


Summations
-
• Resting fMRI: Ketamine was associated with fronto-striatal/ACC connectivity changes (direction varied by circuit and dose).
-
• Task fMRI: Ketamine was associated with higher striatal reward feedback responses and mPFC/ACC modulation.
-
• PET: PET suggested dose-related ACC/PFC metabolic/connectivity effects and region-specific 5-HT1B binding changes.
Limitations
-
• Heterogeneity across modalities and tasks, as each captures different aspects of neural function (evoked versus intrinsic/spontaneous activity), limiting direct comparability.
-
• Methodological variability and participant heterogeneity may have influenced results.
-
• Most imaging assessments were acute/subacute with limited follow-up, restricting inference about durability of neural changes and relationships to longer-term outcomes.
-
• Only a minority of studies used validated anhedonia-specific scales, and many studies had modest sample sizes, limiting symptom-specific interpretation and generalizability.
Introduction
Major depressive disorder (MDD) is one of the leading contributors to global disability and disease burden, with an estimated lifetime prevalence of ~15% worldwide.Reference Rhee, Wilkinson, Steffens, Rosenheck and Olfson1, Reference Rhee, Bommersbach, Rosenheck, Nierenberg and McIntyre2 MDD is characterized by persistent low mood, diminished energy, and anhedonia—marked reduction in the ability to experience pleasure or interest in previously enjoyable activities—which negatively impacts quality of life, interpersonal relationships, and overall homeostatic functioning.Reference Rhee and Steffens3–Reference Wong, Le and Phan9
Replicated neuroimaging evidence suggests that disturbances within reward-related brain regions, particularly the prefrontal cortex and mesocorticolimbic structures, are implicated in individuals with MDD.Reference Le, Wong and Haikazian6, Reference Ng, Alloy and Smith10, Reference Halahakoon, Kieslich, O’Driscoll, Nair, Lewis and Roiser11 The mesocorticolimbic dopamine system, comprising the ventral tegmental area (VTA), nucleus accumbens (NAcc), ventral striatum, and prefrontal cortex (PFC), has a central role in mediating reward-related processes.Reference Marche, Martel and Apicella12–Reference Solomonov, Victoria and Lyons15 Task-based functional magnetic resonance imaging (fMRI) studies have consistently demonstrated reduced activation in the ventral striatum and NAcc during reward paradigms in MDD,Reference Abdallah, Averill and Collins16, Reference Cardona-Acosta and Bolaños-Guzmán17 alongside altered fronto-striatal connectivity during reward processing tasks.Reference Barch, Pagliaccio and Luking18–Reference Borsini, St John Wallis, Zunszain, Pariante and Kempton20
Preclinical studies have revealed activity of the NAcc is dependent on the reward-association learning process, confirming that dopaminergic D1 receptor modulation in medial NAc supports motivation for both natural and drug rewards.Reference Rojo, Cai, Barnhardt and Huang21, Reference Nishitani, Kokume, Taniguchi and Kaneda22 Notwithstanding advances in pharmacotherapy, conventional monoaminergic antidepressants (eg selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors) have limited efficacy in alleviating and/or ameliorating reward-processing, hedonic capacity and motivational drive impairments.Reference Katzman and Sternat23–Reference Lee, Mansur and Brietzke26 The aforementioned limitations are especially pronounced in individuals with treatment-resistant depression (TRD) (ie a subgroup of patients exhibiting an inadequate response to at least two antidepressant trials), underscoring the need for mechanistically informed pharmacotherapeutics that target reward-related pathophysiology.Reference McIntyre, Alsuwaidan and Baune27–Reference Maj, Stein and Parker30 Ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist, has rapid antidepressant effects with unique mechanistic properties in persons with depression.Reference Iadarola, Niciu and Richards31 Importantly, clinical evidence indicates that its efficacy is not inferior to electroconvulsive therapy in treatment-resistant depression, and accumulating data further demonstrate that ketamine rapidly improves outcomes in TRD and confers significant benefits on patient-reported outcomes such as quality of life and functioning.Reference Rhee, Shim, Davoudian, Espinoza and McIntyre32
While the precise mechanisms underlying ketamine’s rapid therapeutic effects remain incompletely elucidated, prevailing hypotheses emphasize modulation of glutamatergic transmission and enhancement of synaptogenesis.Reference Wong, Le and Phan9, Reference Cooper, Rosenblat, Cha, Lee, Kakar and McIntyre33–Reference Gill, Gill and Rodrigues37 Early studies using a subanesthetic dose of ketamine (and esketamine) found a rapid and transient increase in glutamate release in the mPFC, believed to initiate activity-dependent release of brain-derived neurotrophic factor (BDNF) and stimulation of protein synthesis–dependent synapse formation, essential for ketamine’s antidepressant effects.Reference Gerhard, Pothula and Liu38–Reference Xiao, Yin and Teopiz40 Preliminary evidence suggests that ketamine may normalize frontostriatal and limbic network overactivation in TRD, thereby restoring reward responsiveness and alleviating core symptoms such as anhedonia.Reference Mkrtchian, Evans and Kraus41–Reference Le, Wong and Badulescu43 Notwithstanding emerging evidence for ketamine’s pro-hedonic effects,Reference Wong, Le and Phan9, Reference McIntyre, Rosenblat and Nemeroff34 the neurofunctional mechanisms that mediate these therapeutic effects remain incompletely understood. Herein, the present systematic review aims to (1) investigate ketamine’s neurofunctional effects on reward processing circuits across neuroimaging modalities and (2) evaluate how ketamine’s modulation of reward-related brain circuits may inform mechanistic models of anhedonia in depression.
