

1. Introduction
Hepatocellular carcinoma (HCC) is a primary liver malignancy that mainly occurs in patients with chronic liver disease and cirrhosis. HCC is the fifth-leading cause of cancer worldwide, which affected the health of half a million people (Raza & Sood, Reference Raza and Sood2014). From 2002 to 2012, approximately 20,000 new cases were diagnosed in the United States per year. The high incidence of HCC remains a significant threat to public health (Arzumanyan et al., Reference Arzumanyan, Reis and Feitelson2013). Since people below the age of 40 seldom get HCC, the likelihood of HCC reaches its peak at approximately 70 years of age (El-Serag, Reference El-Serag2011). The incidence of HCC among men is two- to four-times higher than among women (El-Serag, Reference El-Serag2011).
The pathogenic mechanism of HCC is complicated, with many of risk factors, mainly including alcoholic fatty liver disease, hepatitis B virus (HBV) or hepatitis C virus (HCV) infection and hereditary hemochromatosis (Lin et al., Reference Lin, Hoffmann and Schemmer2012). The lower-risk factors include autoimmune hepatitis, porphyrias, α1-antitrypsin deficiency and Wilson's disease. However, these risk factors are unevenly distributed across the world depending on racial background, geographic region and environmental factors (El-Serag & Rudolph, Reference El-Serag and Rudolph2007). Worldwide, chronic HBV infection is a major underlying cause of HCC, accounting for approximately 50% of all HCC cases (El-Serag, Reference El-Serag2012). Case–control studies have shown that the incidence of HCC in chronic HBV carriers was 5–15-times greater than that of people without chronic HBV infection, which is particularly applicable to endemic regions such as Asia and Africa. In these areas, HBV is usually transmitted by vertical transmission (from mother to fetus). The high prevalence of HBV strongly contributes to the development of HCC and chronic liver disease (El-Serag & Rudolph, Reference El-Serag and Rudolph2007). In areas with low HCC incidence rates, HBV is mainly spread in adulthood by horizontal transmission (sexual and parenteral routes). More than 90% of acute HBV infections recover spontaneously (El-Serag, Reference El-Serag2012). Both HBV and other aetiological factors are likely to induce chronic liver diseases, such as liver cirrhosis, which is the most common aetiology of hepatic carcinoma (Floyd, Reference Floyd1962).
The prognosis of HCC is one of the worst in cancer, as it is usually detected in its late stages (Feitelson, Reference Feitelson2006). Understand the underlying molecular mechanisms and identifying its potential gene targets will be useful for early detection and clinical therapies (Lambert et al., Reference Lambert, Paliwal, Vaissiere, Chemin, Zoulim and Tommasino2011).
Many efforts have been made to reveal the underlying pathogenetic mechanism of HCC. However, the specific role of HBV in HCC development is not yet completely understood, and the difference between HBV-associated HCC (hereafter referred to as HBVaHCC) and HBV-non-associated HCC (hereafter referred to as HBVnHCC) is still not clear. Here, we explore an integrated method based on Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, which have been widely used to annotate enrichment, to analyse the function of genes (Rhodes & Chinnaiyan, Reference Rhodes and Chinnaiyan2005) and to explore the underlying mechanisms of HBVaHCC and HBVnHCC. Shortest path algorithm was employed to find all shortest paths connecting any two of the known HBVaHCC or HBVnHCC related genes. Genes that occurred in at least one shortest path were regarded as candidates (Floyd, Reference Floyd1962). Further analysis indicated that some candidate genes had been previously reported as HCC-related genes based on literature searches. We hope that our results help to uncover the mechanism of HCC and provide new insights for early diagnosis and novel therapies.
2. Methods
2.1. Encoding genes for classification and prediction
GO (Shaw et al., Reference Shaw, Ashbumer, Blake, Baldarelli, Botstein and Davis1999) is a major bioinformatics initiative aimed at standardizing the description of gene functions and gene products that will facilitate computer-driven information retrieval and the generation of new knowledge from omics-scale data. The KEGG database (Hsieh et al., Reference Hsieh, Su, Wang, Chang, Lei and Lai2004) is high-quality database covering a collection of manually drawn pathway maps representing our knowledge of the molecular interactions and reaction networks of metabolism, genetic information processing, environmental information processing, cellular processes, human diseases and drug development. Gene properties can be depicted by using GO and KEGG pathway annotation. If two genes share a similar function, they will have similar annotations in GO and KEGG pathways. Therefore, we encoded each gene as a numeric vector consisting of an enrichment score for each of the GO and KEGG terms. The GO or KEGG enrichment score is defined as its –log10 of the p-value for examining hyper-geometric tests for one gene or its interaction neighbours (Carmona-Saez et al., Reference Carmona-Saez, Chagoyen, Tirado, Carazo and Pascual-Montano2007; Yang et al., Reference Yang, Chen, Kong, Huang and Cai2014) in a Search Tool for Recurring Instances of Neighbouring Genes (STRING) network. A higher enrichment score means a higher degree of enrichment. As a result, a gene is encoded as an eigenvector with 6242 GO terms and 214 KEGG pathways.
