


Introduction
The M 2(TO4)2Φ8 cyclic tetramer cluster shown in Fig. 1 is one of four clusters of general composition M 2(TO4)2Φn that has been shown by Hawthorne (Reference Hawthorne1983) to have high stability for valency combinations M = 2+, 3+, and T = 5+, 6+ and to thus form the basis for extended hierarchies of mineral structures in phosphate, arsenate and sulfate minerals. In a companion paper (Grey, Reference Grey2025) the structural role of the cluster for M = Fe, Mn, Ca and T = P has been studied in four major primary mineral groups present in granitic pegmatites; triphylite, alluaudite, zwieselite and graftonite. An analysis of the crystal structures of the four groups from the perspective of the connectivity between the metal atom polyhedra and the PO4 tetrahedra for the individual metal atom sublattices has resulted in the identification of structural motifs that are common to the four primary mineral groups. All four structure types contain two-octahedra-wide ribbons formed from corner-linking of the M 2(PO4)2Φ8 cyclic tetramers. The ribbons in the four structure types all have the same topology, but correspond to different geometrical isomers due to different orientations of the tetrahedra and different pairs of octahedral edges involved in the ribbon formation. The same ribbons are also present in arrojadite–dickinsonite group primary phosphates. The persistent presence of the same type of structural unit across several different mineral groups is related to the high flexibility of the cyclic tetramers to adjust to different crystal chemistries by rotation and buckling about the heteropolyhedral corner-linkages, as illustrated in Fig. 1.
(a) and (b) Plan and side view of an ideal M 2(TO4)2Φ8 cluster with mmm point symmetry. (c) and (d) the M 2(TO4)2Φ8 cluster in phosphosiderite, showing rotation and buckling about the corner-linkages. Each cluster comprises two octahedra joined by two tetrahedra. All diagrams were prepared using ATOMS (Dowty, Reference Dowty2004).

Figure 1 Long description
The image A shows a plan view of an ideal (O)Φ cluster with point symmetry. It consists of two octahedra, represented by brown shapes, joined by two green tetrahedra. Blue spheres are located at the vertices of the octahedra and tetrahedra. The image B presents a side view of the same ideal cluster, maintaining the same structural elements and connections. The image C illustrates the (O)Φ cluster in phosphosiderite, showing rotation and buckling about the corner-linkages. The octahedra and tetrahedra are similarly colored, with blue spheres at the vertices. The image D provides a side view of the phosphosiderite cluster, again showing rotation and buckling, with the same color scheme and vertex spheres. Each cluster comprises two octahedra joined by two tetrahedra, demonstrating structural variations between the ideal and phosphosiderite forms.
We present here an extension of the study to secondary phosphate minerals. Moore (Reference Moore1982) has noted that by far the greatest number of hydrothermal and low-temperature (supergene) pegmatite phosphates derive from the triphylite, LiFe2+(PO4)-lithiophilite, LiMn2+(PO4) series of primary minerals, and so the focus will be on alteration of these minerals. The high reactivity of the primary minerals is related to the mobility of the Li+ cation and its ease of removal and replacement; a feature that finds application in synthetic equivalents as cathodes of rechargeable lithium batteries (Padhi et al., Reference Padhi, Nanjundaswamy and Goodenough1997; Yakubovich et al., Reference Yakubovich, Khasanova and Antipov2020).
Triphylite–lithiophilite parent structures
Triphylite, LiFe2+(PO4) and lithiophilite, LiMn2+(PO4), form a solid-solution series with a crystal structure derived from the olivine type, space group Pbnm, with a ≈ 4.7, b ≈ 10.3 and c ≈ 6.0 Å (Losey et al., Reference Losey, Rakovan, Francis and Dyar2004; Lyalina et al., Reference Lyalina, Selivanovai and Hatert2023). The minerals undergo high temperature leaching of lithium and oxidation of Fe2+ and Mn2+ according to the Quensel-Mason series of reactions (Quensel, Reference Quensel1937; Mason, Reference Mason1941) to form heterosite, Fe3+(PO4) – purpurite, Mn3+(PO4) products, which retain the same structure as the parents.
The crystal structure of triphylite/lithiophilite is shown in Fig. 2. (100) layers contain corner-connected cyclic tetramers that form 2-octahedra-wide ribbons along [001]. Adjacent ribbons are linked by octahedral edge-sharing with Li-centred octahedra in the triphylite–lithiophilite minerals, whereas there is no intralayer connection between the ribbons in heterosite–purpurite, where the Li sites are vacant. As outlined in the Introduction, such 2-octahedra-wide ribbons of corner-connected cyclic tetramers are common to several major families of primary phosphate minerals. However, these ribbons do not survive in any alteration products of the primary phosphates. Instead, sheets of trans corner-connected 7 Å octahedral chains, interconnected via cyclic tetramers, are a common structural motif in the alteration products, particularly for those derived from triphylite. In searching for a possible explanation, we re-examined the crystal structure for triphylite–lithiophilite and found that one-octahedra-wide (010) slices of the structure comprise intersecting trans corner-connected octahedral chains oriented along [101] and 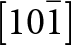 $\left[ {10\bar 1} \right]$, shown in Fig. 3.
$\left[ {10\bar 1} \right]$, shown in Fig. 3.
(100) layer of the structure of triphylite–lithiophilite. Brown circles correspond to octahedral sites occupied by Li. These sites are vacant in heterosite/purpurite and alternately filled and empty in sarcopside.

Figure 2 Long description
The diagram illustrates the crystal structure of triphylite–lithiophilite, featuring octahedral sites represented by brown circles. These sites are arranged in a pattern forming corner-connected cyclic tetramers. The tetramers create two-octahedra-wide ribbons that extend along the direction labeled as [001]. Adjacent ribbons are linked by octahedral edge-sharing, with Li-centered octahedra depicted in brown. The structure is oriented with axes labeled [010] and [001], indicating the spatial arrangement. Green triangles are interspersed between the brown octahedra, contributing to the overall connectivity of the structure. The diagram includes a rectangular outline highlighting a specific section of the structure, emphasizing the arrangement of the octahedra and their connections.
(010) slice through the triphylite structure, showing [101] and  $\left[ {10\bar 1} \right]$ chains of trans-corner-connected octahedra. PO4 tetrahedra (green) share both edges and corners with the octahedra.
$\left[ {10\bar 1} \right]$ chains of trans-corner-connected octahedra. PO4 tetrahedra (green) share both edges and corners with the octahedra.

Figure 3 Long description
The diagram illustrates the crystal structure of triphylite, featuring a network of corner-connected octahedra and PO tetrahedra. The octahedra are arranged in chains oriented along the directions labeled as [101] and [10 dash 1]. The PO tetrahedra, depicted in green, share both edges and corners with the octahedra, forming a complex interconnected pattern. The structure is further defined by the presence of O1, O2 and O3 labels, indicating specific oxygen positions within the arrangement. The diagram includes directional arrows pointing towards [101], [10 dash 1], [001] and [100], providing orientation within the crystal lattice. The overall layout showcases the intricate connectivity and spatial arrangement of the octahedra and tetrahedra within the triphylite mineral structure.
The 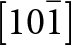 $\left[ {10\bar 1} \right]$ octahedral chains lie in (111) and
$\left[ {10\bar 1} \right]$ octahedral chains lie in (111) and 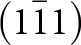 $\left( {1\bar 11} \right)$ planes while the [101] chains are in
$\left( {1\bar 11} \right)$ planes while the [101] chains are in 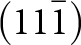 $\left( {11\bar 1} \right)$ and
$\left( {11\bar 1} \right)$ and 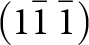 $\left( {1\bar 1\,\bar 1} \right)$ planes. The chain periodicity is 7.5 Å. A (111) slice of the structure is shown in Fig. 4. It shows that the octahedral chains are interconnected via pairs of corner-connected PO4 tetrahedra, forming cyclic tetramers. The resulting
$\left( {1\bar 1\,\bar 1} \right)$ planes. The chain periodicity is 7.5 Å. A (111) slice of the structure is shown in Fig. 4. It shows that the octahedral chains are interconnected via pairs of corner-connected PO4 tetrahedra, forming cyclic tetramers. The resulting 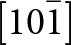 $\left[ {10\bar 1} \right]$ heteropolyhedral ribbons are segments of laueite-type sheets as described by Krivovichev (Reference Krivovichev2004). The 6-polyhedron cluster shown within the red ellipse in Fig. 4 is the smallest repeating segment of the ribbon, comprising a cyclic tetramer plus additional two octahedra forming short segments of the 7 Å chains. As will be shown in the following sections, this cluster is common to virtually all secondary phosphate minerals derived from triphylite–lithiophilite under oxidizing conditions.
$\left[ {10\bar 1} \right]$ heteropolyhedral ribbons are segments of laueite-type sheets as described by Krivovichev (Reference Krivovichev2004). The 6-polyhedron cluster shown within the red ellipse in Fig. 4 is the smallest repeating segment of the ribbon, comprising a cyclic tetramer plus additional two octahedra forming short segments of the 7 Å chains. As will be shown in the following sections, this cluster is common to virtually all secondary phosphate minerals derived from triphylite–lithiophilite under oxidizing conditions.
(111) slice through the triphylite/lithiophilite structure showing ribbons of laueite-like structure.

Figure 4 Long description
The diagram illustrates a structural representation of triphylite/lithiophilite, highlighting interconnected octahedral chains and PO4 tetrahedra. On the left side, the diagram shows octahedral chains arranged vertically, with alternating yellow and green octahedra. These chains are connected by pairs of corner-connected PO4 tetrahedra, forming cyclic tetramers. The tetrahedra are depicted in a lighter shade, contrasting with the octahedra. On the right side, a red ellipse encircles a specific cluster within the structure, indicating the smallest repeating segment of the ribbon. This cluster comprises a cyclic tetramer plus additional two octahedra, forming short segments of the chains. The orientation of the structure is indicated by the axes labeled [100], [010] and [001] on the left side of the diagram, providing a reference for the spatial arrangement of the chains and tetrahedra.
Hydrothermal products tavorite, barbosalite and lipscombite
Studies on pegmatites from numerous localities report tavorite, LiFe3+(PO4)(OH), and barbosalite, Fe2+[Fe3+(PO4)(OH)]2, as early-stage hydrothermal alteration products derived from triphylite–heterosite. Examples are the Tip Top pegmatite, South Dakota, USA (Campbell and Roberts, Reference Campbell and Roberts1986), the Sapucaia and Jocao pegmatites, Brazil (Lindberg and Pecora, Reference Lindberg and Pecora1955; Baijot et al., Reference Baijot, Hatert and Dal Bo2014), the Angarf-Sud pegmatite, Morocco (Fransolet et al., Reference Fransolet, Abraham and Speetjens1985), the Clementine II pegmatite, Namibia (Keller and von Knorring, Reference Keller and von Knorring1989) and the Hagendorf Süd pegmatite, Bavaria (Mücke, Reference Mücke1981). Neither of the minerals contain structural water and they form under oxidizing conditions at temperatures above 250°C. At the Sapucaia pegmatite, massive unaltered triphylite is separated from massive heterosite by a porous region of altered triphylite. Tavorite and barbosalite are found predominantly in a zone between the heterosite and porous triphylite (Lindberg and Pecora, Reference Lindberg and Pecora1955).
Tavorite and related minerals
Tavorite is a member of the amblygonite group with a triclinic crystal structure (Marx et al., Reference Marx, Croguennec, Carlier, Wattiaux, Le Cras, Suard and Delmas2010) and barbosalite is a member of the lazulite group with a monoclinic structure (Redhammer et al., Reference Redhammer, Tippelt, Roth, Lottermoser and Amthauer2000). The unit-cell parameters and space groups are given in Table 1. Moore (Reference Moore1982) has noted that the amblygonite structure is the parent structure type for the 7 Å octahedral corner-sharing structures and the lazulite structure forms the basis of the 5 Å fibre-axis structures in the so-called h-cluster minerals, containing linear trimers of face-sharing octahedra (Moore Reference Moore1970a). Neither of these descriptors, however, take account of a common structural component in both mineral types – kröhnkite-like chains of cyclic tetramers.
Hydrothermal/supergene alteration products of triphylite/lithiophilite listing end-member formulae, space group, unit-cell parameters (Å, °) and the calculated density