Methods
Search strategy and reporting criteria
This systematic review was conducted following the PRISMA guidelines.Reference Lam, Kennedy and Adams28, Reference Page, Moher and Bossuyt44 A comprehensive systematic search was performed across Ovid databases (ie Embase, MEDLINE, PsycINFO), Wiley Cochrane Library, Scopus, Web of Science, and Google Scholar. The search strategy utilized a combination of Medical Subject Headings terms and free-text keywords to capture all relevant studies. Search terms were structured around four key concepts, including depression and anhedonia, ketamine, reward processing, and neuroimaging modalities. No language restrictions were applied. Database searches were conducted from February 2025 (inception) to September 2025, with an update on September 2, 2025. The protocol was not registered; however, eligibility criteria and outcomes were defined a priori and were not modified during screening, extraction, or synthesis.
Inclusion and exclusion criteria
Studies were included if they met the following criteria: (1) human participants aged 18–65 years diagnosed with MDD (including treatment-resistant depression where specified) based on standard clinical assessment or structured diagnostic interviews, (2) single/repeat-dose intravenous, oral, or intranasal ketamine or esketamine administration as the primary intervention, (3) prospective interventional studies, including randomized controlled trials (parallel or crossover), non-randomized controlled trials, and open-label/single-arm interventional studies that used validated clinical scales to assess depression severity, (4) used neuroimaging using fMRI (resting-state and/or task-based), positron emission tomography (PET), fluorodeoxyglucose positron emission tomography (FDG-PET), pharmacological magnetic resonance imaging (phMRI), or related modalities, assessing reward- and/or positive-valence–relevant circuitry during reward/emotion tasks or at rest, and (5) clearly reported timing of imaging in relation to ketamine/esketamine administration (eg during infusion and/or post-dose timepoints). Studies were excluded if they were case reports/series, reviews/meta-analyses, non-human studies, or purely observational designs without ketamine/esketamine administration.
Data extraction
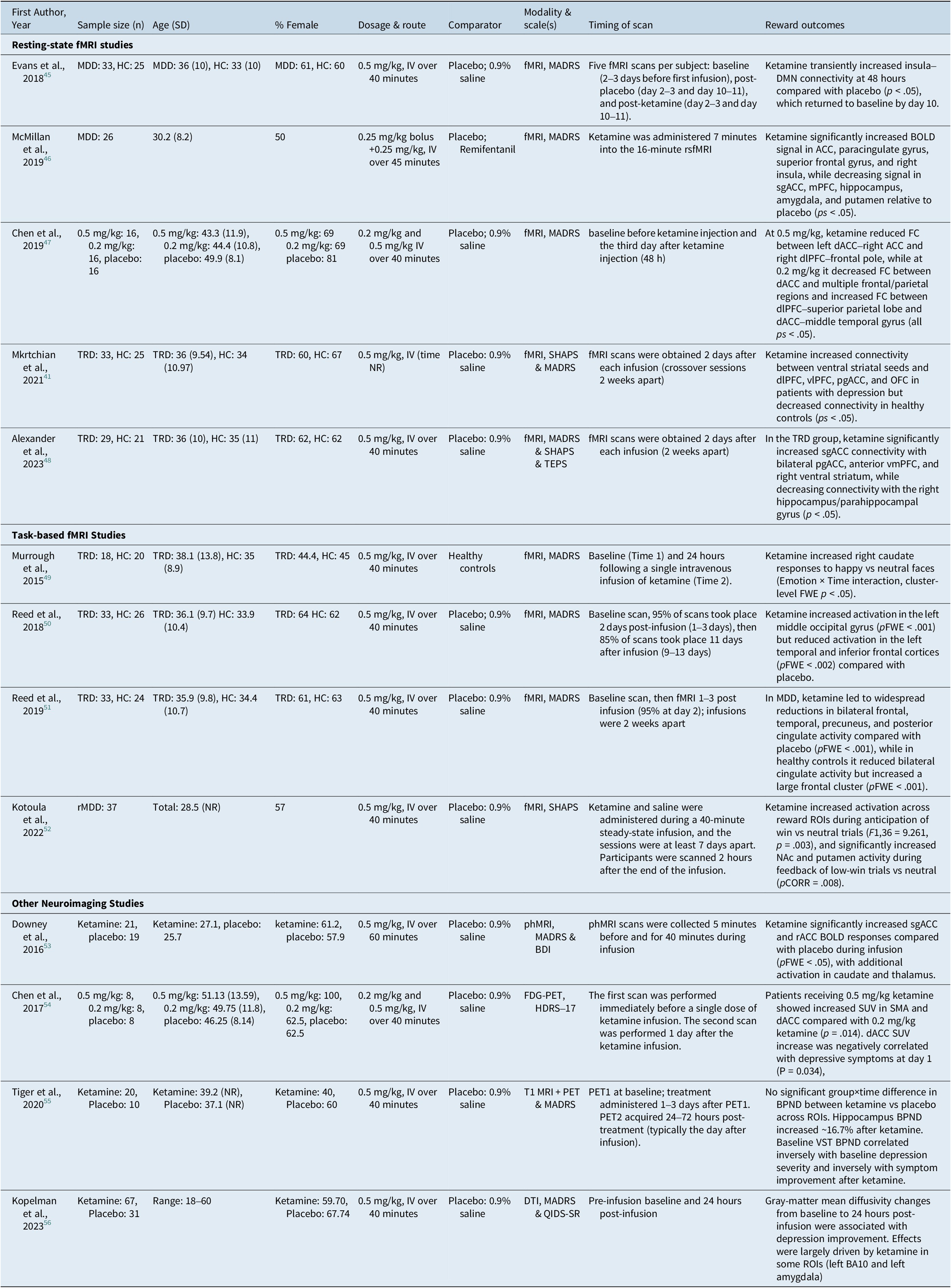
Data extraction was conducted independently by two reviewers (H.F. and W.C.), and discrepancies were resolved through consensus. The following information was extracted from included articles: first author, sample size and demographics, study design, ketamine intervention details (dose, route, and timing), type of neuroimaging modality, reward-related regions of interest, and measures of reward outcomes (Table 1).
