


Ketamine has demonstrated efficacy for treatment-resistant depression (TRD), which affects approximately one third of people with major depressive disorder. Reference Pigott, Kim, Xu, Kirsch and Amsterdam1 Early, proof-of-concept randomised controlled trials (RCTs) demonstrated rapid, but transient improvement after a single dose. Reference Xu, Hackett, Carter, Loo, Gálvez and Glozier2 Trials of repeated dosing have had positive outcomes Reference Nikolin, Rodgers, Schwaab, Bahji, Zarate and Vazquez3,Reference Calder, Kwan, Teopiz, Wong, Rosenblat and Mansur4 with a number needed to treat (NNT) reportedly 7 for a clinical response compared with placebo. Reference Calder, Kwan, Teopiz, Wong, Rosenblat and Mansur4 Two forms of ketamine are increasingly accepted as evidence-based treatments for TRD Reference McIntyre, Alsuwaidan, Baune, Berk, Demyttenaere and Goldberg5 and incorporated into treatment guidelines. Reference Hussain, Gale, Sarma, Smith, Bayes and Loo6 Many jurisdictions have approved a commercially developed ketamine intranasal spray (esketamine). However, the efficacy of off-patent, widely available, generic racemic ketamine, usually using repeated intravenous infusions, has level 1 evidence. Reference Nikolin, Rodgers, Schwaab, Bahji, Zarate and Vazquez3 Given the disabling and chronic nature of TRD, treatment needs to be (cost-)effective, tolerable and safe in the longer term. Understanding the optimal duration and route of treatment, and the effects of longer-term treatment, challenge implementation in clinical practice. Reference Thornton, Wright and Glozier7 Subcutaneous administration has several logistical advantages compared with the more frequently evaluated intravenous infusion route, including being rapid, painless and easily administered. The Ketamine for Adult Depression Study (KADS) showed that adequately dosed, twice-weekly subcutaneous racemic ketamine is feasible, efficacious (compared with active comparator) and safe in treating TRD over a 4- week period. Reference Loo, Glozier, Barton, Baune, Mills and Fitzgerald8 However, the efficacy was short-term, with comparable remission rates in the ketamine and control arms at 4 weeks after the cessation of treatment, as ketamine-treated remitters relapsed. A pilot RCT suggested efficacy of subcutaneous ketamine in older adults Reference George, Galvez, Martin, Kumar, Leyden and Hadzi-Pavlovic9 and there are a few studies of its use in non-blinded treatment, all with small samples (e.g.). Reference Tham, Do, Fridfinnson, Rafizadeh, Siu and Budd10 An uncontrolled study suggested subcutaneous esketamine could be effective. Reference Palhano-Fontes, Cavalcanti-Ribeiro, da Costa Goncalves, de Almeida, Barbosa and de Araujo Ferreira11 These studies of subcutaneous ketamine treatment all have small samples, with short-term outcomes, and/or in people prepared to consent to randomisation, limiting clinical inferences which require larger samples and longer follow-up.
Given the frequency of relapse after a course of effective treatment, Reference McIntyre, Alsuwaidan, Baune, Berk, Demyttenaere and Goldberg5 a key clinical question is whether prior treatment with ketamine, or a patient’s response to this, is indicative of future treatment outcomes. However, these factors have received so little attention in the field that they are not even mentioned in the recent review of personalised ketamine treatment by leading practitioners. Reference Medeiros, Demo, Goes, Zarate and Gould12
We used data from the open label extension (OLE) phase of the KADS RCT Reference Loo, Glozier, Barton, Baune, Mills and Fitzgerald8 to address the two objectives.
-
(a) What is the pattern of response and side-effects of 4 weeks of treatment with subcutaneous racemic ketamine for adults with TRD up to 6 months later?
-
(b) Undertake an exploratory analysis to evaluate the impact of (i) prior treatment with ketamine, or (ii) response in RCT influence effectiveness.