Table 1 Long description
The table lists phosphate minerals related to triphylite and lithiophilite and reports each mineral’s space group, unit-cell edge lengths a, b, and c, unit-cell angles alpha, beta, and gamma, and calculated density in grams per cubic centimeter, with literature references. Two parent minerals are included, followed by hydrothermal alteration products and supergene alteration products. Parent densities are about 3.62 for triphylite and 3.46 for lithiophilite. Hydrothermal products mostly cluster near the upper end of the density range, around 3.23 to 3.68, with heterosite and lipscombite near 3.68 and kidwellite near 3.23. Supergene products are generally lighter, about 2.49 to 3.64, with stewartite the lowest near 2.49 and kryzhanovskite the highest near 3.64. Several minerals list angles on a separate line, indicating non-right unit-cell angles for those structures. Comparisons should be made cautiously because densities are calculated and values come from different sources and crystal systems.
* References: [1] Losey et al. (Reference Losey, Rakovan, Francis and Dyar2004); [2] Eventoff et al. (Reference Eventoff, Martin and Peacor1972); [3] Marx et al. (Reference Marx, Croguennec, Carlier, Wattiaux, Le Cras, Suard and Delmas2010); [4] Redhammer et al. (Reference Redhammer, Tippelt, Roth, Lottermoser and Amthauer2000); [5] Vencato et al. (Reference Vencato, Mattievich and Mascarenhas1989); [6] Grey et al. (Reference Grey, Hochleitner, Kampf, Boer, MacRae, Cashion, Rewitzer and Mumme2023); [7] Grey et al. (Reference Grey, Williams, Kampf, Cashion, Gozukara, MacRae and Keck2018); [8] Kampf et al. (Reference Kampf, Colombo and del Tanago2010); [9] Keck et al. (Reference Keck, Grey, MacRae, Boer, Hochleitner, Rewitzer, Mumme, Glenn and Davidson2022); [10] Kolitsch (Reference Kolitsch2004); [11] Moore (Reference Moore1965); [12] Grey et al. (Reference Grey, Keck, Gable, Mumme, Hochleitner and Glenn2025); [13] Baur (Reference Baur1969); [14] Fanfani et al. (Reference Fanfani, Tomassini, Zanazzi and Zanzari1978); [15] Vrtiska et al. (Reference Vrtiska, Tvrdy, Plasil, Sejkora, Skoda, Chukanov, Massanek, Filip, Dolnicek and Veselovsky2022); [16] Moore and Araki (Reference Moore and Araki1976); [17] Kampf and Moore (Reference Kampf and Moore1976); [18] Moore et al. (Reference Moore, Kampf and Irving1974).
A (100) slice through the tavorite structure is shown in Fig. 5. The M1- and M2-centred octahedra form trans-connected chains parallel to [010], and the chains are interconnected via pairs of PO4 tetrahedra, that link into kröhnkite-type chains along [001], thus forming (100) layers. In Fig. 5 the 6-polyhedron cluster shown within the red ellipse is identical to a segment of triphylite structure shown in Fig. 4, lending support to this cluster being a possible nucleating agent for the formation of triphylite derivative minerals.
(100) slice through the tavorite structure.

Figure 5 Long description
The diagram illustrates a (100) slice through the tavorite structure, featuring M1 and M2-centered octahedra forming trans-connected chains parallel to the direction labeled [010]. These chains are interconnected via pairs of PO4 tetrahedra, which link into kröhnkite-type chains along the direction labeled [001], thus forming (100) layers. The M1 octahedra are colored yellow, while the M2 octahedra are colored orange. The PO4 tetrahedra are depicted in green. Lithium atoms, represented as purple spheres, are interspersed within the structure. A red ellipse highlights a 6-polyhedron cluster, which is identical to a segment of the triphylite structure. This cluster is suggested as a possible nucleating agent for the formation of triphylite derivative minerals. The diagram includes a directional guide with labels [100], [010] and [001] to indicate orientation.
The kröhnkite chains in the (100) layers in Fig. 5 involve the Fe2-centred octahedra in tavorite. Equivalent layers are present parallel to (001), in which the kröhnkite chains involve the Fe1-centred octahedra. These layers are topologically identical to laueite sheets in the laueite supergroup minerals as described by Krivovichev (Reference Krivovichev2004). In laueite the sheets are interconnected via corner-sharing of Mn(H2O)4O2 octahedra with the unshared anions of the PO4 tetrahedra on either side of the sheet, whereas in tavorite the laueite-type layers are fused into a 3D structure by corner-sharing of the tetrahedra in one layer with octahedra in the adjacent layers. The tavorite structure involves the intermeshing of (100) and (001) laueite-type layers, at an angle between the two planes of 75.4°. This is illustrated by the [010] projection of the structure in Fig. 6.
[010] projection of the tavorite structure.
![A diagram of a polyhedral model illustrating a crystal structure with octahedra and tetrahedra, showing direction [100] and [001]. See long description.](https://static.cambridge.org/binary/version/id/urn:cambridge.org:id:binary:20260702065928330-0209:S0026461X26101972:S0026461X26101972_fig6.png?pub-status=live)
Figure 6 Long description
The diagram illustrates a polyhedral model of a crystal structure, featuring interconnected octahedra and tetrahedra. The octahedra are depicted in yellow, while the tetrahedra are shown in green. These polyhedra form a corner-sharing network, where each octahedron shares corners with adjacent tetrahedra, creating a repeating lattice framework. Small nodes mark the vertices of the polyhedra, emphasizing the connectivity between them. The structure is oriented with a direction marker indicating [100] horizontally and [001] vertically, providing spatial orientation within the crystal lattice. This model visually represents the arrangement and connectivity of the polyhedra within the crystal structure, highlighting the geometric and spatial relationships between the components.
The flexibility of the cyclic tetramers connecting the 7 Å chains allows large rotations about the 7 Å axes to change the shape of the cavities to incorporate cations of different sizes, as described by Grey et al. (Reference Grey, Lanyon and Stranger1996) for the case of various hydrothermally prepared iron titanium oxysulfates, (Fe,Ti)SO4(O,OH).
The 7 Å chain rotations are compared in Fig. 7 for amblygonite, Li[Al(PO4)F], and lacroixite, Na[Al(PO4)F]. Amblygonite is triclinic, 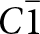 $C\bar 1$, with a = 6.644, b = 7.744, c = 6.910 Å, α = 90.35, β = 117.33 and γ = 91.01° (Groat et al., Reference Groat, Raudsepp, Ercit, Sherriff and Hartman1990) while lacroixite is monoclinic, C2/c, with a = 6.414, b = 8.207, c = 6.885 Å and β = 115.47° (Lahti and Pajunen, Reference Lahti and Pajunen1985). Successive octahedra along the 7 Å chains are rotated in opposite directions in both structures. The combined rotation angle increases from 23° for amblygonite to 36° for lacroixite to provide a more suitable coordination environment for the larger Na+. In contrast to tavorite, where the laueite-type layers are parallel to (100) and (001), Fig. 6, the layers in lacroixite and amblygonite are parallel to (110) and
$C\bar 1$, with a = 6.644, b = 7.744, c = 6.910 Å, α = 90.35, β = 117.33 and γ = 91.01° (Groat et al., Reference Groat, Raudsepp, Ercit, Sherriff and Hartman1990) while lacroixite is monoclinic, C2/c, with a = 6.414, b = 8.207, c = 6.885 Å and β = 115.47° (Lahti and Pajunen, Reference Lahti and Pajunen1985). Successive octahedra along the 7 Å chains are rotated in opposite directions in both structures. The combined rotation angle increases from 23° for amblygonite to 36° for lacroixite to provide a more suitable coordination environment for the larger Na+. In contrast to tavorite, where the laueite-type layers are parallel to (100) and (001), Fig. 6, the layers in lacroixite and amblygonite are parallel to (110) and 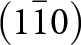 $\left( {1\bar 10} \right)$, with the kröhnkite chains orientated along
$\left( {1\bar 10} \right)$, with the kröhnkite chains orientated along 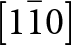 $\left[ {1\bar 10} \right]$ and [110], respectively, as shown in Fig. 7.
$\left[ {1\bar 10} \right]$ and [110], respectively, as shown in Fig. 7.
[001] projections of structures of (a) amblygonite and (b) lacroixite. Small pink circles are Li and large pink circles are Na.

Figure 7 Long description
The image consists of two projections labeled as (a) and (b). Image (a) shows the structure of amblygonite, featuring yellow octahedra connected in a chain with green triangular elements. Small pink circles represent lithium ions and blue circles are positioned at the vertices of the octahedra. A red line indicates a rotation angle of twenty-three degrees. Image (b) depicts the structure of lacroixite, with similar yellow octahedra and green triangular elements. Large pink circles represent sodium ions and blue circles are again at the vertices. A red line shows a rotation angle of thirty-six degrees. Between the two images, an arrow indicates the orientation directions labeled as left bracket zero one zero right bracket and left bracket one zero zero right bracket.
The flexibility and stability of these framework structures built from 7 Å chains and interconnecting kröhnkite chains is illustrated by the diverse range of chemistries of the minerals that adopt the structures that include tilasite, Ca[Mg(AsO4)F, titanite, Ca[Ti(SiO4)O] and kieserite □[Mg(SO4)H2O] and related triclinic and monoclinic minerals as listed by Speer and Gibbs (Reference Speer and Gibbs1976) and Hawthorne et al. (Reference Hawthorne, Groat and Raudsepp1987).
Barbosalite
The  $\left( {11\bar 1} \right)$ layer of the barbosalite structure is shown in Fig. 8. A comparison of the polyhedral connectivity with that for tavorite in Fig. 5 shows that the laueite-type layers in both minerals are topologically identical. The 7 Å chains of trivalent M2-centred octahedra along [101] in barbosalite are interconnected through cyclic tetramers that join into kröhnkite-like chains along
$\left( {11\bar 1} \right)$ layer of the barbosalite structure is shown in Fig. 8. A comparison of the polyhedral connectivity with that for tavorite in Fig. 5 shows that the laueite-type layers in both minerals are topologically identical. The 7 Å chains of trivalent M2-centred octahedra along [101] in barbosalite are interconnected through cyclic tetramers that join into kröhnkite-like chains along  $\left[ {1\bar 10} \right]$. As in tavorite, equivalent laueite-type layers also occur parallel to
$\left[ {1\bar 10} \right]$. As in tavorite, equivalent laueite-type layers also occur parallel to  $\left( {\bar 111} \right)$, approximately orthogonal to those shown in Fig. 8, in which the kröhnkite-like chains are orientated along [110]. The divalent Fe2+ cations in barbosalite occupy M1 sites at the centres of half of the 6-sided cavities, where they are octahedrally coordinated as the central atom of h clusters (Moore, Reference Moore1970a). With the M1 sites occupied, the layer shown in Fig. 8 can be described as a ‘stuffed laueite sheet’. In contrast to the Fe at M1 sites in barbosalite, the Li atoms in tavorite are strongly displaced from the centres of the cavities as shown in Fig. 5 to take up 5-coordination.
$\left( {\bar 111} \right)$, approximately orthogonal to those shown in Fig. 8, in which the kröhnkite-like chains are orientated along [110]. The divalent Fe2+ cations in barbosalite occupy M1 sites at the centres of half of the 6-sided cavities, where they are octahedrally coordinated as the central atom of h clusters (Moore, Reference Moore1970a). With the M1 sites occupied, the layer shown in Fig. 8 can be described as a ‘stuffed laueite sheet’. In contrast to the Fe at M1 sites in barbosalite, the Li atoms in tavorite are strongly displaced from the centres of the cavities as shown in Fig. 5 to take up 5-coordination.
The  $\left( {11\bar 1} \right)$ layer through the structure of barbosalite.
$\left( {11\bar 1} \right)$ layer through the structure of barbosalite.