Summary of Included Studies Evaluating Ketamine’s Effects on Reward Processing Across Neuroimaging Modalities

MDD = major depressive disorder, HC = healthy controls, TRD = treatment-resistant depression, rMDD = remitted major depressive disorder, IV = intravenous, fMRI = functional magnetic resonance imaging, rsfMRI = resting-state functional magnetic resonance imaging, phMRI = pharmacological magnetic resonance imaging, FDG-PET = fluorodeoxyglucose positron emission tomography, PET = positron emission tomography, T1 MRI = T1-weighted magnetic resonance imaging, DTI = diffusion tensor imaging, MADRS = Montgomery–Åsberg Depression Rating Scale, BDI = Beck Depression Inventory, HDRS-17 = 17-item Hamilton Depression Rating Scale, QIDS-SR = Quick Inventory of Depressive Symptomatology–Self-Report, SHAPS = Snaith-Hamilton Pleasure Scale, TEPS = Temporal Experience of Pleasure Scale, BOLD = blood-oxygen-level-dependent, FC = functional connectivity, DMN = default mode network, ACC = anterior cingulate cortex, dACC = dorsal anterior cingulate cortex, sgACC = subgenual anterior cingulate cortex, pgACC = pregenual anterior cingulate cortex, rACC = rostral anterior cingulate cortex, mPFC = medial prefrontal cortex, vmPFC = ventromedial prefrontal cortex, dlPFC = dorsolateral prefrontal cortex, vlPFC = ventrolateral prefrontal cortex, OFC = orbitofrontal cortex, NAc = nucleus accumbens, ROI/ROIs = region(s) of interest, FWE = family-wise error, pFWE = family-wise error–corrected p value, pCORR = corrected p value, SUV = standardized uptake value, SMA = supplementary motor area, BPND = binding potential (non-displaceable), VST = ventral striatum, BA10 = Brodmann area 10, NR = not reported, mg/kg = milligrams per kilogram.
Results
Search results
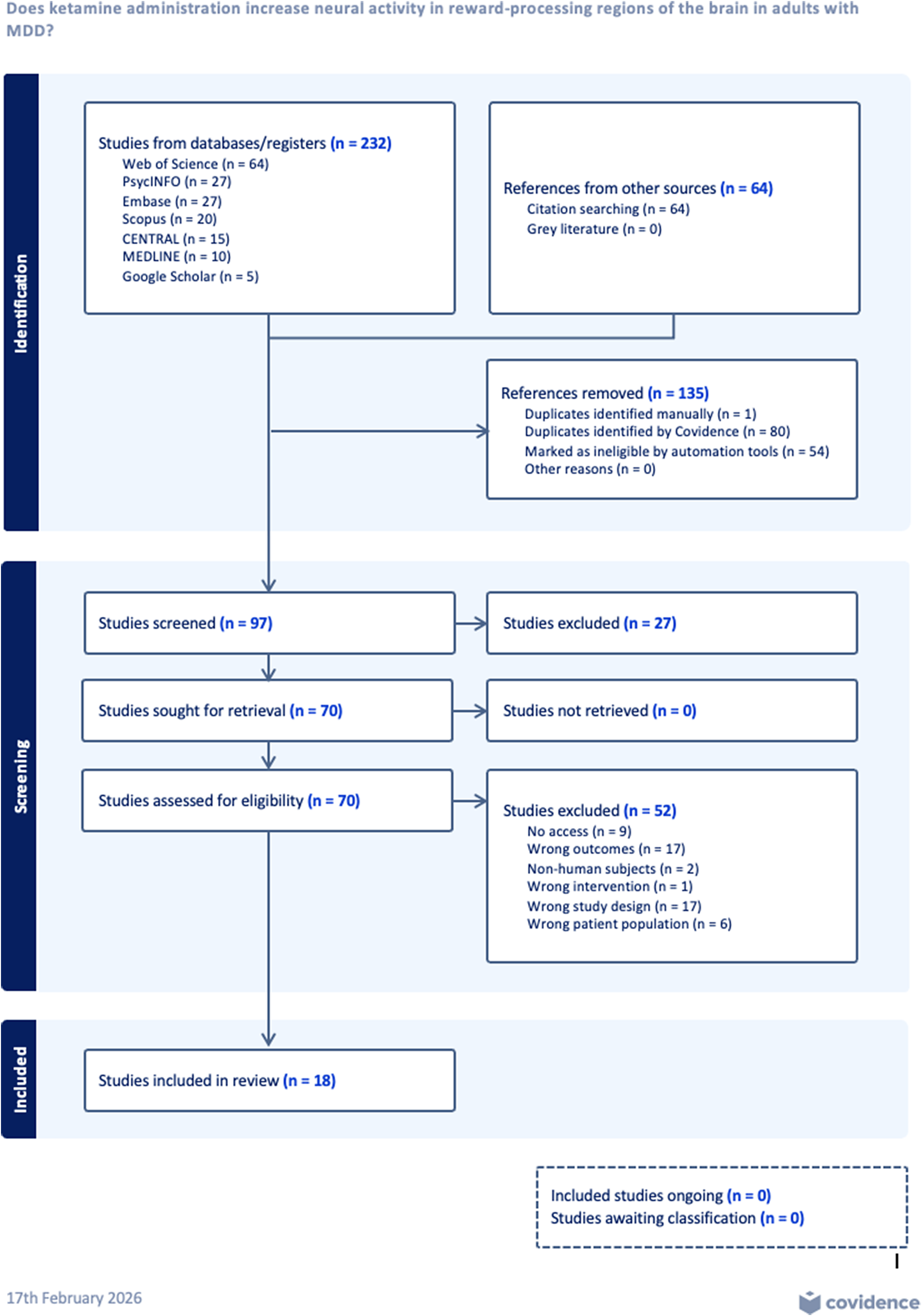
The initial search identified a total of 232 articles (Figure 1). Additionally, 64 studies were identified through citation searching. After the removal of 135 duplicate and ineligible records, title and abstract screening were conducted for 97 articles (n = 27 articles excluded). A total of 70 studies were sought for full-text review, and 52 studies were excluded for the following reasons: inaccessibility (n = 9), wrong outcomes assessing other brain circuits or using irrelevant clinical measures (n = 17), non-human subjects (n = 2), wrong intervention (n = 1), wrong study design (n = 17), and wrong patient population (n = 6). Subsequently, 18 studies met eligibility criteria and were assessed for risk of bias; 13 studies were included in data extraction and qualitative synthesis (Table 1).