Method
Participants and setting
In the KADS study, adult (age > 18) participants in a 4-week, randomised, double-blind, active-control, parallel group, multicentre phase 3 trial Reference Loo, Glozier, Barton, Baune, Mills and Fitzgerald8 conducted in 7 specialist mood disorder centres in Australasia were offered the option of entering a 4-week open label extension (OLE) of treatment if, at 4-week post-treatment follow-up, they scored ≥20 on the Montgomery–Åsberg Depression Rating Scale (MADRS), Reference Montgomery and Åsberg13 regardless of their short-term response in the RCT phase. At recruitment to the OLE phase, participants and trial staff were still blinded to the participant’s RCT assignment (ketamine or midazolam). At baseline, all were ascertained to have TRD (defined as major depressive disorder) of at least 3 months’ duration, confirmed by the Structured Clinical Interview for DSM-5 Research Version, insufficient response to at least 2 adequate trials of antidepressant medications during this episode and scored ≥20 on the MADRS. The dose of any concurrent antidepressant or augmenting medication needed to be stable for ≥4 weeks prior to, and during, the study. These same criteria and written consent applied for entry into the OLE treatment phase.
Intervention
The intervention procedures, outcome and safety assessments, and programme were identical to the RCT phase, Reference Loo, Glozier, Barton, Baune, Mills and Fitzgerald8 except that in the OLE all participants and study staff were aware they were only receiving subcutaneous racemic ketamine, for 8, twice-weekly treatments, 4 weeks after ceasing their allocation trial medication. The initial trial protocol involved a ‘fixed dose’ regimen administering 0.5 mg/kg ketamine subcutaneously into the abdominal wall twice-weekly for 4 weeks, with at least 3 days between treatments. After 51 participants had completed the RCT phase, the Data Safety Monitoring Board recommended a ‘flexible’ response-guided dosing regimen for both RCT and OLE. If participants’ MADRS had not improved by ≥50% from OLE baseline using the 0.5 mg/kg dose, dose was increased to 0.6, 0.75 and 0.9 mg/kg ketamine when assessed at sessions 2, 4 and 6 respectively. Participants were observed for 4 h after the first OLE treatment (and 2 h at other visits). Participants were not allowed to drive after treatment and were discharged after clinical review. There was no other specific treatment provided, although participants could continue current medication and ongoing psychological therapy.
Effectiveness assessments
Primary: Depression was evaluated using the MADRS Reference Montgomery and Åsberg13 prior to each treatment, 3–4 days after the eighth treatment at 4 weeks (the primary OLE treatment course end-point), then 4 weeks and 6 months later.
Secondary: Clinical Global Impression-Severity and Improvement scales Reference Guy14 were assessed at baseline, end of treatment, and 4 weeks and 6 months later.
Safety and side effects
Suicidality was assessed at each treatment with the Columbia Suicide Severity Rating Scale (C-SSRS), Reference Posner, Brown, Stanley, Brent, Yershova and Oquendo15 with risk managed by site study physicians. Urinary symptoms were assessed with the Bladder Pain/Interstitial Cystitis Symptom Score. Reference Humphrey, Arbuckle, Moldwin, Nordling, van de Merwe and Meunier16 Psychotomimetic and ketamine-specific effects were assessed with the Clinician Administered Dissociative States Scale Reference Bremner, Krystal, Putnam, Southwick, Marmar and Charney17 and Ketamine Side Effect Tool (KSET) Reference Short, Dong, Gálvez, Vulovic, Martin and Bayes18 at each treatment.
Blood pressure and heart rate measurements were taken pre-injection, and at 15, 30, 60 and 120 min post-injection on all occasions.
All assessments were undertaken by trial and clinic staff, with central standardisation of MADRS ratings.
Statistical analysis
The two OLE regimen’s characteristics were described and compared. Potential attrition bias was assessed by comparing the baseline demographic and OLE entry clinical characteristics of the sample at each follow-up.
