3650 were engaged full-time or as external contractors in the evaluation of medical devices for conformity assessments (Team–NB 2023). Using concurrent estimated populations in the USA and EU, respectively, of 345 and 449 million, and staff totals including those engaged in supporting activities, these figures equate to one medical device ‘regulator’ per 154,708 inhabitants in the USA and at least one per 85,248 citizens in the EU (not counting those employed by the notified bodies who are not members of Team–NB; or those in competent authorities). By the end of 2024 there were 50 notified bodies designated for the MDR, of whom 44 are members of Team–NB.
Concerns about the EU regulatory system for devices thus cannot be attributed to staff numbers in regulatory bodies; even allowing for many assumptions in the calculations, there are more staff in the EU than in the USA. Instead, the inefficiency of the system can be attributed to its structure. From a clinical perspective, it seems illogical for the EU to have such different regulatory systems for drugs and devices, but for the historical reasons outlined in this review, the system for devices has evolved so that different organizations are responsible for approving clinical study designs, certifying market access, and monitoring safety. Coordination is theoretically possible but unlikely to be successful when each organization is responsible for only one part of the system. In 2017, the European Competent Authorities for Medical Devices (an association of national regulatory agencies of member states) wanted to create guidance documents (Competent Authorities for Medical Devices 2017), but found that there was no forum for determination of basic scientific principles to support their development.
Ethics and Regulatory Convergence
All proposals for major legal amendments to assure safety and effectiveness have been followed by claims from industry that increasing standards for evidence would hamper the introduction of innovative technologies. Claims of a ‘drug lag’ were made while the Kefauver-Harris amendment was being debated, on the basis that Europe would have earlier access to medicines than the USA. When the MDR was being negotiated, the European trade association Eucomed launched a campaign entitled ‘Don’t lose the 3’, claiming that creation of an agency to regulate devices would lead to loss of a 3-year competitive access to innovative technologies in Europe. The FDA set up its pathway for accelerated approval of ‘breakthrough’ products to reverse these time lags.
Every attempt at improving regulatory frameworks may introduce unanticipated challenges. For drugs in the US, the requirement to demonstrate efficacy led to many medicines for children being withdrawn. Similar challenges have been faced in the EU for devices for children or orphan indications. Legislators introducing health policies, and regulators with public health roles, have an ethical responsibility to the public, and similar standards should apply worldwide. There may always be some risks from ‘high-risk’ devices, but the extreme examples cited in this review should be avoidable by appropriate pre-market requirements. The impact of other unexpected issues can be minimized only by collaborations between regulatory authorities who are willing to share their expertise, insights, and regulatory judgements.
Conclusions
The current EU laws for medical devices, the MDR (European Parliament 2017a) and the IVDR (European Parliament 2017b), and in particular their clinical evidence requirements, were the subject of protracted negotiations to agree their final texts during one of the longest ever trilogues between the European Council, Parliament and Commission. There is likely to be limited political appetite to revise the basic legislation, but it appears unavoidable that the fundamental objectives of the regulations need to be reconsidered if they are to achieve the stated aims of protecting public health and supporting innovation (see Recital 1 in the MDR).
The EU directives established criteria for device performance rather than effectiveness, protected intellectual property over the need for transparency, and supported innovation more than safety. The framework was adapted from legislation designed for manufacturing sectors, using inspection bodies that had been developed to meet the needs of industry, and applying standards from international bodies that were established by industry. The MDR retained the same basic structure and was introduced without scientific analysis or testing in practice, as would be optimal now if adopting an alternative ‘regulatory science’ approach. A similar re-analysis is needed for the IVDR but was not a focus of this review. Legislative frameworks should be ‘evidence-based’, applying what has been shown by research to provide the best outcomes for patients.
Acknowledgements
We thank Alan J. Howard, Dario Pirovano and Jeremy Tinkler most sincerely, for generously sharing their personal experience and insights. The opinions in this manuscript, however, remain the responsibility of the authors.
We are grateful also for colleagues who checked the details of their country’s regulatory framework before 1990, including Dario Pirovano (Italy), Steve Eglem (Belgium), Dimitris Panidis (Greece), Eamonn Hoxley and Jeremy Tinkler (UK), Camille Mesnage (France), and Carmen Ruiz-Villar Fernandez-Bravo (Spain).
We are grateful to John P. Swann from the US FDA History Office for providing the Cooper report and supporting material.
Funding
None.
About the Authors
Alan G. Fraser is Emeritus Professor of Cardiology at Cardiff University, UK, and Visiting Professor in the Department of Cardiovascular Sciences at KU Leuven in Belgium. Since 2012 he has represented clinicians at meetings of European medical device regulators, organized by the European Commission in Brussels. He established and led Regulatory Affairs committees, first for the European Society of Cardiology and then for the Biomedical Alliance in Europe. He was scientific coordinator of the EU CORE–MD project (Coordinating Research and Evidence for Medical Devices).
Rita F. Redberg, MD, MSc, is a cardiologist and Professor of Medicine at the University of California, San Francisco and Core Faculty, Philip R Lee Institute for Health Policy Studies. Dr Redberg served on the Medicare Payment Advisory Commission, which advises Congress on Medicare payment issues and the Food and Drug Administration Cardiovascular Devices Expert Panel. She also served two terms on the Medicare Evidence, Development and Coverage Advisory Committee, one as Chairwoman. She has given Congressional testimony multiple times in hearings related to the issue of balancing safety and innovation in medical device approvals.
Tom Melvin is Associate Professor of Medical Device Regulatory Science at the University of Galway. Tom is Chair of the Biomedical Alliance Regulatory Affairs Committee and a member of the European Medicines Agency Expert Panels for medical devices, the European Society of Cardiology Regulatory Affairs Committee and the National Research Ethics Committee for Medical Devices. Tom previously worked at Trinity College Dublin and was a senior medical officer for medical devices at the Health Products Regulatory Authority and Chair of the Clinical Investigation and Evaluation Working Group of the European Commission.



 Open access
Open access