PRISMA flow diagram showing the study selection process.

Risk of bias assessment
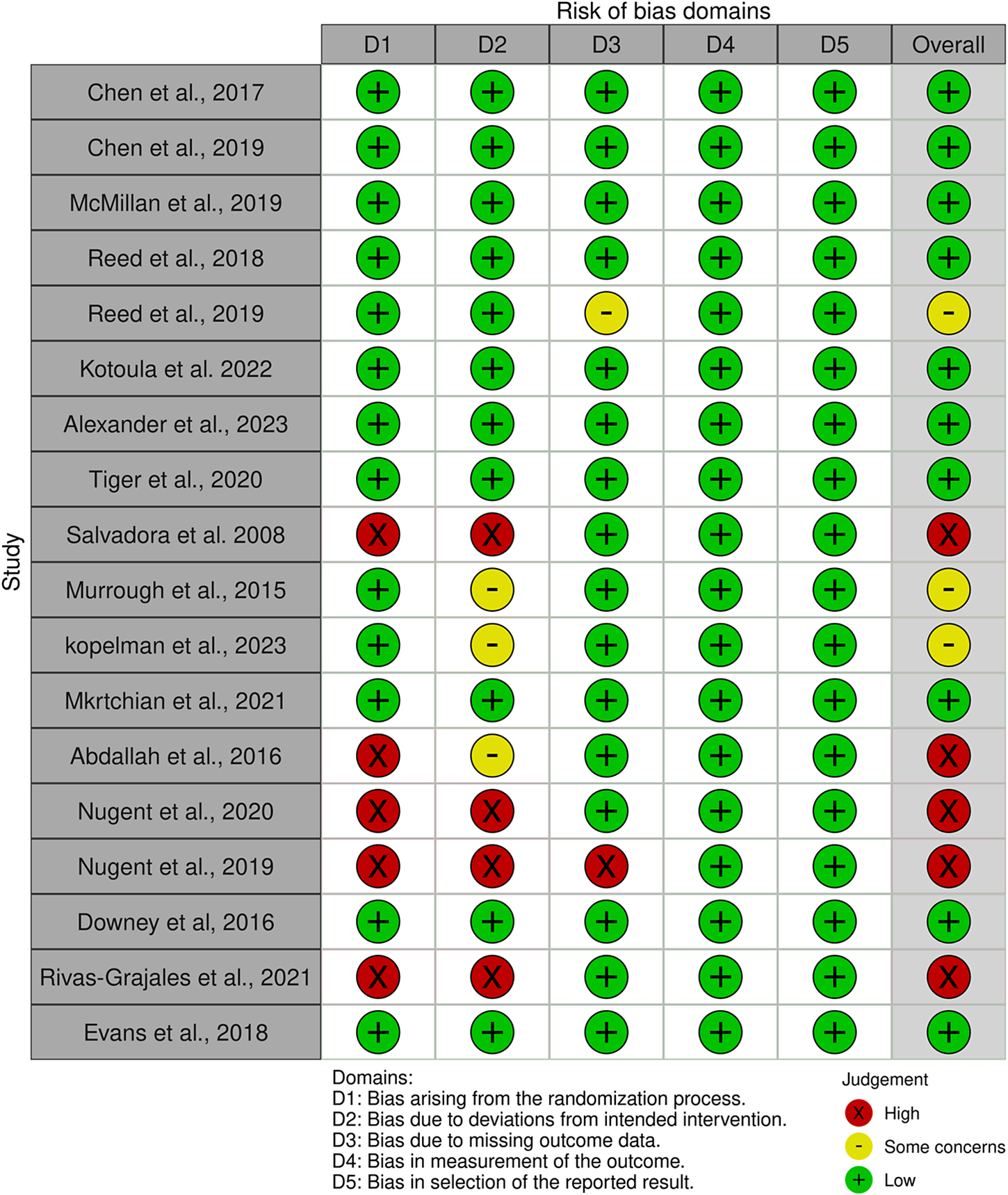
Most of the studies included in this systematic review were randomized, double-blind, placebo-controlled crossover trials, and the majority of them were evaluated as having an overall low risk of bias. Risk of bias was assessed using the Cochrane Risk of Bias 2.0 (RoB 2.0) tool by two reviewers (H.F. and W.C.) for all studies that met eligibility criteria (n = 18). Most studies were determined to have low risk of bias across domains, particularly concerning missing outcome data, measurement of outcomes, and selection of reported results (domains 3–5). However, five studiesReference Abdallah, Averill and Collins16, Reference Salvadore, Cornwell and Colon-Rosario57–Reference Nugent, Ballard, Gilbert, Tewarie, Brookes and Zarate60 were judged to have a high risk of bias, primarily due to concerns related to the randomization process and/or blinding and were therefore excluded from data extraction and synthesis in accordance with our approach to prioritize internal validity. Figure 2 summarizes RoB assessments for all studies assessed (n = 18), including those excluded from synthesis due to high risk of bias.
Risk of bias assessment of included studies using Cochrane’s RoB 2.0 tool and visualized using the RobVis R package (McGuinness and Higgins, 2021)Reference McGuinness and Higgins61.