Aim (a): Changes in the primary outcome measure (MADRS) were described using the continuous measure of change in the MADRS from OLE entry, and the proportions achieving response (≥50% reduction in MADRS), two definitions of remission (MADRS ≤ 10, the KADS RCT primary outcome and MADRS ≤ 12). ‘Nonresponse’ was defined as MADRS change from OLE baseline of ≤25%. For clinical implementation, we chose the definition of ‘response’ as the main outcome rather than change in MADRS, which is information-rich, but not readily understood by clinicians or patients.
Aim (b): The change in MADRS score from the start of the OLE (baseline) to OLE (end) was estimated from a linear mixed-effects model with repeated measures, that included all OLE baseline-to-session scores as repeats (with an unstructured covariance matrix), and session, RCT responder status, MADRS OLE entry score, site and the session × responder status interactions as covariates. We also included the session × baseline MADRS interactions to provide baseline-adjusted estimates. We compared MADRS changes over the OLE between a priori defined groups:
-
(a) prior ketamine treatment in RCT (ketamine versus midazolam arm)
-
(b) RCT responder versus non-responder.
Results
Of the 151 RCT participants who presented for the four-week post-RCT assessment, 130 (86%) entered the OLE phase ( ≥ 20 on the MADRS). Of the 23 who had ‘responded’ at the final assessment immediately after the 4-week RCT phase, 15 (65%) had a MADRS > 20 one month later and entered the OLE phase.
Of 48 OLE participants who underwent the RCT fixed-dose regimen (24 in each RCT arm), 32 were given the same regimen in the OLE phase. For the RCT flexible-dose regimen, 82 participants entered the OLE, 40 (50%) of whom were treated with ketamine in the RCT. Thus, 98 participants underwent the flexible-dose regimen (0.5 mg/kg to 0.9 mg/kg) in the OLE phase, and 32 the fixed-dose regimen (Supplementary Fig. S1 available at https://doi.org/10.1192/bjp.2026.10692).
Those receiving the fixed-dose regimen were more likely to have higher depression and anxiety scores at OLE entry than those in the flexible-dose regimen (Table 1), potentially reflecting an ‘early adopter’ selection bias of initial trial recruitment of more unwell participants. At entry to the OLE, 44 (34%) participants reported some form of active suicidal thoughts (C-SSRS score ≥2).
Open label extension (OLE) baseline characteristics of the participants in fixed and flexible dose regimens

Table 1 Long description
The table presents baseline characteristics of participants in fixed and flexible dose regimens of an Open Label Extension study. It includes data on age, gender, weight, site identity, unsuccessful antidepressant trials, current failed electroconvulsive therapy, melancholic features, MADRS total score, HAM total score, and C-SSRS score. The table has 8 rows and 6 columns, with columns for OLE fixed, OLE flexible, Difference, and p-value. Notable trends include higher depression and anxiety scores in the fixed-dose regimen group, with 44 participants reporting some form of active suicidal thoughts at OLE entry. The table provides mean values, percentages, and statistical comparisons between the two regimens.
a. ECT, electroconvulsive therapy; SCID-5, Structured Clinical Interview for DSM-5; MADRS, Montgomery–Åsberg Depression Rating Scale; HAM-A Hamilton Anxiety Scale; C-SSRS, Columbia-Suicide Severity Rating Scale.
Welch Two Sample t-test; 2-sample test for equality of proportions with continuity correction.
Open label extension attrition
Of the 130 OLE participants, 2 did not commence treatment, 116 (90%) were assessed after completing 4 weeks of ketamine treatment, and 110 (85%) and 73 (56%) assessed at 4-week and 6-month follow-up, respectively (Table S1), with no indication of attrition bias by baseline characteristics. One participant in the flexible regimen withdrew due to poor tolerability of acute ketamine effects during treatment and three were known to have been withdrawn because of lack of improvement or deterioration. Loss to follow-up reasons are mostly unknown, with no response despite multiple contact attempts.
Effectiveness – depression
Response rates over the course of the OLE
The overall response, remission and non-response rates in both regimens over the OLE are shown in Table 2. Thirty percent (35/116) had responded after 4 weeks of treatment, and 4 weeks later 17% (19/110) were still showing a response. Approximately one quarter were in remission after 4 weeks of treatment. Five months later only 11% (8/73) were still classified as showing a response. Over half were non-responders at each time point. Of the 6 lost between end of OLE and 4 weeks, 2 were in remission, and one other was a responder. At 6 months, 19/36 (53%) of the short-term responders and 54/80 non-responders (68%) were assessed, suggesting no attrition of those with poorer outcomes.