2.2. Selecting optimal features from positive and negative samples
A series of feature selection methods was adopted, such as minimum redundancy maximum relevance (mRMR) (Peng et al., Reference Peng, Long and Ding2005), incremental feature selection (IFS) (Peng et al., Reference Peng, Long and Ding2005) and a random forest (RF) algorithm (Ge & Zhang, Reference Ge and Zhang2014; Mao et al., Reference Mao, Raj Kumar, Guo, Zhang and Liang2014).
Specifically, the mRMR method is based on the maximum relativity and minimum redundancy for sorting the features to be screened. Then, IFS was applied in order to make the processing accord with the order that mRMR returns. Once each additional feature is added, classifications were performed in order to evaluate the results of these known features. A classifier is used together with the RF algorithm (using Weka3·6·4 [Bouckaert et al., Reference Bouckaert, Frank, Hall, Holmes, Pfahringer and Reutemann2010] in the RF algorithm). In classification evaluation, ten-fold cross-validation was used in order to simultaneously calculate the true-positive (TP) rate, the true-negative (TN) rate, the false-positive (FP) rate and the false-negative (FN) rate. Then, Matthews's correlation coefficient (MCC) based on the TP, TN, FP and FN rates was used in order to summarize the performance of the classification. When the maximum MCC first appeared, the combination of the features was regarded as the optimal feature set, and they were the maximum relevance and minimum redundancy features of osteoarthritis-related genes. The MCC value was calculated by:
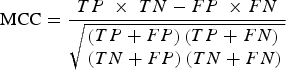 $${\rm MCC} = \displaystyle{{TP\; \times \;TN - FP\; \times FN} \over {\sqrt {\matrix{\left( {TP+ FP} \right)\left( {TP + FN} \right) \hfill \cr \left( {TN + FP} \right)\left( {TN + FN} \right) \hfill}} }}$$
$${\rm MCC} = \displaystyle{{TP\; \times \;TN - FP\; \times FN} \over {\sqrt {\matrix{\left( {TP+ FP} \right)\left( {TP + FN} \right) \hfill \cr \left( {TN + FP} \right)\left( {TN + FN} \right) \hfill}} }}$$
2.3. Predicting novel candidate genes related to HBVaHCC and HBVnHCC
The optimal features of HBVaHCC and HBVnHCC were selected and employed in the RF classifier in order to predict related genes. All of the annotated genes in the Ensemble database were also encoded by GO and KEGG and used as the test samples. The known HBVaHCC- or HBVnHCC-related genes were used as the training samples, respectively. The RF classifier was used in order to predict novel HBVaHCC and HBVnHCC genes, respectively.
2.4. Identifying the shortest-path genes based on a STRING network
Previous studies (Deng et al., Reference Deng, Zhang, Mehta, Chen and Sun2003; Ng et al., Reference Ng, Ciou and Huang2010; Hu et al., Reference Hu, Huang, Liu and Cai2011; Mathews et al., Reference Mathews, Cabarcas, Hurt, Zhang, Jaffee and Farrar2011) have indicated that genes that could interact with each other share similar functions. Thus, we constructed a network based on the gene–gene interactions from STRING (Jensen et al., Reference Jensen, Kuhn, Stark, Chaffron, Creevey and Muller2009). Each interaction in STRING was evaluated with the standard of interaction confidence score ranging from 1 to 999 in order to measure the likelihood of interaction. Dijkstra's algorithm was applied to the R package ‘igraph’ so as to identify the shortest path between each gene pair. In order to find all of the shortest paths connecting any two genes, we selected the genes that occurred in at least one shortest path as candidate genes through applying the shortest-path algorithm to this network. Then, a randomization test was employed in order to filter these candidate genes.