Study characteristics
Thirteen human neuroimaging studies were included in data extraction and synthesis. Most used randomized, double-blind designs (predominantly placebo-controlled) (n = 10), including several double-blind crossover trials with ketamine versus 0.9% saline, active placebo (remifentanil) (n = 1), and one trial comparing ketamine with lanicemine and saline. Across studies, ketamine was administered almost exclusively intravenously, most commonly 0.5 mg/kg over ~40 minutes, with a small number of dose/infusion variations (eg 0.2 mg/kg arms in two studies; bolus+infusion protocols totaling 0.5 mg/kg; and a 60-minute infusion in one study). Across studies, the most common imaging modality was fMRI (n = 9). Studies also utilized phMRI (n = 1), FDG-PET (n = 2), T1-weighted MRI (n = 1), and diffusion tension imaging (n = 1) to assess changes in reward-processing regions post-ketamine. Task-based paradigms were used in four studies, while resting-state or non-task approaches were used in the remaining studies. The total sample size across the included studies was 623 participants (62% female), with 482 participants diagnosed with a depressive disorder diagnosis (MDD/TRD /remittent MDD) undergoing ketamine administration and 141 healthy controls (Table 1). Various clinical diagnostic tools were used to assess depressive symptoms (Montgomery–Åsberg Depression Rating Scale [MADRS], Beck Depression Inventory, Quick Inventory of Depressive Symptomatology – Self-Report, and Hamilton Depression Rating Scale (17-item version)) as well as anhedonia symptoms (Snaith-Hamilton Pleasure Scale [SHAPS] and Temporal Experience of Pleasure Scale [TEPS]). Only a minority of studies (n = 3) employed validated anhedonia-specific measures, whereas the majority relied on global depression scales. Therefore, not all reported neural changes can be directly attributed to changes in anhedonia, and interpretations in the review distinguish between these measurement approaches. Neuroimaging timepoints clustered into three main windows: during infusion (phMRI/rsfMRI), acute post-infusion assessments ranging from ~2 hours to ~48 hours, and a smaller subset with extended follow-up out to approximately 10–11 days post-infusion. Although esketamine was eligible, no neuroimaging studies administrating esketamine met inclusion criteria. Characteristics and findings of included studies assessing ketamine’s effects on neural reward processing in adults with MDD, with some studies involving a healthy control group.
Findings from resting-state fMRI
Most rsfMRI studies used 0.5 mg/kg IV ketamine infused over 40 minutes in crossover designs, with scanning conducted at baseline and approximately 2 days post-infusion.Reference Mkrtchian, Evans and Kraus41, Reference Evans, Szczepanik, Brutsché, Park, Nugent and Zarate45, Reference Alexander, Hawkins, Evans, Mehta and Zarate48 One additional longitudinal resting-state study in TRD compared 0.5 mg/kg vs 0.2 mg/kg vs placebo, with scans acquired at baseline and day 3 post-infusion.Reference Chen, Lin and Tu47 In TRD, ketamine was associated with increased sgACC connectivity to bilateral pgACC, bilateral anterior vmPFC, and right ventral striatum relative to placebo, while decreasing sgACC connectivity to the right hippocampus/parahippocampal gyrus (p < .05).Reference Alexander, Hawkins, Evans, Mehta and Zarate48 Within the TRD subgroup, changes in sgACC–pgACC connectivity following ketamine versus placebo were associated with improvement in SHAPS scores, suggesting a symptom-relevant link between sgACC coupling and hedonic function. Similarly, ketamine increased functional connectivity between ventral striatal seeds and the dorsolateral prefrontal cortex (dlPFC), ventrolateral PFC (vlPFC), pregenual ACC, and orbitofrontal cortex (OFC) in patients with depression, whereas connectivity decreased in healthy volunteers.Reference Mkrtchian, Evans and Kraus41 Within TRD, improvement in SHAPS scores at day 2 was associated with increased dorsal caudate–right vlPFC connectivity, while day 10 SHAPS improvement was associated with increased dorsal caudate–pgACC connectivity. Findings across multiple studies also indicated altered connectivity within the default mode network (DMN), salience network (SAL), and central executive network (CEN), which encompass reward-related prefrontal and cingulate regions.Reference Evans, Szczepanik, Brutsché, Park, Nugent and Zarate45, Reference Kopelman, Keller and Panny56 In TRD patients randomized to different ketamine doses, the standard-dose group (0.5 mg/kg) demonstrated reduced functional connectivity of the left dACC and right dlPFC with other frontal regions, while the low-dose group (0.2 mg/kg) showed a broader reduction in bilateral dACC connectivity with frontal and parietal regions.Reference Chen, Lin and Tu47 Within the standard-dose group, reductions in suicidal ideation were negatively correlated with reductions in connectivity between the left dACC and right ACC, indicating symptom-linked connectivity associations. Within the low-dose group, reductions in suicidal ideation were positively correlated with increased connectivity between the right dlPFC and left superior parietal region, suggesting dose-sensitive connectivity–symptom relationships. In a simultaneous EEG/fMRI infusion study, ketamine was associated with increased BOLD signal in ACC, paracingulate gyrus, left frontal pole, superior frontal gyrus, parahippocampal gyrus, and right insula.Reference McMillan, Sumner and Forsyth46 Ketamine was also associated with decreased BOLD signal in sgACC, mPFC, hippocampus, right amygdala, putamen, and somatomotor cortex regions. Ketamine-related changes in right insula BOLD signal were significantly associated with antidepressant response, further suggesting symptom-linked engagement of salience–reward integration circuitry. Across studies, ketamine doses ranged from 0.2 to 0.5 mg/kg, and imaging time points varied widely (during infusion to 2–48 hours, with a few extending to 7–10 days). These differences should be considered when interpreting cross-study patterns.Reference Mkrtchian, Evans and Kraus41, Reference Evans, Szczepanik, Brutsché, Park, Nugent and Zarate45–Reference Alexander, Hawkins, Evans, Mehta and Zarate48
Findings from task-based fMRI