Montgomery–Åsberg Depression Rating Scale (MADRS) response profile at the end of open label extension (OLE) and follow-ups

Table 2 Long description
The table presents data on the Montgomery–Åsberg Depression Rating Scale (MADRS) response profile at the end of Open Label Extension (OLE) and follow-ups. It includes three columns: END-OLE with 116 participants, OLE - 4-week follow-up with 110 participants, and OLE - 6-month follow-up with 73 participants. The rows detail remission rates, response rates, and non-response rates. Remission is defined by MADRS TOTAL scores of 10 or less and 12 or less. Response is defined by a 50 percent or greater change in MADRS scores. Non-response is defined by a 25 percent or less change in MADRS scores. The table shows a decrease in remission and response rates over time, with an increase in non-response rates. The mean MADRS levels also increase over the follow-up periods.
Change in depressive symptom severity over 6 months
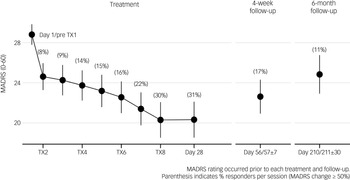
The combined regimen MADRS scores at each time point (Fig. 1) indicate a mean decline over OLE treatment of 8.6 points on the MADRS from 28.8 (s.d. 5.96) at baseline to 20.3 (s.d. 9.83) at the end of the OLE, 23.0 (s.d. 9.04) 4 weeks later and 24.8 (s.d. 8.33) at 6-month follow-up. The most substantial effect was seen after the first dose.
Mean (±95% CI) Montgomery–Åsberg Depression Rating Scale (MADRS) score prior to each treatment session over 4 weeks of treatment and up to 6 months after commencing the Open Label Extension. TX, treatment session.

Fig. 1 Long description
The line graph illustrates the mean Montgomery–Åsberg Depression Rating Scale (MADRS) score, with a 95% confidence interval, prior to each treatment session over 4 weeks of treatment and up to 6 months after commencing the Open Label Extension. The x-axis represents the timeline, starting from Day 1/Pre TX1, through various treatment sessions (TX2, TX4, TX6, TX8), Day 28, Day 56/57±7, and Day 210/211±30. The y-axis represents the MADRS score ranging from 20 to 28. The graph shows a decreasing trend in MADRS scores from Day 1/Pre TX1 to Day 28, with percentage responders indicated at each session. The scores slightly increase at the 4-week follow-up and 6-month follow-up points. All values are approximated.
Comparative effectiveness of dosing regimens
There were no consistent significant differences in the change in MADRS from OLE baseline between the two dosing regimens at any time point (Fig. S2). The improvement in MADRS score from OLE baseline was significantly more than zero at each treatment for both dosing regimens, and still significantly lower than at the start of treatment at the 4-week and 6-month follow-up assessments, despite symptoms increasing after ketamine cessation.
Impact of prior ketamine treatment
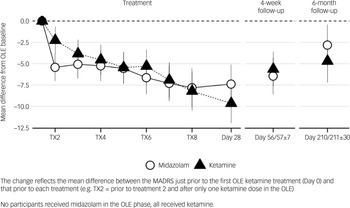
Participants who had received ketamine in the RCT demonstrated less response after their first OLE treatment (i.e. at pre-treatment 2 assessment (T × 2)) (mean MADRS difference 3.2 (s.e. 1.12), p < 0.005) than those who had received midazolam, but at no other time points, indicating that prior ketamine treatment may possibly attenuate the effect of the first re-treatment, but neither enhanced nor attenuated the effect of repeat dosing (Fig. 2, Table S2).
Change in Montgomery–Åsberg Depression Rating Scale (MADRS) from open label extension (OLE) baseline to each treatment and at follow-up – stratified by ketamine exposure in a randomised controlled trial. TX, treatment session.