2.5. HCC specimens and quantitative real-time RT-PCR
Thirty HCC tumour tissues and 20 matched adjacent normal liver tissues were obtained from surgical resection at Panyu District Central Hospital (Guangzhou, China). Routine in situ hybridization for HBV-encoded RNA was performed in order to classify HBVaHCC or HBVnHCC. There were 14 HBVaHCC tissues and 16 HBVnHCC tissues in our tumour tissues. Total RNA was extracted from the snap-frozen tumour tissues or adjacent normal liver tissues with TRIzol reagent (Life Technology). cDNA was obtained by reverse transcription of RNA. Quantitative RT-PCR was performed on an ABI 7900 HT Sequence Detection System (Applied Biosystems) using TaqMan Gene Expression Master Mix (4369016, Applied Biosystems). The expression values were shown as relative expression levels to GAPDH. The primer sequences are shown in Supplementary Table 1 (available online).
2.6. Ethics statement
Prior written informed consent was obtained from each patient, and the study was conducted in accordance with the principles of the Declaration of Helsinki and approved by Panyu District Central Hospital.
3. Results
3.1. Known HBVaHCC- or HBVnHCC-related genes in databases
The Genetic Association Database (Zhang & Lu, Reference Zhang and Lu2015) is a comprehensive database of complex human diseases that includes the disease genes of complex diseases annotated by the reported literature and genome-wide association study experimental data. When searching the database with “hepatitis B hepatocellular carcinoma” as the keyword, we obtained 52 HBVaHCC-related genes. Moreover, by searching with “hepatocellular carcinoma” as the keyword and removing the HBV-associated genes, we obtained 129 HBVnHCC-related genes.
The DisGeNET Database (Sun & Karin, Reference Sun and Karin2008) annotates disease genes through the integration of public databases and gene–disease relationships in the reported literature. Currently, the database contains 381,056 gene–disease relationships (covering 16,666 genes and 13,172 kinds of disease). When searching the database with “hepatitis B virus-related hepatocellular carcinoma” (umls: C1333977) as the keyword, we obtained 16 genes. In the same way, we found the HBVnHCC-related genes from the DisGeNET and Catalogue of Somatic Mutations in Cancer (COSMIC) (Forbes et al., Reference Forbes, Bindal, Bamford, Cole, Kok and Beare2011) databases.
We found a total of 63 HBVaHCC-related genes and 152 HBVnHCC-related genes (no duplicates) through database searches, whose specific gene names and database sources are shown in Supplementary Table 2.
3.2. Optimal features for distinguishing HBVaHCC-related and HBVnHCC-related genes
The HBVaHCC-related and HBVnHCC-related genes collected from the databases were encoded by GO and KEGG. In order to identify the features that could describe the differences between HBVaHCC and HBVnHCC, we took HBVaHCC genes as the positive samples and HBVnHCC genes as the negative samples. We employed a series of feature selection procedures in order to identify the optimal features that could distinguish these samples. As a result, these optimal features between these two types of HCC were different, containing 52 GO terms and two KEGG terms (as shown in Table 1).
The optimal distinct genetic features between hepatitis B virus-associated and -non-associated hepatocellular carcinoma

The GO terms consist of three main types: the biological process, the cellular component and the molecular function GO terms. In order to exemplify the profile of the huge GO terms in the optimal set, we grouped them into the children of three root GO terms (as shown in Fig. 1). The children terms briefly described the different GO terms for distinguishing HBVaHCC-related and HBVnHCC-related genes. The KEGG terms are pathways of “O mannosyl glycan biosynthesis” and “melanogenesis”.
The distribution of the Gene Ontology (GO) terms for the optimal distinct features between hepatitis B virus-associated and -non-associated hepatocellular carcinoma. The GO terms are grouped into the children of three root GO terms.

3.3. Optimal feature sets for distinguishing tumour-related genes in HBVaHCC and HBVnHCC
We also compared HBVaHCC-related genes with other genes and HBVnHCC-related genes with other genes in order to investigate the characteristics of HBVaHCC- and HBVnHCC-related genes. The tumour genes were taken as the positive samples and other genes were randomly selected as the negative samples. Specifically, HBVaHCC had 63 tumour-related genes as positive samples and 252 other genes randomly selected as negative samples. HBVnHCC had 152 tumour-related genes as positive samples and 508 other genes randomly selected as negative samples. Because the number of negative samples was much larger than that of the positive samples, the negative samples were randomly divided into four groups. The four negative sample sets and their corresponding positive samples were formed as four training sets, HBVaHCC and HBVnHCC respectively. The feature selecting classifier identified the optimal features in the four HBVaHCC and four HBVnHCC training sets, respectively. The corresponding number of features was considered to be the optimal feature set in the eight groups when the MCC value reached the maximum. Finally, we took the union of four optimal feature sets in HBVaHCC and HBVnHCC as the final optimal feature set of HBVaHCC and HBVnHCC, respectively.