Task-based fMRI studies typically administered 0.5 mg/kg IV ketamine over 40 minutes, with scanning conducted either 2 hours post-infusion, 24 hours post-infusion, or 2 days post-infusion, depending on the paradigm.Reference Murrough, Collins and Fields49–Reference Kotoula, Stringaris and Mackes52 Importantly, not all task paradigms were reward-specific. Dot-probe and emotional face/emotion-processing tasks primarily index attentional bias and affective salience/emotion processing, which engage salience and affective control systems (eg insula, dorsal ACC, amygdala) and may influence reward processing indirectly. In contrast, reward-specific paradigms such as the monetary incentive delay (MID) task more directly probe reward anticipation and feedback processes typically involving striatal circuitry. Reward-related outcomes were assessed using tasks involving attentional bias to affective cues, explicit emotional processing, facial emotion perception, and monetary reward anticipation and receipt. Using the dot-probe test with emotional stimuli, ketamine produced significantly greater activation in the left middle occipital gyrus compared with placebo (pFWE <0.001), while reducing activation in the left temporal and inferior frontal cortices (pFWE <0.002).Reference Reed, Nugent, Furey, Szczepanik, Evans and Zarate50 Across conditions, angry faces elicited greater activation than happy faces in bilateral dACC, putamen, thalamus, and posterior cingulate, but no main group effects were observed. In an emotional processing paradigm, ketamine compared with placebo led to widespread reductions in activity across bilateral frontal, temporal, precuneus, and posterior cingulate regions in patients with depression (pFWE <0.001).Reference Reed, Nugent, Furey, Szczepanik, Evans and Zarate51 No regions showed increased activation post-ketamine in this group. In healthy controls, ketamine reduced activity in the bilateral cingulate cortex but increased activation in a large frontal cluster (pFWE <0.001). Reward anticipation and outcome processing were assessed using the MID task.Reference Kotoula, Stringaris and Mackes52 Ketamine increased activity during the anticipation of win versus neutral trials across predefined reward-related ROIs (F1,36 = 9.261, p = .003), although effects in NAc and caudate during high-win anticipation did not survive correction for multiple comparisons. During the feedback phase of low-win trials, ketamine significantly increased activity in the NAc and putamen compared with placebo (pCORR = .008). Additional effects across ROIs were observed during no-win trials (F1,36 = 5.467, p < .001) but did not survive ROI-level correction. VTA activation during low-win feedback significantly correlated with plasma (2R,6R)-HNK levels 2 hours post-infusion (pFDR = .03). Emotional face processing tasks further revealed modulation of medial prefrontal and cingulate activity. During infusion, ketamine significantly reduced BOLD responses in the medial prefrontal cortex (mPFC) and posterior cingulate/precuneus compared with placebo (p < .05). Positive emotion processing further demonstrated ketamine-related modulation of striatal activity. In a task contrasting happy versus neutral faces, responses in the right caudate increased following ketamine, whereas neutral responses remained stable (cluster-level FWE p < .05).Reference Murrough, Collins and Fields49 A significant interaction was also observed in the left middle frontal gyrus during negative emotion trials, though effects were less robust.
Other imaging findings
In a phMRI study using an emotional faces paradigm, ketamine significantly increased BOLD signal in the subgenual ACC (sgACC) and rostral ACC (rACC) relative to placebo (all pFWE < 0.05).Reference Downey, Dutta and McKie53 Responses followed a gradual increase that plateaued after approximately 24 minutes of infusion. Ketamine also increased activity in the mid and posterior cingulate cortex, bilateral thalamus, and caudate, peaking within the first 4 minutes of infusion. Using FDG-PET, Chen et al. (2017) reported that TRD patients receiving 0.5 mg/kg ketamine had significantly increased standardized uptake values (SUV) in the supplementary motor area and dorsal ACC compared with patients receiving 0.2 mg/kg (p = 0.014).Reference Chen, Li and Lin54 No significant main effects of time or group were observed in ANOVA, though the increase in dACC SUV was statistically significant. In a PET study of SSRI-resistant depression using a 5-HT1B receptor ligand, ketamine was not associated with significant ROI-wide binding changes relative to placebo, but hippocampal binding increased after the first ketamine infusion (p = .036).Reference Tiger, Veldman, Ekman, Halldin, Svenningsson and Lundberg55 Baseline ventral striatal 5-HT1B binding was negatively correlated with depressive symptom severity and also negatively correlated with magnitude of symptom reduction following ketamine, supporting a predictive association involving reward-related serotonergic markers.
In a diffusion MRI trial, ketamine showed no significant main effect on mean diffusivity (MD) change across examined regions at 24 hours, but regional MD changes moderated symptom outcomes.Reference Kopelman, Keller and Panny56 Decreases in MD in left BA10 and left amygdala were associated with greater improvement in depression scores in the ketamine group, indicating microstructural–clinical associations in prefrontal–limbic circuitry relevant to reward valuation and affect regulation. In contrast, increases in hippocampal MD were associated with greater improvement in MADRS within the ketamine group, suggesting region-specific directionality in microstructural associationsReference Kopelman, Keller and Panny56.
Discussion
The present systematic review synthesizes evidence from functional neuroimaging studies to evaluate the associations between ketamine administration and neural reward processing in adults with MDD/TRD. In addressing our first objective, the included studies generally demonstrated associations between ketamine and alterations in neural activity or connectivity within ventral striatal, ACC, and prefrontal regions. Regarding our second objective, multimodal neuroimaging findings suggest that these neural patterns often co-occur with rapid antidepressant improvement; however, given the correlational nature of the data, these patterns should not be interpreted as direct evidence that ketamine causes specific circuit-level modifications or synaptic changes. Only a minority of studies directly measured anhedonia using validated scales such as SHAPS or TEPS, whereas most relied on global depression instruments. As a result, the reviewed findings should be understood as reflecting broader reward-related and/or antidepressant-associated processes, rather than definitive evidence of ketamine-specific anti-anhedonic mechanisms.