Fig. 2 Long description
The line graph presents the mean difference from OLE baseline in MADRS scores, stratified by ketamine exposure, during treatment and follow-up periods. The x-axis represents different time points, including treatment sessions (TX2, TX4, TX6, TX8), Day 28, Day 56/57, and Day 210/211. The y-axis shows the mean difference from OLE baseline, ranging from 0.0 to -12.5. Two data lines are depicted: one for Midazolam (represented by open circles) and one for Ketamine (represented by filled triangles). The graph indicates a general decrease in MADRS scores over time for both treatments, with Ketamine showing a more pronounced reduction. Error bars indicate variability in the data. All values are approximated.
Impact of prior response
The comparison of mean change in MADRS over OLE demonstrated by RCT responders (n = 15, 3 of whom were in the control arm), and non-responders (n = 115) is shown in Fig. S3 and Table S3. RCT responders tended to exhibit greater change in MADRS than RCT non-responders after week 3 and by the end of the OLE, the reduction in MADRS was significantly greater in RCT responders, and remained so at 6 months.
Sensitivity analysis of prior responsiveness
To evaluate whether this repeated response to treatment reflected a participant’s response to the non-specific aspects of the care provided, we assessed whether non-responders to the active placebo in the RCT remained non-responsive in the OLE arm. Of the 58 participant midazolam non-responders in the RCT, 13 (22%) met MADRS response criteria when subsequently given ketamine in addition to the same supportive RCT care (Table S4). However, a similar proportion, 11/44 (25%), of non-responders to ketamine in the RCT responded after 4 weeks of ketamine treatment in the OLE. Most of the RCT ketamine responders (82%) responded again in the OLE.
Clinical global impression
Severity
The proportion of participants rated as having a ‘mild illness or better’ (≤3) increased from 8% at OLE baseline to 46% at end of OLE and 31% at 4-week follow-up. 6% were consistently rated as severely ill at all time points (Fig. S4).
Improvement
By the end of treatment, 45% were rated by clinicians as ‘very much or much’ improved, but this fell to 32% and 17% at 4 weeks and 6 months. Only 7, 9 and 14% worsened with treatment at end of OLE, 4-week and 6-month follow-ups, respectively (Fig. S5).
Safety and side-effects
There were no deaths or self-harm events requiring admission reported throughout the OLE. Three severe adverse events were reported, all in the flexible-dose regimen, which were judged as being unlikely to be related to ketamine, e.g. voluntary admission for depression 87 days after the last ketamine dose.
Blood pressure and heart rate data are displayed in Figs. S6, S7. Clinically relevant blood pressure increases occurred in 4/31; (12.9%) fixed-dose and 11/97 (11.3%) in the flexible-dose regimen. No participant required rescue medication. Fourteen (11%) reported at least one new urological symptom, but none had a clinically significant BPICC_SS score (≥19) after the treatment course.
Psychomimetic and other ketamine-specific effects
There were expected non-significant transient increases in dissociative symptoms during each treatment session (Fig. S8). The most frequent acute symptoms elicited using the KSET (Table S5) reflect expected effects of ketamine and with similar frequencies to the trial. Of the acute symptoms reported by >10% of participants, 4 (dissociation, reduced concentration, anxiety and palpitations) were more frequently experienced by those treated with higher doses in the flexible-dose regimen, and only dissociation after correcting for multiple testing. There were no differences in treatment-emergent side-effects between the regimens (Table S4).
Craving and recreational use
Four participants responded ‘yes’ once to the question ‘Have you craved ketamine?’ during treatment, all during the first two weeks. All rated this as ‘mild’, occurring the day after treatment, and no one reported this more than once or after treatment 3. One participant reported 3 episodes of recreational ‘ketamine/drug mix use with friends’ in the 2 months after ceasing treatment. The participant did not use ketamine again in the 4 months prior to the 6-month follow-up or report craving on any of the 11 times asked.