Therefore, the optimal features for predicting HBVaHCC-related genes included 1768 GO terms and 70 KEGG pathways (as shown in Supplementary Table 3), while the optimal features for predicting HBVnHCC included 1202 GO terms and 59 KEGG pathways (as shown in Supplementary Table 4). The feature terms of the two types of HCC were different for the most part, which was coherent with the distinct mechanism of cancer development. It was demonstrated that we could adopt these features in order to distinguish these two types of HCC.
3.4. Novel HBVaHCC and HBVnHCC genes predicted by optimal GO and KEGG pathway terms
Based on the above optimal features of HBVaHCC and HBVnHCC genes, we predicted novel related genes from all of the annotated genes. The RF algorithm acted as the prediction machine in order to help us determine the genes that had similar functions to the known HBVaHCC- and HBVnHCC-related genes. We predicted 1290 genes as being related to HBVaHCC and 1005 genes related to HBVnHCC (as shown in Supplementary Table 5). Among them, 1236 and 881 genes were novel genes, respectively.
3.5. Shared genes related to both HBVaHCC and HBVnHCC
In the genes that were predicted by the classifier, there were 639 shared genes between HBVaHCC and HBVnHCC (as shown in Supplementary Table 6), accounting for approximately 50% of the total number of predicted genes. As these two types were both related to HCC development, it was expected that they would share most of their genes. We also employed another approach, the shortest-path analysis, in order to determine the HBVaHCC-related and HBVnHCC-related genes in the gene interaction network and collect the shared genes. We consequently obtained the candidate related genes and determined the betweenness of them. In the shortest-path network, the betweenness of genes indicates the number of shortest paths containing the gene as an inner node and connecting all pairs. Therefore, it is possible that genes with higher betweenness were more likely to have been related to cancer genes than those with lower betweenness. Through the shortest-path analysis, we identified the related genes and found 108 shared genes for both types (as shown in Supplementary Table 7). In total, there were 679 shared genes as found by combining the predicted genes through the classifier and shortest-path genes. In addition, there were 69 overlaps between the predicted genes and the genes obtained from the shortest-path analysis (as shown in Table 2). These genes were considered to have similar functions to the known related genes and, indeed, they interacted with these known related genes, which was worthy of further investigation.
The 69 overlaps between the predicted genes and the genes obtained from shortest-path analysis

3.6. Expression verification of the identified genes in tumour and normal tissues
In order to confirm the presence of these overlapped genes in HCC specimen, we compared the expression of the top ten relevant genes for both HBVaHCC and HBVnHCC between tumour tissues and normal tissues by quantitative real-time RT-PCR. We verified six significantly up-regulated genes and two significantly down-regulated genes in HBVaHCC tissues (Fig. 2(a)) and verified seven significantly up-regulated genes and one significantly down-regulated gene in HBVnHCC tissues (Fig. 2(b)). These genes have similar expression patterns in the two types of HCC. CASP3, HDAC1, IRS1, RAF1, RIPK1 and SYK were significantly up-regulated in both HBVaHCC and HBVnHCC tissues. FYN was significantly down-regulated in both tissues. This result suggested that our system of biological measurement for identifying HCC-related genes was reliable.
Gene expression verification of the novel identified genes by quantitative real-time RT-PCR. (a) Expression of the top ten overlapped identified genes in hepatitis B virus-associated hepatocellular carcinoma tissues. (b) Expression of top ten overlapped identified genes in hepatitis B virus-non-associated hepatocellular carcinoma tissues. *p < 0·05, **p < 0·01.