Modulation of reward circuitry
Converging evidence indicates that subanesthetic dosing of ketamine is associated with increased neural activity in key reward-processing regions. Specifically, in a monetary incentive task, ketamine (0.5 mg/kg) was linked to significantly higher BOLD activation in the NAcc and putamen during reward feedback just 2 hours post-infusion, compared to placebo.Reference Kotoula, Stringaris and Mackes52 Because symptom assessments and imaging time points varied across studies, these neural differences may occur prior to, alongside, or shortly before measurable clinical improvement, but they should not be interpreted as temporally proving symptom change. Task-based fMRI studies consistently indicate that ketamine can acutely normalize or heighten responses in ventral striatal regions that are often blunted in depression, such as enhancing neural reactivity to positive/reward cues, which may relate to, but do not definitively establish, ketamine’s anti-anhedonic properties.Reference Kotoula, Stringaris and Mackes52, Reference Lally, Nugent, Luckenbaugh, Niciu, Roiser and Zarate62 Ketamine was also associated with differences in FC within the broader reward network, particularly between the PFC and striatum, similar to findings in other study designs.Reference Alexander, Hawkins, Evans, Mehta and Zarate48, Reference Abdallah, Roache and Gueorguieva63 Resting-state fMRI studies 24 hours after ketamine show increased connectivity in fronto-striatal circuits, with responders exhibiting increased connectivity between the lateral PFC and striatal regions like the caudate.Reference Abdallah, Roache and Gueorguieva63 In persons with depression, ketamine increased fronto-striatal connectivity (interpreted as a normalization of circuit hypoactivity associated with anhedonia), whereas healthy volunteers showed slight connectivity reductions post-ketamine, suggesting an association between ketamine treatment and modulation of cortico-striatal loops implicated in motivation and reward processing.
In addition, PET imaging provides complementary evidence wherein ketamine was associated with region-specific changes in 5-HT1B receptor binding (eg hippocampus), and baseline ventral striatal 5-HT1B binding was associated with symptom severity and symptom change in depressed patients.Reference Tiger, Veldman, Ekman, Halldin, Svenningsson and Lundberg55 The aforementioned electrophysiological findings accord with the fMRI results by highlighting ketamine’s rapid enhancement of neural network responsiveness. Taken together, task-activation studies, connectivity analyses, and molecular imaging indicate that ketamine is associated with modulation of reward circuitry, which supports emerging literature suggesting that modulation of the positive valence system may be a mechanistic target for personalized treatment strategies in depression.Reference McIntyre, Rosenblat and Nemeroff34
It is important to note that the observed variability across studies may be partly attributable to heterogeneity in ketamine dosing (0.2–0.5 mg/kg), and timing of imaging. Differences in study populations—particularly TRD versus non-resistant MDD and the inclusion of healthy controls—likely further contribute to divergent findings. Analytic choices, including ROI-based versus whole-brain approaches, also shape the reported neural patterns.
Biological mechanisms of ketamine’s action
Findings indicate that ketamine is associated with increased availability of serotonin 1B receptors in depressed patients.Reference Tiger, Veldman, Ekman, Halldin, Svenningsson and Lundberg55 This is mechanistically interesting because 5-HT₁_B receptors are autoreceptors and heteroreceptors that modulate neurotransmitter release: their activation tends to inhibit serotonin release but facilitates dopamine release in the striatum.Reference Du Jardin, Liebenberg and Cajina64 Increased FC between the mPFC and the NAcc and heightened ventral striatal activation during reward tasks have been interpreted as potentially reflecting underlying synaptic potentiation driven by glutamatergic plasticity.Reference Morris, Costi, Tan, Stern, Charney and Murrough42, Reference Chen, Lin and Tu47, Reference Alexander, Hawkins, Evans, Mehta and Zarate48, Reference Reed, Nugent, Furey, Szczepanik, Evans and Zarate50, Reference Kotoula, Stringaris and Mackes52 Electrophysiological evidence further supports this interpretation, as ketamine-induced increases in gamma-band oscillations—markers of cortical excitation and local circuit reorganization—were observed shortly after infusion.Reference Le, Wong and Badulescu43, Reference McMillan, Sumner and Forsyth46, Reference Lundin, Sepe-Forrest and Gilbert65
On a systems level, ketamine may act as a “network reset” agent, disrupting pathological brain states commonly observed in depression. The acute network desynchronization observed in EEG and fMRI studies immediately following ketamine infusion disrupts maladaptive connectivity patterns, such as hyperconnectivity of the default mode network and hypoconnectivity of reward circuits.Reference McMillan, Sumner and Forsyth46, Reference Lundin, Sepe-Forrest and Gilbert65 Over time, as NMDA receptor blockade subsides, brain networks appear to reconfigure into healthier, more interconnected states characterized by restored fronto-striatal and executive network integration.Reference Ardalan, Wegener, Rafati and Nyengaard66 Additionally, research has found that higher plasma levels of the ketamine metabolite HNK were associated with greater activation of the VTA, consistent with HNK’s role as an AMPA receptor facilitator promoting continued activation of dopaminergic reward pathways.Reference Kotoula, Stringaris and Mackes52, Reference Nogo, Jasrai and Kim67 Ketamine’s ability to rapidly reactivate dormant synapses, enhance neurotrophic signaling, amplify dopaminergic activity, and reconfigure network dynamics offers a multi-dimensional mechanism that distinguishes it from traditional monoaminergic antidepressants and explains its unique efficacy in rapidly restoring interest, motivation, and pleasure in depression. While preclinical models robustly demonstrate mechanisms such as AMPA-mediated synaptic strengthening, BDNF-dependent neuroplasticity, and dendritic spine formation, these cellular processes cannot be directly measured using human neuroimaging techniques. Thus, references to “synaptic potentiation” or “network reconfiguration” in humans should be understood as theoretical interpretations, supported by animal literature and consistent with observed human imaging correlates, but not directly demonstrated in the included studies.