Figure 8 Long description
The diagram illustrates the barbosalite structure, highlighting the arrangement of polyhedral units. At the top, labeled M1, are red spheres representing divalent Fe2+ cations occupying M1 sites. Below them, green and yellow polyhedra are interconnected, forming chains. The green polyhedra are labeled M2, indicating trivalent M2-centered octahedra. These chains run along the direction labeled as [101] and [1-10], showing the orientation of the kröhnkite-like chains. The polyhedra are connected through cyclic tetramers, forming a network. Labels a, b and c are present, indicating specific points or connections within the structure. Blue spheres are located at the vertices of the polyhedra, representing coordination points. The diagram visually represents the connectivity and orientation of the layers within the barbosalite structure.
The barbosalite unit cell can be transformed using the matrix  $(1\bar 10$ 110
$(1\bar 10$ 110  $\bar 10\bar 1$) to give a C-centred triclinic cell with a = b = 10.48, c = 7.55 Å, α = β = 110.61 and γ = 91.26°. A comparison with the tavorite cell in Table 1 shows very similar parameters, but with a doubling of the two 5 Å axes due to ordering of divalent cations and vacancies at the M1 site.
$\bar 10\bar 1$) to give a C-centred triclinic cell with a = b = 10.48, c = 7.55 Å, α = β = 110.61 and γ = 91.26°. A comparison with the tavorite cell in Table 1 shows very similar parameters, but with a doubling of the two 5 Å axes due to ordering of divalent cations and vacancies at the M1 site.
Lipscombite
Baijot et al. (Reference Baijot, Hatert and Dal Bo2014) describe the replacement of triphylite by lipscombite, Fe2+[Fe3+(PO4)(OH)]2, a dimorph of barbosalite. In contrast with the monoclinic and triclinic minerals described above, its symmetry is tetragonal, P43212, with a = 7.310 and c = 13.212 Å (Vencato et al., Reference Vencato, Mattievich and Mascarenhas1989). Its structure is usually described in terms of h-clusters that form rods oriented along [110] and along  $\left[ {1\bar 10} \right]$ in alternate (001) planes (Moore, Reference Moore1970a; Vencato et al., Reference Vencato, Mattievich and Mascarenhas1989). The h-clusters involve trimers of face-shared octahedra, M2–M1–M2, alternating with empty sites, where M1 and M2 correspond to the formula M12+[M23+(PO4)(OH)]2.
$\left[ {1\bar 10} \right]$ in alternate (001) planes (Moore, Reference Moore1970a; Vencato et al., Reference Vencato, Mattievich and Mascarenhas1989). The h-clusters involve trimers of face-shared octahedra, M2–M1–M2, alternating with empty sites, where M1 and M2 correspond to the formula M12+[M23+(PO4)(OH)]2.
Despite the different symmetry and unit-cell geometry, the crystal structure of lipscombite is closely related to those of tavorite and barbosalite, in being composed of two families of stuffed laueite-related layers. These are mutually orthogonal in lipscombite, but only approximately orthogonal in the other minerals. One family of layers in lipscombite, parallel to (110), is shown in Fig. 9. It has  $\left[ {1\bar 10} \right]$ kröhnkite-type chains located at z = 0, ½, that are interconnected via corner-connected M2-octahedra at z = ¼, ¾. These latter octahedra form kröhnkite-type chains along [110], consistent with the tetragonal symmetry.
$\left[ {1\bar 10} \right]$ kröhnkite-type chains located at z = 0, ½, that are interconnected via corner-connected M2-octahedra at z = ¼, ¾. These latter octahedra form kröhnkite-type chains along [110], consistent with the tetragonal symmetry.
(110) heteropolyhedral layer in lipscombite.

Figure 9 Long description
The diagram illustrates the crystal structure of lipscombite, featuring a network of interconnected polyhedra. The structure includes M1 and M2 octahedra, with M2 labeled at the top center and M1 at the right center. The polyhedra are arranged in a repeating pattern, forming kröhnkite-type chains. These chains are oriented along the direction labeled as left bracket one one zero right bracket. The diagram also includes directional arrows indicating the crystallographic axes: left bracket zero zero one right bracket pointing upwards, left bracket one zero zero right bracket pointing to the right and left bracket zero one zero right bracket pointing downwards. The structure is composed of orange and green polyhedra, with small blue spheres at the vertices, representing the atomic connections within the crystal lattice.
The [001] chains of M2-centred octahedra in lipscombite represent an interesting extension of the ligand isomerism in corner-connected octahedral chains discussed by Moore (Reference Moore1970b). Moore’s study was restricted to two-octahedra chain periodicities of ∼7 Å, whereas lipscombite has a four-octahedra repeat of 13.2 Å = 2 × 6.6 Å. The chains are located at the 43 screw axes at 0, ½, z and ½, 0, z, and the screw axis operation results in the cis-pairs of anions of each octahedron that are involved in the intrachain tetrahedral bridging being rotated by 90° in each successive octahedron along [001], resulting in a spiral chain. The 90° rotations of successive octahedra result in them being more strongly tilted than in the case of the 7 Å chain structures, resulting in a considerable shortening of the chain periodicity. The involvement of cis-pairs of anions of each octahedron in intrachain tetrahedral bridging in lipscombite differs from the trans-pairs of anions forming intrachain bridges in tavorite and barbosalite, and so the layers, as shown in Fig. 9, are a topological modification of laueite-type layers.
Rockbridgeite group and related 5 Å fibrous minerals
Rockbridgeite is ubiquitous as a triphylite alteration product, considered by Moore (Reference Moore1973) to be second only in abundance to the parent triphylite. The rockbridgeite-group minerals have the structural formula M1(M2)2(M30.5□0.5)4(PO4)3X 5, where □ = vacancy, X = OH, H2O, M1 and M3 = Fe3+. The different group members are determined by the M2 site dominant cations, ferrorockbridgeite (Fe2+), manganrockbridgeite (Mn2+), plimerite (Zn), rockbridgeite (Fe2+0.5Fe3+0.5), frondelite (Mn2+0.5Fe3+0.5) and ferrirockbridgeite (Fe3+0.67□0.33) (Grey et al., Reference Grey, Kampf, Keck, Cashion, MacRae, Gozukara, Peterson and Shanks2019). The minerals with heterovalent occupation at M2 have X = OH, whereas the other members have (OH)4(H2O). The low or absent water content is consistent with the minerals being late-stage hydrothermal alteration products, with Moore (Reference Moore1973) locating their formation temperature at either side of 200°C.
All rockbridgeite group members except manganrockbridgeite have orthorhombic symmetry, Bbmm, with a ≈ 13.9, b ≈ 17.0 and c ≈ 5.2 Å. Moore (Reference Moore1970a) described rockbridgeite as a member of the h-cluster family of minerals, containing linear face-shared trimers (M2M1M2) of octahedra. The clusters share edges, giving chains along [010]. The chains are interconnected along [001] via corner-connected PO4 tetrahedra in (100) planes and via corner-connection with M3-centred octahedral dimers along [100]. The [001] chains of M3-centred octahedra are disordered with half-occupancy of the M3 sites, as first established by Moore (Reference Moore1970a) for rockbridgeite. Manganrockbridgeite has monoclinic symmetry, P21/m, with a = 5.20, b = 16.94, c = 7.45 Å and β = 110.17° (Grey et al., Reference Grey, Hochleitner, Kampf, Boer, MacRae, Cashion, Rewitzer and Mumme2023), with ordering of M3 cations and vacancies along the 5.2 Å axis direction. In the following discussion we will consider first the manganrockbridgeite crystal structure, with ordering of M3 cations and vacancies.
A different perspective on the crystal structure of manganrockbridgeite, that links it to the triphylite structure, is made evident by viewing an (010) slice through the structure as illustrated in Fig. 10a. This shows that the slice corresponds to a laueite sheet, with trans-corner-connected M2 and M3-centred octahedral chains along [101] and with kröhnkite-type chains of cyclic tetramers involving M2-centred octahedra along [100]. The layer shown in Fig. 10a is topologically identical to (100) layers in tavorite (Fig. 5) and  $\left( {11\bar 1} \right)$ layers in barbosalite (Fig. 8).
$\left( {11\bar 1} \right)$ layers in barbosalite (Fig. 8).
Laueite-related (010) heteropolyhedral layers in (a) manganrockbridgeite and (b) rockbridgeite, Pnma model for ordering at M3 sites.

Figure 10 Long description
The image consists of two sub-images labeled (a) and (b). In sub-image (a), the structure shows a series of interconnected polyhedra forming a layer. The polyhedra are arranged in chains labeled as [101] chains, with M2 and M3 sites marked within the structure. The polyhedra are connected at corners, forming a repeating pattern. The axes are labeled with 'a' and 'c'. In sub-image (b), a similar arrangement is shown, but with a twin plane indicated between the two structures. The M2 and M3 sites are again labeled and the axes are marked as 'a' and 'c'. The twin plane is shown as a dividing line between the two mirrored structures, illustrating the symmetry and connectivity of the layers.
The manganrockbridgeite structure is viewed in projection along the 5.2 Å axis in Fig. 11a. This shows that pairs of laueite-type layers are interconnected along [010] via PO4 tetrahedra that form cyclic tetramers with the M3-centred octahedra. These are orthogonal to the [100] kröhnkite chains involving the M2-centred octahedra, so the double layer represents a small slab of barbosalite-type structure. These slabs are related by the mirror operation at y = ¼ and ¾. The manganrockbridgeite crystal structure can thus be described as unit-cell twinning of barbosalite-type structure.
Projection along the 5.2 A axis of the structures of: (a) manganrockbridgeite; (b) Pnma model for rockbridgeite; (c) gayite; and (d) kenngottite.