Discussion
This report of the OLE phase of a clinical trial describes the medium-term outcomes and safety of the largest cohort receiving repeated dose subcutaneous racemic ketamine for TRD. The setting is closer to real-world treatment (i.e. non-randomised and unblinded) conditions. We believe this provides useful information for clinical practice. Further, and for the first time, we provide data on whether prior ketamine treatment influences the effectiveness of subsequent treatment.
Overall, we observed a clinically meaningful average effect (MADRS change >8 points) with treatment, similar to that seen in the RCT (8) flexible-dose arm (8.8, s.d. 10.2). This average change masks the variability clinicians need when informing patients considering ketamine treatment, where data about the likelihood of response or non-response is desired. We found that up to a quarter of participants (18 and 26% – defined by a MADRS of ≤10 or ≤12) experienced short-term remission, and 30% were responders at end of OLE, again similar to the flexible-dose ketamine arm of the KADS trial Reference Loo, Glozier, Barton, Baune, Mills and Fitzgerald8 (19, 22 and 29%, respectively). As in the RCT, these benefits often dissipated rapidly after treatment cessation; 4 weeks after OLE ketamine treatment fewer than 1 in 5 were responders. However, attrition was higher in responders, possibly reflecting treatment burden, so these later response rates may be an under-estimate.
The treatment was well tolerated with no reports of severe adverse events, or self-harm events either during, or within 2 months of ceasing, ketamine treatment. There were expected, transient effects, in blood pressure and acute symptoms, with no observed adverse cumulative sequalae up to 6 months later.
Influence of prior clinical factors on response to ketamine retreatment
The study design also enabled a preliminary exploration of whether prior ketamine treatment influences the outcome of a second course. Beyond a differential first dose effect, prior treatment did not influence the response trajectory over the subsequent 6 months, suggesting this factor should not influence the selection of, or information for, patients. Acknowledging that the study was not powered for this analysis, the observed differences over the course of treatment of <2.5 points on the MADRS, and lack of an observable differential pattern, does not support a clinically meaningful differential effect.
We also undertook a limited assessment of any impact of prior response. Of the 11 prior ketamine responders who relapsed after RCT treatment cessation, 9 responded again. This may indicate prior response could be used to select or prioritise patients. Conversely, prior non-response to ketamine was less informative. One quarter of ketamine non-responders in the RCT were classified as responders when retreated. We do not know whether this is best explained by some kind of ‘cumulative effect’, the negative impact of randomisation and treatment under blinded conditions, or the influence of external factors, e.g. positive or negative life events, all worth exploring as ketamine treatment becomes mainstream. The therapeutic effects of ketamine, at least for some, are bolstered by the observation that over one fifth of those who did not respond to 4 weeks of midazolam in the RCT subsequently responded to ketamine treatment in the OLE.
Comparison with previous research
An important clinical question when evaluating treatment outcomes is to what extent the samples in different studies are comparable. It might seem that the response rates from this programme were suboptimal, with lower response rates than those reported in other studies of subcutaneous or intravenous out-patient ketamine treatment of c.50%. Reference Anand, Mathew, Sanacora, Murrough, Goes and Altinay19 However, the definition of TRD is contested (5) and the variability in severity, chronicity comorbidity etc. of people who meet its fairly low threshold is enormous.