4. Discussion
4.1. Different features of HBVaHCC and HBVnHCC
As a unique kind of hepatocellular cancer, HBVaHCC is triggered by HBV and has some distinct features. Based on GO and KEGG enrichment analysis, we obtained 54 GO and KEGG features in the optimal feature list (as shown in Table 1), which included 31 biological process terms, 12 cellular component terms, nine molecular function terms and two KEGG terms. Some GO terms were shown to be highly related to the differences between HBVaHCC and HBVnHCC. For example, inclusion of GO:0002839 (positive regulation of the immune response to tumour cell), GO:0060330 (regulation of response to interferon-γ) and GO:0001909 (leukocyte-mediated cytotoxicity) revealed that the immune antiviral response to the HBV proteins played an important role in HBVaHCC (Chen et al., Reference Chen, Wei, Sun and Tian2005).
GO:0034136 (negative regulation of the Toll-like receptor 2 [TLR2] signalling pathway) and GO:0038061 (NIK/NF-κB signalling) might contribute to distinguishing HBVaHCC from HBVnHCC. NF-κB transcription factors and Toll-like receptors are crucial regulators of danger recognition and immune responses. Activation of the TLR3 pathway plays an important role in suppressing HBV replication in the liver, and TLR ligands might act as the promising therapeutics for chronic HBV infections (Zhang & Lu, Reference Zhang and Lu2015). The NF-κB signalling pathway has great relevance to several liver diseases, including chronic HBV infection, fibrosis, liver cirrhosis and HCC, and has an important hepatoprotective function. The NF-κB pathway might serve as a useful target for the treatment of HCC (Sun & Karin, Reference Sun and Karin2008).
GO:0016638 (oxidoreductase activity, acting on the CH–NH2 group of donors) and GO:0015002 (haem–copper terminal oxidase activity) has been associated with oxidative reactions, and the pre-S mutant of HBsAg has been reported to contribute to the oxidative stress and DNA damage of hepatocytes (Hsieh et al., Reference Hsieh, Su, Wang, Chang, Lei and Lai2004).
GO:0005585 (collagen type II) refers to the process by which type II collagen triply forms fibrils and is highly associated with HBVaHCC. It has been reported that HBV infection may have an indirect effect on the occurrence of HCC through the fibrosis process (Mantych et al., Reference Mantych, Devaskar, deMello and Devaskar1991). Hepatocellular fibrosis will induce architecture derangement and portal hypertension. The irreversible rearrangement of the fibrils may cause cirrhosis (Chu & Liaw, Reference Chu and Liaw2006).
Some terms, including GO:0006117 (acetaldehyde metabolic process), GO:0047844 (deoxycytidine deaminase activity), GO:0016011 (dystroglycan complex), GO:0015012 (heparan sulphate proteoglycan biosynthetic process), GO:0045263 (proton-transporting ATP synthase complex), coupling factor F(o) and ‘KEGG: O mannosyl glycan biosynthesis’ are believed to be involved in metabolism and cellular biosynthesis. Compared with normal tissues, tumours have many important alternations in metabolic pathways supporting the rapid growth and proliferation of cancer cells by providing rapid ATP generation and increasing the biosynthesis of macromolecules. It is clear that genetic mutations and the tumour microenvironment can alter cellular metabolism (Cairns et al., Reference Cairns, Harris and Mak2011). It is known that various tumours caused by different molecular mechanisms may have many common but unique alterations and metabolic profiles (Baenke et al., Reference Baenke, Peck, Miess and Schulze2013). The inclusion of these terms indicates that HBVaHCC and HBVnHCC may have some distinct underlying features of metabolism.
GO:0072091 (regulation of stem cell proliferation) and GO:0017145 (stem cell division) refer to the process by which stem cells proliferate and divide in order to provide cells that can develop into mature and functional cells.
These terms are associated with cell growth and proliferation, which are common hallmarks of cancer and may play an important role in HCC (Xiao et al., Reference Xiao, Li and Huang2004).
4.2. Analysis of the optimal features characterizing HBVaHCC-related genes
We selected the top 15 GO children terms ranked by relevance to HBVaHCC for further analysis. They covered 12 biological process terms, one cellular component term and two molecular function GO terms. Several GO terms were shown to be highly related to HBVaHCC. HBVaHCC had several shared but distinctive characteristics compared with HBVnHCC.
GO:0009636 (response to toxic substances), GO:0032870 (cellular response to hormone stimuli) and GO:0034059 (response to anoxia) were all related to the cellular stress response to stimuli. It is known that HBV can infect liver cells and activate several important reactions that can contribute to the development of HCCs. For example, the HBV X protein (HBx) has pleiotropic effects, including cell responses to genotoxic stress, cell division and apoptosis (Zhang et al., Reference Zhang, Zhang and Ye2006).