Figure 11 Long description
This diagram presents crystallographic projections of four mineral structures, labeled a through d. Sub-image a shows the manganrockbridgeite structure, highlighting a barbosalite slab and a mirror plane. The structure includes M1, M2 and M3 sites, with red circles representing specific atomic positions. Sub-image b illustrates a model for rockbridgeite, featuring interconnected layers with labeled points a, b and c and red circles indicating atomic sites. Sub-image c depicts the gayite structure, with M2, M3, M4 and Na sites and points a, b and c. Purple circles denote specific atomic configurations. Sub-image d shows kenngottite, characterized by triple layers of twinned-laueite, with points a, b and c and red circles marking atomic sites. Each sub-image provides a view along the 5.2 angstrom axis, demonstrating structural differences such as the presence of a mirror plane in a, the model nature of b, the Na site in c and the triple-layer twinning in d. No numeric scales or units are shown, focusing on the spatial arrangement and structural relationships between the minerals.
Although the orthorhombic rockbridgeite group members have long-range disorder at the M3 sites, there is evidence for short-range ordering in the form of diffuse streaks in diffraction patterns in positions that violate the extinction conditions for the Bbmm space group (Röska et al., Reference Röska, Park, Behal, Hess, Günther, Benka, Pfleiderer, Hoelzel and Kimura2018). A model for the local order, that is consistent with electron diffraction patterns, has been established in space group Pnma (Grey et al., Reference Grey, Williams, Kampf, Cashion, Gozukara, MacRae and Keck2018). An (010) slice of the ordered Pnma structure is shown in Fig. 10b, where it can be compared with the corresponding (010) section in manganrockbridgeite, shown in Fig. 10a. The Pnma structure has the same laueite-like arrangement of kröhnkite chains along the 5.2 Å axis connecting trans-corner-connected octahedral chains. However, the octahedral chains have a zig-zag form with a doubling of the periodicity to 13.8 Å relative to the 7 Å chains in manganrockbridgeite. The zig-zag chains result from unit-cell-scale twinning of the 7 Å chains at (100) twin planes passing through the M2-centred octahedra. The twin operation is a two-fold rotation about the 5 Å c axis, as shown in Fig. 10b. We refer to layers as shown in Fig. 10b as twinned laueite-type layers.
The M2-centred octahedra, which participate in the [001] kröhnkite chains, have cis pairs of anions involved in intrachain connections to PO4 tetrahedra while the M3-centred octahedra have trans pairs of anions forming intrachain links. For comparison both the M2- and M3-centred octahedra in the 7 Å chains in manganrockbridgeite have trans pairs of anions participating in intrachain connections. As in manganrockbridgeite, pairs of laueite-like sheets are interconnected along [010] via cyclic tetramers to give slabs of barbosalite-like structure, and the slabs are related by mirror planes at y = ¼ and ¾, to complete the 3D framework, shown in Fig. 11b.
Numerous fibrous iron phosphate minerals have, in common with orthorhombic rockbridgeite, unit cells with two of the axes being ∼5.2 and 13.8 Å. All such minerals contain twinned laueite-like sheets identical to that shown in Fig. 10b, comprising intersecting kröhnkite chains along the 5.2 Å axis and zig-zag trans-corner-connected octahedral chains along the 13.8 Å axis. These include the dufrenite-group minerals, with general formula XM1M2(M3)2(M4)2(PO4)4(OH)6·2H2O, X = □, Na+, Ca2+, and with monoclinic unit cells, having a ≈ 25.8, b ≈ 5.15, c ≈ 13.8 Å and β ≈ 111.5° (Elliott and Kampf, Reference Elliott and Kampf2024). An [010] projection of the structure of the dufrenite-group mineral gayite (Kampf et al., Reference Kampf, Colombo and del Tanago2010) is shown in Fig. 11c. This shows that ∼9 Å wide (100) slabs of barbosalite-like structure as in rockbridgeite are interconnected along [100] by corner-sharing with M1-centred octahedra.
The fibrous mineral kenngottite, Mn2+3Fe3+4(PO4)4(OH)6(H2O)2, is commonly found coating nodules of rockbridgeite-group minerals at Hagendorf Süd (Keck et al., Reference Keck, Grey, MacRae, Boer, Hochleitner, Rewitzer, Mumme, Glenn and Davidson2022). It has monoclinic symmetry, P2/a, with a ≈ 13.9, b ≈ 5.2, c ≈ 12.2 Å and β ≈ 98.8°. Its structure has (001) sheets of laueite-type, as shown for rockbridgeite in Fig. 10b. The sheets are interconnected along [001] via cyclic tetramers to form ∼9 Å wide barbosalite-like slabs, as for rockbridgeite and the dufrenite-group minerals. The structure is completed by the insertion of a (001) laueite-type sheet between adjacent barbosalite slabs as shown in Fig. 11d.
The fibrous iron phosphate minerals kidwellite and ‘laubmannite’ also have pairs of axes 13.9 and 5.2 Å (Kolitsch, Reference Kolitsch2004). On the basis that this pair of axes is common to minerals containing h-clusters, Moore assumed that ‘laubmannite’ and kidwellite (labelled ‘Mineral A’ by Moore, Reference Moore1970a) both contained the linear trimers of face-shared octahedra. The structure refinements of the minerals by Kolitsch (Reference Kolitsch2004), however, showed that h-clusters were not present in either mineral. Instead, one can identify sheets of twinned laueite type, as shown for rockbridgeite in Fig. 10b, in the published structures of both minerals, consistent with the pairs of axes 13.9 and 5.2 Å.
Low-temperature secondary alteration minerals
At temperatures below ∼250°C, where coordinated water and additional hydroxyls can be structurally accommodated, the alteration of triphylite/lithiophilite and their hydrothermal products produces a plethora of secondary minerals. The number and variety of species formed is increased by the incorporation of additional cations from the hydrothermal solutions. For example at Hagendorf Süd (Mücke, Reference Mücke1981; Birch et al., Reference Birch, Grey, Keck, Mills and Mumme2018) and at Palermo #1 pegmatites (Segeler et al., Reference Segeler, Ulrich, Kampf and Whitmore1981; Kampf et al., Reference Kampf, Falster, Simmons and Whitmore2013), Zn2+ in solution from the decomposition of sphalerite together with Ca2+ react with associated triphylite and its metasomatic products to form numerous hydrated secondary species including phosphophyllite, scholzite, nizamoffite, jungite, schoonerite-group minerals, jahnsite-group minerals, fairfieldite, whitlockite, steinmetzite, mitridatite and secondary apatite. Similarly, the incorporation of alkalis and Al3+ from solution leads to secondary species such as leucophosphite, whiteite-group minerals and crandallite. In general, the crystal structures of these secondary minerals containing additional cations have little in common with that of the parent triphylite/lithiophilite structures and so in this study, which focuses on crystal-chemical relationships, they will not be discussed further. Instead, we consider the secondary minerals that contain only the cations present in the parent minerals, Fe2+ and Mn3+ and their oxidized forms. Moore (Reference Moore1973) and Nizamoff (Reference Nizamoff2006) separate the low temperature alteration reactions of triphylite into two groups, involving oxidizing and non-oxidizing/reducing conditions. The minerals belonging to each group have structural features in common but with no strong structural links between the groups and so they are considered separately below.
Low temperature alteration under non-oxidizing conditions
The most common hydrated secondary minerals formed from triphylite/lithiophilite under non-oxidizing conditions are hureaulite, (Mn,Fe)2+5(PO4)2(PO3OH)2(H2O)4, ludlamite, (Fe,Mn)2+3(PO4)2(H2O)4, phosphoferrite, (Fe,Mn)2+3(PO4)2(H2O)3 and vivianite, Fe2+3(PO4)2(H2O)8 (Moore, Reference Moore1973; Nizamoff, Reference Nizamoff2006). Their unit-cell parameters and space groups are given in Table 2. The anhydrous mineral sarcopside, Fe2+3(PO4)2 (Moore, Reference Moore1972) is included in Table 2. Sarcopside and its Mn analogue zavalíaite occur most commonly as exsolution lamellae in triphylite and lithiophilite, respectively (Peacor, Reference Peacor1969; Hatert et al., Reference Hatert, Roda-Robles, de Parseval and Wouters2012) with ordered replacement of 2Li+ by M 2+ + □. However, sarcopside has been synthesised by hydrothermal treatment of vivianite above 250°C (Mattievich and Danon, Reference Mattievich and Danon1977) so it may also play a role as an intermediate phase in the alteration of triphylite.
Crystallographic parameters (Å, °), density and mean oxygen volume for hydrated secondary phosphates formed from triphylite/lithiophilite under reducing conditions

Table 2 Long description
The table lists seven phosphate minerals and compares their crystal system information (space group and unit cell lengths a, b, c and, where applicable, the beta angle) along with calculated density and mean oxygen volume. Densities range from 2.70 grams per cubic centimeter for vivianite to 3.95 for sarcopside, with triphylite also high at 3.62. Hydrated phases tend to be less dense than the anhydrous entries: ludlamite is 3.23, phosphoferrite 3.32, and vivianite 2.70, compared with sarcopside 3.95 and zavalíaite 3.84. Mean oxygen volume spans a narrow band from 18.1 cubic angstroms for triphylite to 19.2 for zavalíaite and vivianite, suggesting only modest variation across the group. Several minerals share the same space group type (notably sarcopside, zavalíaite, and ludlamite), but their beta angles differ, indicating structural differences despite similar symmetry labels. Some cell dimensions are presented as doubled values for certain axes, so direct comparisons of a, b, and c should be made cautiously. Values are compiled from multiple literature sources, so small differences may reflect differing measurement conditions or refinements rather than true mineral-to-mineral changes.
* References: [1] Losey et al. (Reference Losey, Rakovan, Francis and Dyar2004); [2] Moore (Reference Moore1972); [3] Hatert et al. (Reference Hatert, Roda-Robles, de Parseval and Wouters2012); [4] Ito and Mori (Reference Ito and Mori1951); [5] Moore and Araki (Reference Moore and Araki1976); [6] Moore and Araki (Reference Moore and Araki1973); [7] Mori and Ito (Reference Mori and Ito1950).
None of the minerals in Table 2 have structures containing 7 Å corner-connected octahedral chains that are common to the structures of the hydrothermal alteration minerals described above. Instead, the common structural feature is the presence of strings of edge-shared octahedra, ranging from octahedral dimers in vivianite, trimers in sarcopside and ludlamite, pentamers in hureaulite to stepped chains in phosphoferrite. The edge-shared octahedral strings lie in approximately close packed layers that have a stacking periodicity of ∼4.7 Å (sarcopside, ludlamite and vivianite) or ∼9.4 Å = 2× 4.7 Å (phosphoferrite and hureaulite). Views of the structures of sarcopside, ludlamite, phosphoferrite and hureaulite are shown edge-on to the close-packed layers in Fig. 12.
Edge-shared octahedral strings interconnected via PO4 tetrahedra in (a) sarcopside, (b) ludlamite, (c) phosphoferrite and (d) hureaulite.