Importantly, our OLE sample design selected for greater ‘treatment-resistant’, enriching for poorer prognosis by excluding those showing a sustained response after the RCT. Further, the KADS participants seem particularly resistant to placebo effects. The placebo response rate at 4 weeks in the KADS trial was small; 5.6% (5/88), much lower than the 21% seen in other TRD trials. Reference Matsingos, Wilhelm, Noor, Yildiz, Rief and Hofmann20 Further, the within-group MADRS effect sizes in the RCT active control arm were −0.60 and −0.58 in the 2 dosing regimens, a magnitude lower than the pooled within-group placebo group effect size of −1.85 reported in systematic reviews of ketamine RCTs Reference Jones, Razza, Weissman, Karbi, Vine and Mulsant21 and −1.08 for other treatments for TRD. Reference Matsingos, Wilhelm, Noor, Yildiz, Rief and Hofmann20
Some trials of ketamine show low doses may be ineffective, Reference Smith-Apeldoorn, Veraart, Kamphuis, Spijker, van der Meij and van Asselt22 whereas some clinical cohort or file review studies suggest high levels of effectiveness (e.g. Li et al Reference Li, Zhou, Liu, Wang, Lan and Zhang23 reported that 80% of patients were classified as responders 9 months after 6 infusions of low-dose ketamine). It may be that higher doses or longer treatment with subcutaneous ketamine than 0.9 mg/kg are required to improve response rates as there was no superiority seen in the flexible-dose regimen (Fig. S1). The observation of the improving MADRS trajectory when treatment was stopped in the flexible-dose regimen, whereas the MADRS had plateaued in the fixed-dose regimen, supports this interpretation. Second, the flexible dose induced more acute dissociation symptoms and there is some, Reference Niciu, Shovestul, Jaso, Farmer, Luckenbaugh and Brutsche24 but by no means consistent, Reference Sajid, Galfalvy, Keilp, Burke, Mann and Grunebaum25 evidence suggesting that dissociation may be associated with more antidepressant effect.
Up to 50% more participants were described as ‘much or very much improved’ or having ‘mild depression’ or better by their clinicians at treatment end, and 4 weeks later, than when defined by MADRS standards. Clinicians may incorporate other information than just depressive symptoms, e.g. function or relationships, in their determination of improvement. We are undertaking further work to evaluate how best we can make person-centric evaluations of improvement for clinical use rather than reliance upon regulatory approval bodies’ insistence on only symptom metrics.
Limitations
The limitations of this study in part reflect some of the strengths emerging from the OLE design, such as selecting a highly treatment-resistant sample. As an investigator-initiated study, with fewer exclusions than many industry trials, the patients may be more generalisable to clinical practice. However, this method excluded those unwilling to be subject to randomisation in the RCT, which introduces possible selection bias. We do not know if this led to any over- or under-estimation of effects or side-effects. Although the OLE assessments were unblinded, the raters had been trained for the RCT, and were unaware of RCT arm allocation, reducing the potential for information bias to explain the novel observation regarding prior treatment with ketamine.
As different ketamine treatment approaches are scaled up and expanded, many questions remain. What is an optimum dosing protocol (e.g. frequency, maximum dose, rate of up-titration, duration) of subcutaneous ketamine remains to be clarified. Is prior response to ketamine a replicable predictor of future response? Does this translate across types of ketamine and delivery systems? We also need to know if longer treatment improves outcomes, or sustains effects for longer once ceased, and if ketamine should be considered a long-term treatment for this chronically unwell population.
Implications
This study has several implications for research and clinical practice. In this highly treatment-resistant sample, we observed similar remission and response rates after a 4-week open label course of subcutaneous racemic ketamine to those seen in the RCT phase, Reference Tham, Do, Fridfinnson, Rafizadeh, Siu and Budd10 but response declining substantially after treatment cessation. We observed no clinically significant adverse safety profile at doses up to 0.9 mg/kg, and importantly, no suicide or self-harm requiring admission reported up to 6 months after ceasing treatment. We found no evidence that prior ketamine treatment either attenuated or enhanced the effectiveness of a second course of treatment, and that people who responded to treatment were likely to respond to retreatment, clinically relevant findings that need (dis)confirmation.
Supplementary material
The supplementary material is available online at https://doi.org/10.1192/bjp.2026.10692
Data availability
De-identified data from this study may be requested from the corresponding author, subject to approval from the Trial Steering Group and the approving Human Research Ethics Committee.
Acknowledgement
We sincerely thank all the participants who were involved in the study, and all investigators and staff at the study centres.