The inclusion of GO:0010884 (positive regulation of lipid storage) and GO:0010957 (negative regulation of the vitamin D biosynthetic process) met our expectations. As we discuss in Section 4.1, there is increasing evidence supporting the implications of metabolism in hepatocellular cancer development (Montella et al., Reference Montella, Crispo and Giudice2011; Jump et al., Reference Jump, Depner, Tripathy and Lytle2015).
The inclusion of GO:0097200 (cysteine-type endopeptidase activity involved in the execution phase of apoptosis) implied that cell apoptosis was associated with HBVaHCC. HBx is a key regulator of HBV replication that plays an important role in the development of HBVaHCC (Ha & Yu, Reference Ha and Yu2010). HBx modulates a variety of cellular pathways, including apoptosis. Thus, deregulation of normal apoptotic pathways might influence the development and progress of HCC (Rawat et al., Reference Rawat, Clippinger and Bouchard2012).
The inclusion of GO:0006999 (nuclear pore organization), GO:0030118 (clathrin coat) and GO:0045837 (negative regulation of membrane potential) in the top feature list might provide us with new insights for understanding the mechanisms of HBVaHCC.
4.3. Analysis of the optimal features characterizing HBVnHCC-related genes
We chose the top 15 features associated with HBVnHCC for further analysis. The top 15 feature list contained 11 biological process terms, two molecular function terms and two cellular component terms. Some GO terms were related to HBVnHCC.
GO:0038186 (lithocholic acid receptor activity) was also highly related to the development of cancer. Lithocholic acid was effective at killing several types of cancer cells, including some brain tumours and breast cancers. It is reported that lithocholic acid specifically targeted cancer cells without killing normal cells, which might improve existing chemotherapy drugs (Arlia-Ciommo et al., Reference Arlia-Ciommo, Piano, Svistkova, Mohtashami and Titorenko2014). Findings have shown that lithocholic acid has a broad anti-tumour effect on several kinds of cancer cells derived from different tissues (Goldberg et al., Reference Goldberg, Beach, Davies, Harkness, Leblanc and Titorenko2011).
GO:0070664 (negative regulation of leukocyte proliferation) refers to any process that reduces or prevents the extent of leukocyte proliferation, which is highly related to hepatocellular cancer. HCC is a typical kind of inflammation-related cancer. Several studies have indicated that the tumour microenvironment and leukocyte infiltrate play important roles in the development and progress of inflammation-related cancers by secreting cytokines, chemokines and growth factors (Capece et al., Reference Capece, Fischietti, Verzella, Gaggiano, Cicciarelli and Tessitore2013). Immune responses participate in different stages of cancer, such as tumour initiation, malignant conversion and metastasis. It has been generally accepted that inflammation plays an essential role in tumorigenesis; therefore, an inflammatory microenvironment is required for all kind of tumours (Grivennikov et al., Reference Grivennikov, Greten and Karin2010).
GO:0097057 (TRAF2–GSTP1 complex) was involved in disrupting TNF signalling and regulating inflammatory responses. Monocytes and many other immunological cells secrete TNF cytokines, which have been implicated in tumorigenesis (Balkwill, Reference Balkwill2006). By binding to receptors, TNF could activate various downstream signalling cascades, including inflammatory, apoptotic and stress pathways, such as the NF-κB, MAPK and JNK pathways in tumours (Roderburg et al., Reference Roderburg, Gautheron and Luedde2012).
GO:0001541 (ovarian follicle development) and GO:0001542 (ovulation from ovarian follicles) were associated with the progression of the ovarian follicle. Ovaries might be related to HCC. It has been reported that a small fraction of HCCs can spread to the ovaries (de Groot et al., Reference de Groot, Dukel, Chadha-Ajwani, Metselaar, Tilanus and Huikeshoven2000; Papp et al., Reference Papp, Pal and Hurst2003).
4.4. Shared genes related to both HBVaHCC and HBVnHCC
Although HBVaHCC and HBVnHCC differ greatly in many aspects, they also have a lot of characteristics in common. Previous research has demonstrated that all kinds of cancers share several common features, including cell growth, proliferation, apoptosis and metastasis, which contribute to the malignant transformation of normal cells. Based on the analysis of the shortest-path algorithm, we found a number of genes that might be associated with HBVaHCC and HBVnHCC, several of which were subsequently confirmed by experimental data from previously published research.