Figure 12 Long description
The image consists of four sub-images labeled (a) to (d), each depicting edge-shared octahedral strings interconnected via PO tetrahedra. In sub-image (a), the structure of sarcopside is shown with octahedral trimers labeled M1 and M2, arranged in layers with a stacking periodicity of approximately four point seven angstroms. Sub-image (b) illustrates ludlamite, also featuring octahedral trimers labeled M1 and M2, similarly arranged in layers. Sub-image (c) presents phosphoferrite, where the octahedral strings are labeled M1 and M2, forming a more complex network. Finally, sub-image (d) depicts hureaulite, with octahedral pentamers labeled M1, M2 and M3, arranged in a stepped chain configuration. Each sub-image shows the structures edge-on to the close-packed layers, highlighting the periodicity and arrangement of the octahedral strings.
The sarcopside structure is a slight monoclinic distortion of the orthorhombic triphylite structure (Moore, Reference Moore1972). The structure contains linear trimers, M2–M1–M2 of edge-shared octahedra that are interconnected along [010] via pairs of PO4 tetrahedra (Fig. 12a). Each tetrahedron shares an edge and a corner with M1-centred octahedra above and below to give tetramers that extend as chains along [010]. The three-octahedra-wide [010] columns join with other columns along [102] by corner-sharing of the M2-centred octahedra to form  $\left( {\bar 201} \right)$ layers as shown in Fig. 12a.
$\left( {\bar 201} \right)$ layers as shown in Fig. 12a.
Ludlamite, with end-member composition Fe2+3(PO4)2(H2O)4, has a crystal structure (Ito and Mori, Reference Ito and Mori1951) that is closely related to that of sarcopside, as evident from the same space group and similar unit-cell parameters given in Table 2. The ludlamite structure comprises the same type of linear trimers of edge-shared octahedra, M2–M1–M2, as in sarcopside, formed into columns along [010] by linking with PO4 tetrahedra, and into  $\left( {\bar 201} \right)$ layers by corner-sharing of the M2-centred octahedra in adjacent columns, as shown in Fig. 12b. The major difference between the
$\left( {\bar 201} \right)$ layers by corner-sharing of the M2-centred octahedra in adjacent columns, as shown in Fig. 12b. The major difference between the 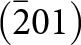 $\left( {\bar 201} \right)$ layers in the two minerals concerns the orientation of the PO4 tetrahedra. A 180° rotation about [010] of the tetrahedra in ludlamite, relative to sarcopside, changes the octahedral–tetrahedral linkage from edge-sharing to corner-sharing. As a result, in ludlamite the PO4 tetrahedra and the M1-centred octahedra form cyclic tetramers that extend as kröhnkite chains along the 4.7 Å axis direction. The same reorientation of the tetrahedra to give cyclic tetramers that extend into kröhnkite chains occurs in the other alteration products formed in non-oxidizing conditions, as shown in Fig. 12.
$\left( {\bar 201} \right)$ layers in the two minerals concerns the orientation of the PO4 tetrahedra. A 180° rotation about [010] of the tetrahedra in ludlamite, relative to sarcopside, changes the octahedral–tetrahedral linkage from edge-sharing to corner-sharing. As a result, in ludlamite the PO4 tetrahedra and the M1-centred octahedra form cyclic tetramers that extend as kröhnkite chains along the 4.7 Å axis direction. The same reorientation of the tetrahedra to give cyclic tetramers that extend into kröhnkite chains occurs in the other alteration products formed in non-oxidizing conditions, as shown in Fig. 12.
Phosphoferrite, (Fe2+,Mn2+)3(PO4)2(H2O)3, has the orthorhombic unit-cell parameters given in Table 2. It has a structure (Moore and Araki, Reference Moore and Araki1976) based on linear trimers of edge-shared octahedra, M2–M1–M2 as in ludlamite. However, in contrast to corner-linking of the trimers in ludlamite, the trimers in phosphoferrite are edge-connected to form stepped chains along [001]. The octahedral chains are interconnected along [100] by corner linkage with PO4 tetrahedra. As shown in Fig. 12c, cyclic tetramers involving an M1-centred and an M2-centred octahedron are linked in pairs to form short segments of kröhnkite chains along [100].
Hureaulite, Mn2+5(PO4)2(PO3OH)2(H2O)4, has a more complicated arrangement of edge-shared octahedral trimers, M2–M1–M2, with further edge-sharing to M3-centred octahedra at either end, giving Z-shaped pentamers along [100] (Moore and Araki, Reference Moore and Araki1973). The pentamers are interconnected along both [010] and [001] by corner-sharing between M2-centred and M3-centred octahedra. As for ludlamite and phosphoferrite, the octahedra in the M2–M1–M2 trimers are corner-connected to PO4 tetrahedra to form cyclic tetramers. In hureaulite, all three octahedra of the trimers are involved in forming kröhnkite chains along [001], as shown in Fig. 12d.
Vivianite, Fe2+3(PO4)2(H2O)8, is the most highly hydrated and lowest density secondary phosphate mineral formed under non-oxidising conditions from triphylite. Its crystallographic parameters are given in Table 2 (Mori and Ito, Reference Mori and Ito1950). Edge-shared M2-centred octahedral dimers corner-share with PO4 tetrahedra to form kröhnkite-like columns along [001]. The heteropolyhedral columns are interconnected along [100] into (010) layers by corner connection of the PO4 tetrahedra with M1(H2O)4O2 octahedra. The layers are held together by H-bonding. Vivianite is the highest known member (n = 8) of the homologous series of compounds Fe2+3(H2O)n(PO4)2 described by Moore and Araki (Reference Moore and Araki1976), that include phosphoferrite (n = 3) and ludlamite (n = 4). The ubiquitous presence of cyclic tetramers, extended into kröhnkite-like chain segments, is in evidence in all minerals in Table 2 having a unit-cell parameter of n × 4.7 Å, n = 1,2.
An interesting crystallochemical aspect of the structures of hureaulite and phosphoferrite is the co-existence of both M 2(TO4)2Φ7 and M 2(TO4)2Φ8 clusters. The two minerals have octahedral corner-sharing between adjacent strings of edge-shared octahedra, and the pairs of corner-shared octahedra are bridged by pairs of tetrahedra to form tetrameric M 2(TO4)2Φ7 clusters. Hawthorne (Reference Hawthorne1979) has noted that the unpolymerized cluster occurs in the mineral morinite, and that polymerized morinite-type clusters occur in several phosphate minerals including hureaulite, phosphoferrite and whitmoreite.
Low temperature alteration under oxidizing conditions
The most commonly reported hydrated phosphate minerals derived from triphylite/lithiophilite and their metasomatic alteration phases under oxidizing conditions include laueite, pseudolaueite, stewartite, strunzite, beraunite, bermanite, kryzhanovskite, whitmoreite/earlshannonite and phosphosiderite (Moore, Reference Moore1973; Mücke, Reference Mücke1981; Segeler et al., Reference Segeler, Ulrich, Kampf and Whitmore1981; Campbell and Roberts, Reference Campbell and Roberts1986; Nizamoff, Reference Nizamoff2006; Kreinik, Reference Kreinik2022). The crystal structures of the first four minerals are based on different combinatorial polymorphs of heteropolyhedral layers (laueite-type sheets) of corner-connected octahedra and tetrahedra as described by Moore (Reference Moore1975) and Krivovichev (Reference Krivovichev2004). The layers in the four minerals are compared in Fig. 13 and their crystallographic parameters are given in Table 1.
Laueite-type sheets in (a) laueite, (b) stewartite, (c) strunzite and (d) pseudolaueite.

Figure 13 Long description
The image A shows laueite-type sheets in laueite, characterized by a grid-like arrangement of octahedra and tetrahedra. The octahedra are depicted in orange, while the tetrahedra are in green, forming a repeating pattern. The image B illustrates stewartite, where the octahedra and tetrahedra are similarly arranged but with a slight tilt, creating a more angled structure. The octahedra are orange and the tetrahedra are green, connected at their corners. The image C represents strunzite, featuring a more complex interlocking pattern of yellow octahedra and green tetrahedra, forming a dense network. The image D shows pseudolaueite, with yellow octahedra and green tetrahedra arranged in a circular pattern, creating a distinct layered structure. Each mineral's sheet is composed of corner-connected octahedra and tetrahedra, illustrating the combinatorial polymorphs of heteropolyhedral layers.
The layers in laueite/stewartite (Figs 13a,b) and strunzite/pseudolaueite (Figs 13c,d) correspond to different topological isomers. In the laueite and stewartite layers, two independent octahedra have corner-connections to two, and four PO4 tetrahedra, respectively whereas in the strunzite and pseudolaueite layers there is only one type of octahedron and it shares corners with three PO4 tetrahedra. The layers in stewartite and pseudolaueite are geometrical isomers of the layers in strunzite and laueite, respectively, due to different types of octahedral–tetrahedral corner-connections. This is manifested in different orientations of the PO4 tetrahedra decorating the octahedral chains. In strunzite the tetrahedra in each chain all have the same orientation (unshared apices all pointing + + + or − − − relative to the layer) whereas in pseudolaueite the unshared tetrahedral apices alternate + − + − along the octahedral chains. In laueite the orientation of the unshared apices of the intrachain PO4 tetrahedra is also alternately + − + − along the chain, whereas in stewartite the sequence is + + − −, giving a doubling of the chain periodicity (Grey et al., Reference Grey, Keck, Gable, Mumme, Hochleitner and Glenn2025).
The layer structures in the four minerals all contain the six-member clusters that are present in {111} planes in triphylite/lithiophilite (Fig. 4), and the same trans-connectivity of the [001] octahedral chains. In laueite and stewartite, the cyclic tetramers are connected into kröhnkite chains along [100], whereas in strunzite and pseudolaueite, each cyclic tetramer is connected to four surrounding cyclic tetramers via octahedral–octahedral and octahedral–tetrahedral corner-sharing. It is interesting that the structure of phosphosiderite, Fe(PO4)H2O)2 (Moore, Reference Moore1966), also contains cyclic tetramers that corner-connect to four surrounding cyclic tetramers. In this case the eight corner-shared pairs per cluster are all octahedral–tetrahedral, giving a three-dimensional framework (Fig. 14).
[100] projection of the structure of phosphosiderite.