Limitations
A crucial consideration is the presence of inconsistent methods across imaging modalities. Each modality and task address different aspects of neural function, which complicates direct comparison. For example, a monetary reward task may specifically index ventral striatal phasic activity, whereas resting connectivity focuses on intrinsic synchronization.Reference Nogo, Jasrai and Kim67 Divergence in results may arise due to one capturing evoked activity and the other spontaneous activity. In addition, analytic choices vary, wherein some analyses were ROI-driven (regions like NAcc or sgACC), while others were whole-brain or connectome-based. Other methodological differences that may affect our interpretation of the results include the sociodemographic differences (eg illness duration, comorbidities, additional antidepressant treatments) in the enrolled population. Most imaging assessments were acute; limited data exist on whether these brain changes correlate with longer-term outcomes (eg 1–2 weeks or relapse timing). The short follow-ups (often 24 h or 7 days at most) are a limitation if we aim to tie neural changes to lasting clinical remission. A formal assessment of publication bias could not be conducted due to the limited number of included studies and substantial heterogeneity in imaging modalities and task paradigms, which violate the assumptions of statistical bias tests such as funnel plots or Egger’s regression. As a result, the presence of publication bias cannot be ruled out. Neuroimaging studies in particular are susceptible to small-sample reporting and selective publication of significant findings, which should be considered when interpreting the results. Finally, the protocol was not prospectively registered. Although our eligibility criteria and outcomes were established a priori and did not change during the review, the lack of public registration may reduce transparency relative to best-practice standards.
Future directions
Future research should involve larger, adequately controlled neuroimaging trials and utilize machine learning models to predict ketamine response for personalized treatments.Reference Xu, Vekaria and Wang68 Longitudinal studies tracking multiple time points are needed to elucidate ketamine’s sustained neural effects. Combining PET, fMRI, EEG, and MR spectroscopy can clarify interactions across molecular, functional, and electrophysiological domains.Reference Gerhard, Pothula and Liu38, Reference McMillan, Sumner and Forsyth46 Future studies should prioritize anhedonia and reward function as primary outcomes, integrating behavioral measures of real-world reward experiences to better translate brain changes into clinical recovery.Reference Lally, Nugent, Luckenbaugh, Niciu, Roiser and Zarate62
Conclusion
This systematic review found that ketamine administration is associated with neurofunctional changes in reward-related circuitry in adults with MDD/TRD, most consistently involving the ventral striatum/NAc and connected ACC–prefrontal networks. Given heterogeneity in methods and timing—and because few studies directly measured anhedonia—these findings should be interpreted as reflecting broader reward- and antidepressant-associated processes rather than definitive ketamine-specific anti-anhedonic mechanisms. Larger longitudinal neuroimaging studies that prioritize validated anhedonia outcomes are needed to clarify the consistency, time course, and clinical relevance of these associations.Reference Cao, Zhu and Zuckerman25
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1017/S109285292610087X.
Acknowledgements
The authors would like to thank Dr. Roger S. McIntyre for giving me the opportunity to work on this paper and for fostering an environment that has enhanced my knowledge in this field. They are also deeply grateful to Gia Han Le for her thoughtful review and continuous support throughout the writing process and the various stages of this paper.
Author contribution
Conceptualization: H.F., G.H.L., A.T.H.K. Methodology: H.F., A.T.H.K. Validation: G.H.L., W.C., H.F., B.S. Data curation: H.F., G.H.L., W.C., B.S. Investigation: H.F., B.S. Visualization: H.F., G.H.L. Project administration: H.F., G.H.L., C.D. Supervision: R.S.M., G.H.L. Writing (original draft): H.F. Writing (review & editing): H.F., G.H.L., A.T.H.K., W.C., B.S., S.W., B.C., T.G.R., S.B., H.K.Y.L., H.F.G.-B., R.S.M.
Financial support
This research paper was not funded by any organization and entity.
Disclosures
T.G.R. was supported in part by the National Institute on Aging (#R21AG070666; R21AG078972; R01AG088647), National Institute of Mental Health (#R01MH131528), National Institute on Drug Abuse (#R21DA057540), and Health Resources and Services Administration (#R42MC53154-01-00). T.G.R. serves as a review committee member for the National Institutes of Health (NIH), Patient-Centred Outcomes Research Institute (PCORI) and Substance Abuse and Mental Health Services Administration (SAMHSA) and has received honoraria payments from NIH, PCORI, and SAMHSA. T.G.R. has also served as a stakeholder/consultant for PCORI and received consulting fees from PCORI. T.G.R. serves as an advisory committee member for the International Alliance of Mental Health Research Funders (IAMHRF). H.F.G.-B. has received research grant support from the Ministry of Science, Technology, and Innovation (MinCiencias) in Colombia; UKRI in the United Kingdom; and speaker fees from Roche, Pfizer, Abbott, and Synergy R&D. R.S.M. has received research grant support from CIHR/GACD/National Natural Science Foundation of China (NSFC) and the Milken Institute; speaker/consultation fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Neurawell, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular, NewBridge Pharmaceuticals, Viatris, Abbvie, Bristol Myers Squibb (BMS) Teva, Adhere Tech, GH Research, Autobahn Theapeutics and Atai Life Sciences.
Competing interest statement
The authors have no conflicts of interest to disclose.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used ChatGPT to brainstorm key words for the search strategy. ChatGPT was also used to generate the full version of the abbreviations used in Table 1 (which was then validated by the authors). After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the published article.



 Open access
Open access