The protein encoded by the BAD gene belongs to the BCL-2 family, which is known for module cell apoptosis. Phosphorylated BAD protein has been reported to be down-regulated in HCC cells compared with normal hepatocytes. The loss of phosphorylated BAD results in deregulation of cell apoptosis, which might play a key role in liver tumourigenesis (Yoo et al., Reference Yoo, Lee, Jeong, Soung, Nam and Lee2006). The CD38 gene encodes a multifunctional ectoenzyme that is widely expressed in different tissues, especially in leukocytes. The CD38 molecule is strongly associated with cancer development and has been reported to be up-regulated on the cell surfaces of leukemic B cells (Bauvois et al., Reference Bauvois, Durant, Laboureau, Barthelemy, Rouillard and Boulla1999). The protein encoded by CREBBP is a transcriptional co-activator and important regulator of eukaryotic gene expression. It is involved in the transcriptional regulation of many important transcription factors, including P53 (Grossman, Reference Grossman2001). Dysfunction of this gene probably contributes to cancer development (Mullighan et al., Reference Mullighan, Zhang, Kasper, Lerach, Payne-Turner and Phillips2011). The PROM1 gene encodes a transmembrane glycoprotein located on membrane protrusions. It has been reported to be involved in suppressing cell differentiation and maintaining stem cell properties. PROM1 is known to be highly related to HCC and has often served as a marker of hepatoblast and liver cancer stem cells (Katoh & Katoh, Reference Katoh and Katoh2007; Andrisani et al., Reference Andrisani, Studach and Merle2011).
The NOS2 gene is also associated with HCC. The protein encoded by NOS2 is a nitric oxide synthase expressed in liver. As a reactive free radical, nitric oxide participates in several processes, including anti-microbial and anti-cancer pathways (Levesque et al., Reference Levesque, Hobbs, Anstey, Chancellor, Misukonis and Granger2001). The levels of nitric oxide in HCC are significantly down-regulated compared with adjacent non-tumour liver tissues (p < 0·001). Compared with adjacent normal liver tissues, the expression levels of NOS2 and nitric oxide in HCC are significantly down-regulated in HCC. These decreased levels of nitric oxide/NOS2 might contribute to HCC development and progression (Zhou et al., Reference Zhou, Wang, Tian, Yang and Yang2012).
Note that the HBVnHCC-related genes constitute the HCC-related gene set that was not associated with HBV in order to cater to our design. Therefore, the HBVnHCC genes used here could include genes that are involved in other causes of HCC, and the HBVnHCC genes in this study constituted a complex group. HCV infection is another main risk factors for HCC incidence. We also searched the HCV-related HCC genes using the keywords “hepatitis C virus-related hepatocellular carcinoma” or other synonyms in the databases. There were only 19 genes that were related to HCV in the HBVnHCC genes. Removing these 19 genes, we again analysed the remaining 133 HBVnHCC genes. The optimal features for distinguishing the new HBVnHCC genes from the other genes constituted 1097 terms, 164 fewer than that of the previous HBVnHCC group. These 164 optimal features included 159 GO terms and five KEGG terms. The significant differences of these optimal features compared with the previous ones were the absence of terms for antioxidant activity, receptor regulator activity, protein binding transcription factor activity, extracellular regions and cell junctions. Using the new optimal features, the 209 genes that had been previously predicted to be HBVnHCC-related genes now were predicted to be non-related ones.
In this study, we collected the known HBVaHCC- or HBVnHCC-related genes from public databases and analysed their genetic features. Comparing the features between two types of HCC, we identified 52 GO terms and two KEGG terms for distinguishing them. Comparing the features of HBVaHCC- or HBVnHCC-related genes to the features of genes with no relation to HCC, we identified the optimal features for extracting HBVaHCC- and HBVnHCC-related genes and predicted 1236 and 881 novel genes for them, respectively. We further analysed the gene interaction network and also found some genes that might be involved in HCC by using the shortest-path algorithm. Combing the predicted genes through classifier and shortest-path genes, we identified 679 shared genes for the two types of HCC. These findings might provide genetic targets for studying the roles of HBV in HCC, as well as useful insights for understanding the pathogenesis of HCC with or without HBV.
This work was supported by Guangdong Province Science and Technology Planning Project (No. 20130319C) and Guangzhou University of Chinese Medicine Pass-it-on Project (No. XH20140117).
5. Declaration of interest
None.
6. Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0016672316000124.