Figure 14 Long description
The diagram illustrates the structure of phosphosiderite, featuring cyclic tetramers connected through octahedral-tetrahedral corner-sharing. The tetramers are arranged in a repeating pattern, forming a three-dimensional framework. Each tetramer is connected to four surrounding tetramers, creating a network of interconnected units. The diagram includes directional indicators labeled as [001] and [010], showing the orientation of the structure. The tetramers are depicted with alternating colors, highlighting the distinct octahedral and tetrahedral connections. The overall arrangement forms a complex lattice, emphasizing the structural connectivity within the mineral.
Phosphosiderite, along with its dimorph strengite, are listed by Moore (Reference Moore1973) as oxidized alteration products of triphylite/lithiophilite, but they are also commonly reported as alteration products derived from zwieselite/triplite and fluorapatite. For example at pegmatite mines at El Criollo, Argentina; Hillside, Arizona and Sitio do Castelo, Portugal, they occur as large, well-formed crystals grown from solution in vugs of altered zwieselite/triplite and fluorapatite (Demartin et al., Reference Demartin, Gay, Gramaccioli and Pilati1997; Peacor et al., Reference Peacor, Dunn and Simmons1984, Kampf et al., Reference Kampf, Grey, Alves, Mills, Nash, MacRae and Keck2017). It is tempting to suggest that the M 2(TO4)2Φ8 cyclic tetramer cluster in phosphosiderite is present in solution, analogous to the suggestion by Hawthorne (Reference Hawthorne1979) that the M 2(TO4)2Φ7 cluster in morinite and as a polymerized form in several hydrated phosphate minerals is present in the fluid phase that the minerals crystallised from.
The minerals kryzhanovskite, (Fe3+,Mn2+)3(PO4)2(OH,H2O)3, bermanite, (Mn,Fe)2+Mn3+2(PO4)2(OH)2(H2O)4, and whitmoreite, (Fe,Mn)2+Fe3+2(PO4)2(OH)2(H2O)4, differ from the other alteration products in Table 1 in that their structures are built from octahedral edge-sharing, and they do not contain 7 Å corner-connected chains. Their structures are more closely aligned with those of the alteration minerals formed under reducing conditions in Table 2. In fact, kryzhanovskite is the oxidized form of phosphoferrite in Table 2, with the same space group and similar unit-cell parameters (Moore and Araki, Reference Moore and Araki1976). Its structure is built from stepped chains of edge-shared octahedra along [001] that are interconnected along [100] by corner-linkage with PO4 tetrahedra, forming cyclic tetramers as shown in Fig. 12c for phosphoferrite.
In the same vein, bermanite is reported as forming from the oxidative alteration of hureaulite (Leavens and Simpson, Reference Leavens and Simpson1975) and has a related structure containing chains of edge-shared octahedra. The crystallographic parameters for bermanite, from the structure determination by Kampf and Moore (Reference Kampf and Moore1976) are given in Table 1. The structure is based on heteropolyhedral layers parallel to (010) that are interconnected along [010] by corner-sharing of intralayer PO4 tetrahedra with trans-connected Mn2+O2(H2O)4 octahedra. The heteropolyhedral layers have [101] chains of edge-shared octahedra containing the Mn3+ cations, which are separated by corner-connected PO4 tetrahedra. The octahedral–tetrahedral connections form kröhnkite-like chains, orientated along [100] and [001], as shown in Fig. 15a.
(a) (010) heteropolyhedral layer in bermanite. (b) (100) layer in whitmoreite. A cyclic tetramer is outlined with a red ellipse, and a morinite cluster is outlined with a blue circle.

Figure 15 Long description
The image A shows the (010) heteropolyhedral layer in bermanite. This layer is composed of interconnected polyhedra, with orange and green shapes representing different structural units. The polyhedra are arranged in chains parallel to the [001] and [100] directions. The label M1 is positioned on one of the orange polyhedra, while M2 is on a green polyhedron, indicating specific sites within the structure. The image B illustrates the (100) layer in whitmoreite, featuring a similar arrangement of polyhedra. Yellow and green polyhedra are organized in a grid-like pattern, with directional arrows indicating the [001] and [010] orientations. A cyclic tetramer is outlined with a red ellipse and a morinite cluster is highlighted with a blue circle, emphasizing specific structural features within the layer.
As noted by Kampf and Moore (Reference Kampf and Moore1976), whitmoreite has an identical formula to that of bermanite when Fe is substituted for Mn, and there are similarities in their structures. Both structures comprise heteropolyhedral layers that are interconnected via corner-connections of the intralayer PO4 tetrahedra with interlayer M2+O2(H2O)4 octahedra. The structures of the layers in the two minerals are quite different, however. As shown in Fig. 15b, the (100) heteropolyhedral layers in whitmoreite are built from edge-shared octahedral dimers that each share corners with four adjacent dimers. The PO4 tetrahedra occupy sites above and below the vacant octahedral sites, so that they each corner-share to three octahedra within the layer, and with the fourth vertex normal to the layer where it coordinates to the interlayer octahedra. As observed by Hawthorne (Reference Hawthorne1979), the pairs of corner-connected octahedra in the layer are bridged by two PO4 tetrahedra, forming morinite-type clusters that polymerise into chains along [001]. These chains alternate with chains of cyclic tetramers along [010].
Beraunite is another hydrated phosphate that can form from triphylite via its hydrothermal alteration products under low-temperature oxidizing conditions. At Hagendorf Sud it forms directly from rockbridgeite (Mücke, Reference Mücke1981). The nomenclature for beraunite-group minerals has recently been revised (Vrtiska et al., Reference Vrtiska, Tvrdy, Plasil, Sejkora, Skoda, Chukanov, Massanek, Filip, Dolnicek and Veselovsky2022), with the formula for type beraunite being redefined as the fully oxidized form, Fe3+6(PO4)4O(OH)5(H2O)4·2H2O, and the space group revised from C2/c to Cc (Table 1). The trans-octahedral chains in laueite-type layers in rockbridgeite are not retained in beraunite, but the h-clusters and the kröhnkite chains are common to both minerals.
The (100) heteropolyhedral layer in Fig. 16 is shown in plan (a) and edge-on (b). M4-centred octahedra link with PO4 tetrahedra to form kröhnkite chains along [010]. The chains are interconnected along [001] via corner-sharing with morinite-type clusters containing M1-centred octahedra. The heteropolyhedral layers are interconnected by octahedral–octahedral and octahedral–tetrahedral corner-sharing with M2O2(OH)2(H2O)2 octahedra. The joining of dense heteropolyhedral slabs or layers through corner-connections with hydrated octahedrally coordinated cations is a common feature of the low-temperature oxidized alteration products derived from triphylite/lithiophilite. This tendency was noted by Kampf and Moore (Reference Kampf and Moore1976) as suggesting that insular octahedral species may coexist in the alteration fluid medium with more complex clusters that form the basis for the layers.
(a) (100) heteropolyhedral layer in beraunite. (b) edge-on view of layer.

Figure 16 Long description
The image consists of two parts. The first part, labeled (a), shows a plan view of the (100) heteropolyhedral layer in beraunite. This layer is composed of M4-centered octahedra linked with PO4 tetrahedra, forming kröhnkite chains along the direction labeled [010]. These chains are interconnected along the direction labeled [001] through corner-sharing with morinite-type clusters that contain M1-centered octahedra. The second part, labeled (b), provides an edge-on view of the same layer. In this view, the kröhnkite chain is highlighted, showing its connection with the morinite-type cluster. The M3 and M4 positions are marked, indicating the locations of the octahedra within the structure. The diagram illustrates the complex interconnection of these layers through octahedral and tetrahedral corner-sharing, characteristic of the low-temperature oxidized alteration products derived from triphylite or lithiophilite.
Discussion
Triphylite/lithiophilite stand out from other primary phosphate minerals, including zwieselite/triplite, graftonite and alluaudite groups in their much higher susceptibility to alteration. Mason (Reference Mason1941) has noted that in intimately intergrown triphylite–graftonite and lithiophilite–triplite assemblages, the triphylite/lithiophilite has undergone alteration while the associated graftonite/triplite remain fresh and unaltered. The relative ease of leaching of the large univalent Li+ cation coupled with oxidation of Fe to form heterosite/purpurite must play a role in this; often the presence of these minerals is the only indication that triphylite/lithiophilite were the original primary minerals.
Another factor could be local valence oversaturation associated with the tetrahedral–octahedral edge-sharing anions O3 (see Fig. 3) in triphylite/lithiophilite and oxidized polymorphs, resulting in weakening of bonds and increased susceptibility to leaching. The other primary minerals listed above do not contain octahedral–tetrahedral edge-sharing. In triphylite/lithiophilite the O3 anions coordinate to Li++P5++2M 2+ and the formal valence is 2.09. This increases to 2.25 in heterosite/purpurite that contain M 3+. In sarcopside, O3 coordinates to 3M 2++P5+ and the bond valence is also 2.25. In addition to oversaturation, bond strain could play a role. The octahedral O3–O3 edge shared with PO4 is only 2.4 Å while the opposite edge is 3.6 Å; the associated O3–M–O3 angles are 66° and 119°, so the octahedra are strongly deformed and prone to attack by hydrothermal fluids.
A striking observation concerning the crystallochemical properties of the triphylite/lithiophilite alteration products is that although the density decreases by more than 30% with increasing extent of alteration (Tables 1 and 2), the mean volume per oxygen, V ox, remains relatively constant, with less than 6% change for the alteration products formed under reducing conditions (Table 2) and a 4% change for the hydrothermal products formed under oxidizing conditions (Table 3). Even for the supergene alteration products, the increase in V ox relative to that in triphylite is less than 6%.
Non-standard unit-cell settings (Å, °) to emphasise the structural relationships between the oxidized alteration products, together with the mean oxygen volume, V ox (Å3)

Table 3 Long description
The table lists 13 minerals with their space group, unit-cell lengths a, b, and c in angstroms, cell angles alpha, beta, and gamma in degrees, and mean oxygen volume Vox in cubic angstroms. Vox spans a narrow range from 17.5 in Tavorite to 19.2 in Laueite, with Bermanite at 19.1 and Whitmoreite at 19.0 also high. Several minerals share similar Vox near 18.2, including Barbosalite, Manganrockbridgeite, Gayite, and Beraunite. Many entries show repeated or scaled cell lengths, such as b near 5.2 angstroms and c given as about 6.9 angstroms times two for multiple minerals, indicating related structural settings. Cell angles vary by symmetry: some are all 90 degrees, while others have oblique angles around 110 degrees and, for Laueite, a notably low gamma near 71 degrees.
In their paper on the structures of phosphoferrite and its oxidised equivalent kryzhanovskite, Moore and Araki (Reference Moore and Araki1976) comment that the V ox values of 18.7 Å3 (phosphoferrite) and 16.9 Å3 (kryzhanovskite) correspond to dense-packed structures. The triphylite structure is based on a distorted hexagonal closest-packed (hcp) anion lattice, and the very similar V ox values in Tables 2 and 3 are consistent with all its alteration products having an anion packing density approximately equivalent to a closest packed anion lattice.
In Fig. 17 the anion close packing for an ideal hcp lattice is compared with those for sarcopside and ludlamite. The ideal hcp lattice is hexagonal, P63/mmc, with c = √(8/3)a. With a = 3 Å (= c/2 for triphylite), c = 4.90 Å for ideal hcp and V ox = 19.1 Å3, whereas with c = 4.69 Å (= a for triphylite), a = 2.87 Å for ideal hcp and V ox = 16.7 Å3. These two values span the range of V ox values for triphylite alteration products in Tables 2 and 3.
Plan and side view of anion packing for (a) ideal hcp lattice, (b) sarcopside and (c) ludlamite. The dark blue circles in (c) are H2O.