Author contributions
N.G., C.K.L., P.B.M., P.G., D.H.-P., A.S., M.L.H., A.R., B.T.B., D.B., D.M., G.C., M.B. and S.D.H. were involved in the grant proposal that led to funding of the study. N.G., E.S., C.K.L., A.A., V.D., D.B., B.T.B., N.T.M., P.G., M.L.C., S.S., V.G.-O. and A.J.B. were involved in data collection and investigation. D.H.-P., S.N., D.M., V.D. and R.M. were involved in data curation. N.G., E.S., R.M., C.K.L. and S.N. wrote the early drafts of the manuscript. N.G., R.M., V.D. and S.N. conducted the data analysis. All authors reviewed, edited and approved the manuscript.
Funding
The study was funded by a competitive research grant from the Australian National Health and Medical Research Council (APP1105089). M.B. is supported by a National Health and Medical Research Council (NHMRC) Leadership 3 Investigator grant (2017131). C.K.L. is supported by an NHMRC Leadership Investigator grant (1195651). N.G., R.M. and E.S. were supported (partially or fully) by the Australian Government through the Australian Research Council’s Centre of Excellence for Children and Families over the Life Course (Project ID CE200100025). The lead author and manuscript guarantor (N.G.) affirms that the manuscript is an honest, accurate and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Declaration of interest
In the past 36 months, N.G. has received speaker’s bureau honoraria from Servier Laboratories, Janssen and Lundbeck and served on advisory boards for Servier Laboratories, Esia, Seqirus and Lundbeck. D.B. is a director and part-owner of Neurotrials Victoria Pty Ltd, trading as Neurocentrix and Neurocentrix TMS Pty Ltd; he serves on the advisory board for Eli Lilly and Janssen, and is currently supported by grant funding from Praxis, Janssen, Eli Lilly, Biogen and NHMRC; he has served on speaker panels for Servier, Janssen and Eli Lilly in the past 12 months; he is an investigator on the Janssen Quality of Life Esketamine study. B.T.B. has received grants and served as consultant, advisor or continuing medical education (CME) speaker for AstraZeneca, Bristol-Myers Squibb, Janssen, Lundbeck, Otsuka, Servier, Teva, Boehringer Ingelheim, GH Research, Medscape, Angelini, the NHMRC, DFG, the European Union, EraPerMed, BMB/BMBF, the Wellcome Trust, the Fay Fuller Foundation and the James and Diana Ramsay Foundation. Within the last 36 months, P.G. has attended a Janssen New Zealand advisory board, and is named on a patent for a controlled release ketamine tablet developed by Douglas Pharmaceuticals. In the past 36 months, D.M. has received research consulting fees from Douglas Pharmaceuticals for a clinical trial involving ketamine. In the past 3 years. G.C. has received educational and travel support from Servier, Astra Zeneca, Otsuka Australia, Merck Sharp & Dohme and Janssen-Cilag in the past 5 years; he also served on an advisory board for the AFFINITY trial. A.S. is a director of the Australian Medicines Handbook Pty Ltd (unpaid) and has received funding support from the Australian and New Zealand College of Anaesthetists to investigate ketamine for chronic post-surgical pain. S.D.H. has received speaker and consultancy fees from Janssen and Servier and served on advisory boards for Janssen and Lundbeck. C.K.L. is on the Clinical Advisory Board for Douglas Pharmaceuticals and has received fees for the following: Janssen Cilag advisory board, Douglas Pharmaceuticals advisory board. S.S. received research funding from Douglas Pharmaceuticals as an investigator on a ketamine clinical trial. In the past 3 years, V.G.-O. has received educational and travel support from Lundbeck-Otsuka and Janssen, as well as speaker fees for Janssen. R.M., E.S., A.R., M.B., S.N., P.B.M., N.T.M., D.H.-P., M.L.H., V.D., M.L.C., A.J.B. and A.A.: None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2013. All procedures involving human subjects/patients were approved by Sydney Local Health District (Royal Prince Alfred Hospital (RPAH Zone)) Human Research Ethics Committee (Australia; X16-0146 & HREC/16/RPAH/168) and Southern Health and Disability Ethics Committee (New Zealand; 16/STH/104). The protocol is available at http://doi.org/10.17605/OSF.IO/6FPGU.





 Open access
Open access
eLetters
No eLetters have been published for this article.