Figure 17 Long description
The diagram consists of three labeled sub-images, A, B and C, each showing plan and side views of anion packing. Sub-image A represents the ideal lattice, with light blue spheres connected by brown lines, indicating anions and their bonds. The plan view shows a hexagonal arrangement, while the side view reveals a uniform stacking pattern. Sub-image B illustrates sarcopside, where light blue spheres are mixed with pink and yellow spheres, representing different atomic sites. The plan view displays a more complex arrangement and the side view shows variations in stacking compared to the ideal lattice. Sub-image C depicts ludlamite, featuring light blue and dark blue spheres, with the dark blue circles identified as H2O. The plan view shows a distinct pattern and the side view highlights differences in packing and coordination. The diagram aims to compare how anion packing deviates from the ideal lattice in sarcopside and ludlamite, focusing on structural relationships and geometric motifs such as rows, offsets and layering. No units or scales are present in the image.
Sarcopside has a small monoclinic distortion of the triphylite structure with a hcp oxygen lattice. As shown in Fig. 17b, however, the anion close packing is considerably deformed to adjust to the separate coordination requirements of the small tetrahedrally coordinated P5+ cations, where O–O ≈ 2.4 Å, and the large octahedrally coordinated Fe2+ cations, with O–O ≈ 3 Å. This results in puckering of the anion lattice, shown in Fig. 17b. The polyhedral articulations for hcp, however, are still obeyed, with corner-sharing only for intralayer octahedral–tetrahedral joins and edge-sharing only for interlayer octahedral–tetrahedral joins involving the basal anions of the tetrahedra.
Ludlamite has a more deformed packing of anions than sarcopside, shown in Fig. 17c. Hexagonal close-packing is restricted to local strips that Moore (Reference Moore1971) described as ‘interrupted close packing’. Despite the increased deformation associated with incorporation of H2O into the lattice, the anion packing density is almost unchanged, with V ox = 18.7 Å3 for ludlamite compared with 18.8 Å3 for sarcopside. As a consequence of the anion lattice deformation in ludlamite, the interlayer octahedral–tetrahedral articulation is modified from edge-sharing to corner-sharing, as illustrated in Fig. 12 for ludlamite, phosphoferrite and hureaulite. As a result, the edge-shared octahedral strings in these minerals are interconnected by corner-connections with pairs of PO4 tetrahedra. The resulting cyclic tetramers extend as segments of kröhnkite chains normal to the close-packed layers as discussed in a previous section on low temperature alteration under non-oxidizing conditions.
Mattievich and Danon (Reference Mattievich and Danon1977) have prepared synthetic equivalents of sarcopside, phosphoferrite and ludlamite from hydrothermal treatment of near-neutral solutions containing solid vivianite. Their studies showed that heating vivianite, the n = 8 member of the homologous series, Fe3(PO4)2(H2O)n, gave members with decreasing n values as the temperature was increased; ludlamite (n = 4) formed in the temperature range 100 to 180°C, phosphoferrite (n = 3) up to 200°C and sarcopside above 250°C. These studies suggest that the three minerals can form from triphylite through local dissolution and reprecipitation according to the reaction: 3LiFe(PO4) + n H2O → Fe3(PO4)2(H2O)n + Li3PO4
The anion packings shown in Fig. 17 suggest that the distorted close-packed anion lattice for triphylite (or sarcopside) could provide a suitable nucleating surface for heterogeneous precipitation of progressively lower n members as reaction with the hydrothermal fluids under non-oxidizing conditions occurs at progressively lower temperatures.
The crystal structures of alteration products formed from triphylite/lithiophilite under oxidizing conditions differ markedly from those described above. The major difference is that they are based on corner-connected octahedral chains rather than on strings of edge-shared octahedra. In both cases, however, the octahedral chains are interconnected via corner-connections with PO4 tetrahedra, forming cyclic tetramers. A consistent structural motif, in oxidized alteration products, is the cross-linking of 7 Å octahedral chains with 5.2 Å kröhnkite chains, giving laueite-like layers. In the hydrothermal alteration minerals barbosalite, tavorite and lipscombite two sets of quasi-orthogonal laueite-like layers interpenetrate, while in the lower-temperature hydrothermal alteration minerals including rockbridgeite-group and dufrenite-group minerals, there is only one extended orientation of laueite-like layers and the second orientation is limited to two layers. This is illustrated in Fig. 18 for manganrockbridgeite. A projection along the 7 Å octahedral chain direction is compared to the same projection for barbosalite. It is seen that the manganrockbridgeite structure comprises two-layer wide barbosalite slabs parallel to (010) that are mirrored at y = ¼ and ¾, so it can be described as composed of unit-cell-twinned barbosalite slabs.
(a) is a projection of the manganrockbridgeite structure along the 7 Å chain direction. (b) is a two-layer wide section of (a), while (c) is a projection of the barbosalite structure along the 7 Å chain direction, showing identical topology to (b), so the manganrockbridgeite structure can be described as unit-cell twinning of barbosalite.

Figure 18 Long description
The image consists of three sub-images labeled (a), (b) and (c). Sub-image (a) shows a projection of the manganrockbridgeite structure along the seven angstrom chain direction. It features a series of interconnected polyhedral units, with a directional arrow indicating the orientation labeled as left bracket zero one zero right bracket. Sub-image (b) presents a two-layer wide section of the structure seen in (a), maintaining the same interconnected polyhedral arrangement. Sub-image (c) displays a projection of the barbosalite structure along the same seven angstrom chain direction, showing an identical topology to sub-image (b). The structures in all sub-images are composed of yellow and green polyhedra, interconnected in a repeating pattern, illustrating the structural similarities and differences between manganrockbridgeite and barbosalite.
Similar two-layer wide slabs are present in the orthorhombic rockbridgeite-group minerals and in dufrenite-group minerals, with the difference that the corner-shared octahedral chains in the laueite-like layers are twinned at the unit-cell level, giving sawtooth-shaped chains with a doubled periodicity to ∼14 Å. A further modification is that M atoms can occupy vacant sites in the 5.2 Å chains, forming face-shared octahedral trimers, the h-clusters of Moore (Reference Moore1970a). We have used the terms ‘twinned laueite’ and ‘stuffed laueite’ to describe the layers involving these modifications. In the supergene alteration minerals of the laueite group the central atoms of the h clusters relocate as MO2(H2O)4 octahedra interconnecting the laueite-like layers via corner-sharing with intralayer PO4 tetrahedra. The persistence of intersecting corner-shared octahedral chains and kröhnkite chains in the oxidised alteration products is reflected in the unit-cell parameters as given in Table 3. In this Table the unit cells have, where necessary, been changed to non-standard settings compared to the published cells in Table 1, to better illustrate the relationships for the different minerals. As seen from Table 3, barbosalite, tavorite and lipscombite have two axes involving ∼5.2 Å repeats, reflecting the quasi-orthogonal interpenetrating laueite-like sheets, while all other minerals have just one axis of ∼5.2 Å, corresponding to one extended laueite-type sheet orientation. Barbosalite, tavorite, manganrockbridgeite and laueite have a cell parameter of ∼7–7.5 Å corresponding to trans-connected octahedral chains, whereas the orthorhombic rockbridgeite-group and dufrenite-group minerals have a doubling of the 7 Å axis due to unit-cell twinning of the octahedral chains. Lipscombite also has a doubling of the 7 Å axis, but this is due to a different geometrical connectivity of the PO4 tetrahedra decorating the chain, as occurs also for stewartite (Table 1).
(a)  $\left[ {10\bar 1} \right]$ projection of the triphylite structure. (b) a ribbon excised from (a). (c) [010] projection of the lacroixite structure.
$\left[ {10\bar 1} \right]$ projection of the triphylite structure. (b) a ribbon excised from (a). (c) [010] projection of the lacroixite structure.

Figure 19 Long description
The image A showing a projection of the triphylite structure, featuring interconnected octahedral chains. The chains are aligned along the direction labeled as left bracket zero one zero right bracket. The octahedra are colored in yellow and green, forming a complex lattice. The image B showing a ribbon excised from the triphylite structure, highlighting the octahedral rotations. A red arrow indicates a rotation angle of forty degrees, emphasizing the structural change. The image C showing a projection of the lacroixite structure, with a similar arrangement of octahedra. The octahedra are arranged in a grid-like pattern, maintaining the yellow and green color scheme. The structures in images B and C are compared to confirm the topological identity of the slabs, with interpenetrating sets of quasi-orthogonal layers.
The persistent presence of laueite-like layers in the oxidized alteration products led us to examine the parent triphylite structure for a possible origin. As shown in Fig. 3, the triphylite structure contains two sets of corner-connected octahedral chains, along [101] and  $\left[ {10\bar 1} \right]$. In Fig. 19a is shown a projection of the triphylite structure down one set of the octahedral chains,
$\left[ {10\bar 1} \right]$. In Fig. 19a is shown a projection of the triphylite structure down one set of the octahedral chains, 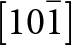 $\left[ {10\bar 1} \right]$. A vertical slab parallel to (101) is separated from the projected triphylite structure in Fig. 19b. It shows that the octahedral rotations about the chain direction are quite large, ∼40°. In Fig. 19c is shown a 7 Å projection for lacroixite, that has a similar large rotation of the octahedra about the chain axis. A comparison of Figs (b) and (c) confirm the topological identity of (101) slabs of triphylite with the family of structures having interpenetrating sets of quasi-orthogonal laueite-like layers. These include tavorite and barbosalite that are the highest temperature alteration products of triphylite/lithiophilite and their oxidized polymorphs. It is tempting to consider that regions of triphylite shown in Fig. 19b act as nucleating sites for the growth of alteration products such as tavorite, barbosalite, rockbridgeite and others. If this is the case, then parent/alteration phase assemblages should show an equal distribution of two orientations of the product, corresponding to the two independent 7 Å chain directions, [101] and
$\left[ {10\bar 1} \right]$. A vertical slab parallel to (101) is separated from the projected triphylite structure in Fig. 19b. It shows that the octahedral rotations about the chain direction are quite large, ∼40°. In Fig. 19c is shown a 7 Å projection for lacroixite, that has a similar large rotation of the octahedra about the chain axis. A comparison of Figs (b) and (c) confirm the topological identity of (101) slabs of triphylite with the family of structures having interpenetrating sets of quasi-orthogonal laueite-like layers. These include tavorite and barbosalite that are the highest temperature alteration products of triphylite/lithiophilite and their oxidized polymorphs. It is tempting to consider that regions of triphylite shown in Fig. 19b act as nucleating sites for the growth of alteration products such as tavorite, barbosalite, rockbridgeite and others. If this is the case, then parent/alteration phase assemblages should show an equal distribution of two orientations of the product, corresponding to the two independent 7 Å chain directions, [101] and 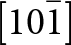 $\left[ {10\bar 1} \right]$. It would be interesting to conduct single-crystal diffraction studies on parent/alteration phase associations to gain further insights on possible paragenesis mechanisms.
$\left[ {10\bar 1} \right]$. It would be interesting to conduct single-crystal diffraction studies on parent/alteration phase associations to gain further insights on possible paragenesis mechanisms.
Conclusions
The triphylite–lithiophilite, Li(Fe2+,Mn2+)PO4, series of minerals with olivine-related structures are exceptional amongst the primary phosphates for their high susceptibility to alteration and their extensive range of alteration products. Their high reactivity is related to the mobility of the Li+ cation and its ease of removal and replacement; a feature that finds application in synthetic equivalents as cathodes of rechargeable lithium batteries (Yakubovich et al. Reference Yakubovich, Khasanova and Antipov2020).
The crystal chemistries of the alteration products are quite different, depending on whether the alteration occurs under reducing or oxidizing conditions. In the former case, the alteration minerals generally conform to the homologous series formula (Fe2+,Mn2+)3(H2O)n(PO4)2, n = 0 (sarcopside) to 8 (vivianite), and their crystal structures are based on strings of edge-shared octahedra, ranging from octahedral dimers in vivianite, trimers in ludlamite and sarcopside, pentamers in hureaulite and stepped chains in phosphoferrite. The edge-shared octahedral strings are interconnected by corner-sharing with PO4 tetrahedra, forming M 2(TO4)2Φ8 cyclic tetramers that are polymerised into kröhnkite-type chains or chain segments.
In contrast, alteration under oxidizing conditions gives rise to corner-connected rather than edge-connected octahedral chains. These chains, with a periodicity of 7n Å, n = 1,2, are cross-linked by M 2(TO4)2Φ8 cyclic tetramers to give laueite-like heteropolyhedral layers.
Under high-temperature hydrothermal alteration conditions, the structures of the alteration products such as tavorite, barbosalite and lipscombite contain two sets of interpenetrating, quasi-orthogonal, laueite-type layers. With decreasing temperature of hydrothermal alteration the structures of alteration products such as rockbridgeite and dufrenite are modified so as to contain only one orientation of the laueite-type layers, in two-layer-wide slabs. Alteration minerals formed under supergene conditions, such as the laueite-group minerals have single laueite-type layers interconnected via corner-sharing with hydrated octahedrally coordinated divalent cations. An interesting observation is that although the densities of the triphylite alteration products decrease by more than 30% with increasing extent of alteration, the mean volume per oxygen remains relatively constant over the full range of alteration conditions, from 17.5 Å3 for tavorite to 19.2 Å3 for laueite, c.f. 18.1 Å3 for triphylite. Thus the anion packing density remains approximately equivalent to that for a closest-packed anion lattice for all alteration products.
The persistent presence of laueite-like layers in all oxidized alteration products led to an examination of the parent triphylite structure for a possible origin. As shown in Fig. 19, the triphylite structure contains (101) slabs of corner-connected octahedra and tetrahedra that are topologically identical with the interpenetrating sets of quasi-orthogonal laueite-like layers in the structures of the high-temperature alteration products, tavorite and barbosalite. This suggests that such (101)triphylite slabs could act as nucleating sites for the growth of the hydrothermal alteration phases.
Acknowledgements
I thank Rupert Hochleitner, Christian Rewitzer, Frank Hawthorne and William Mumme for providing helpful comments on improving the manuscript.
Competing interests
The author declares none.
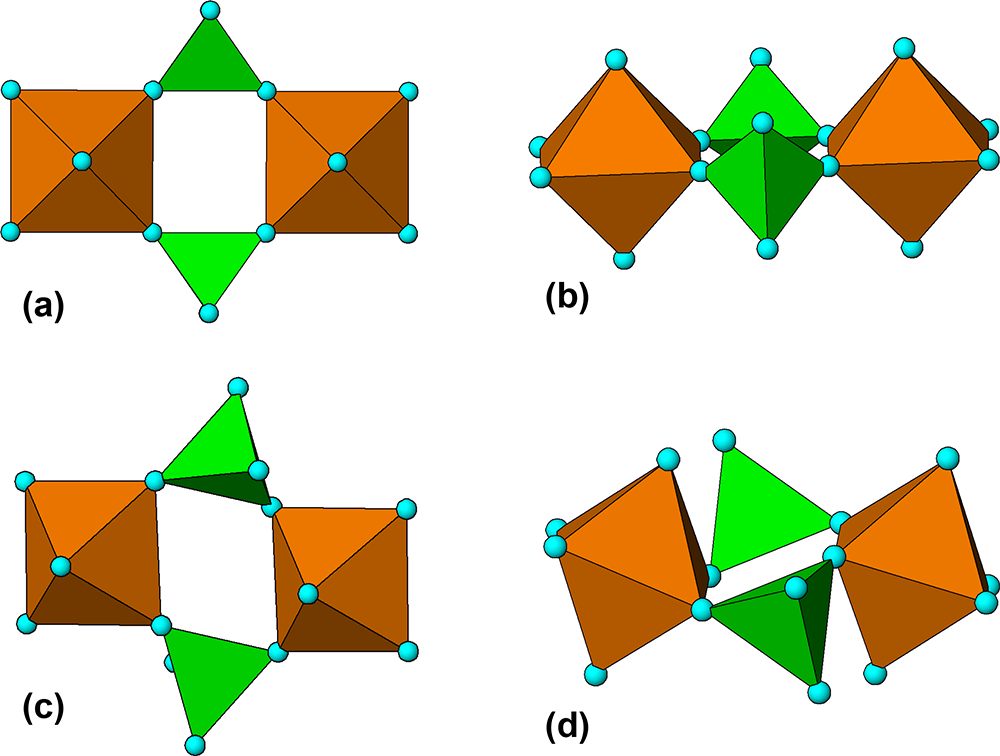
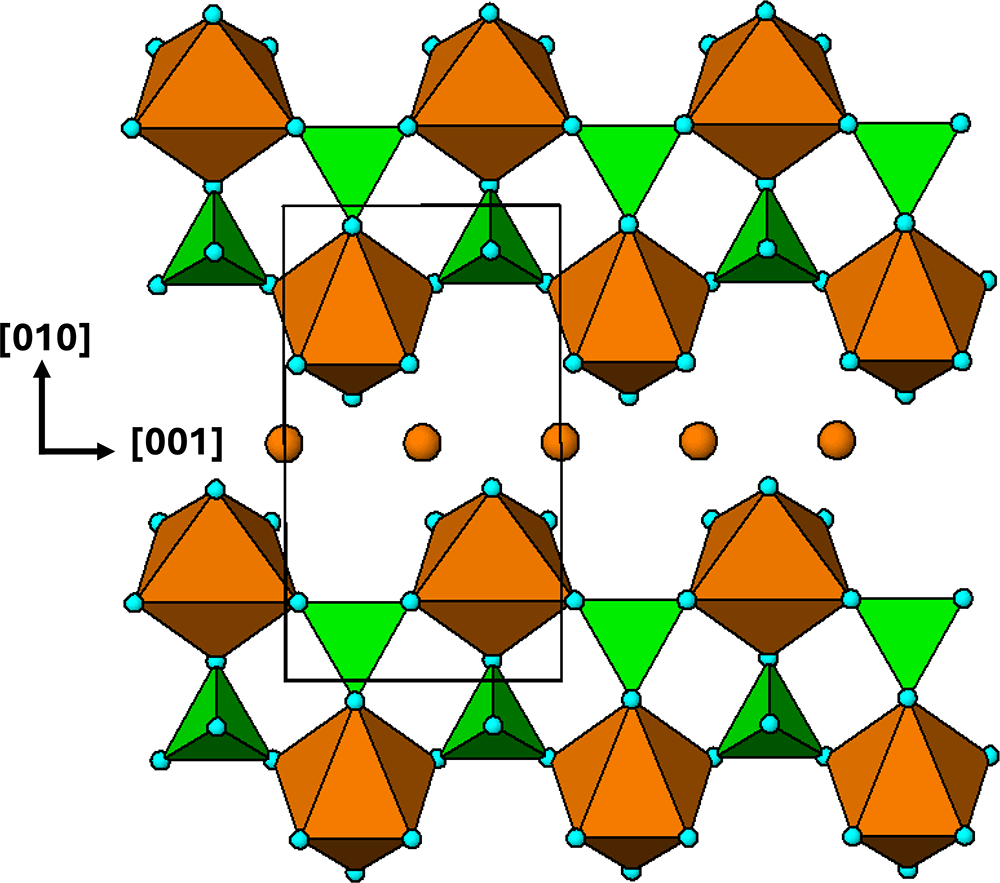
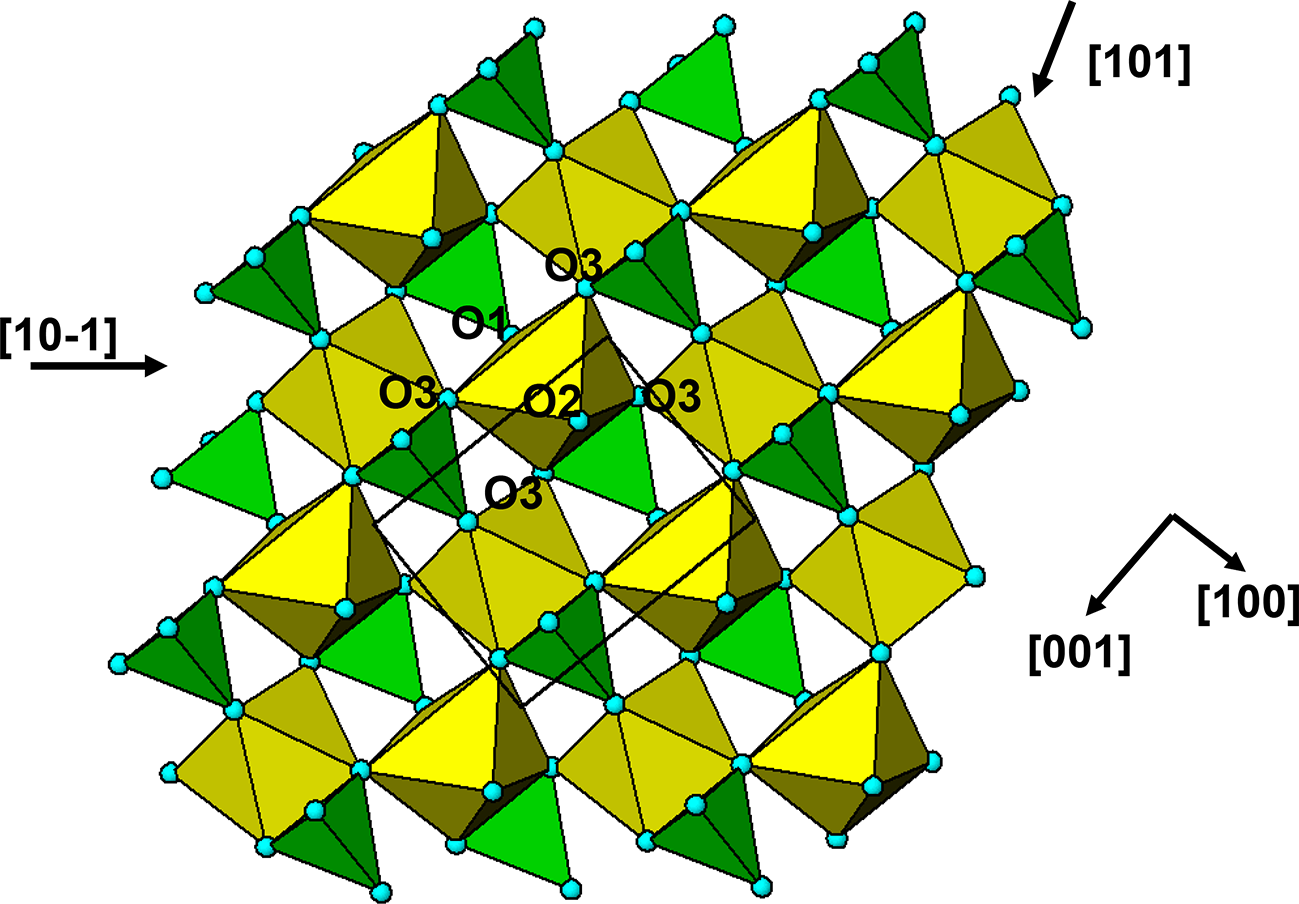

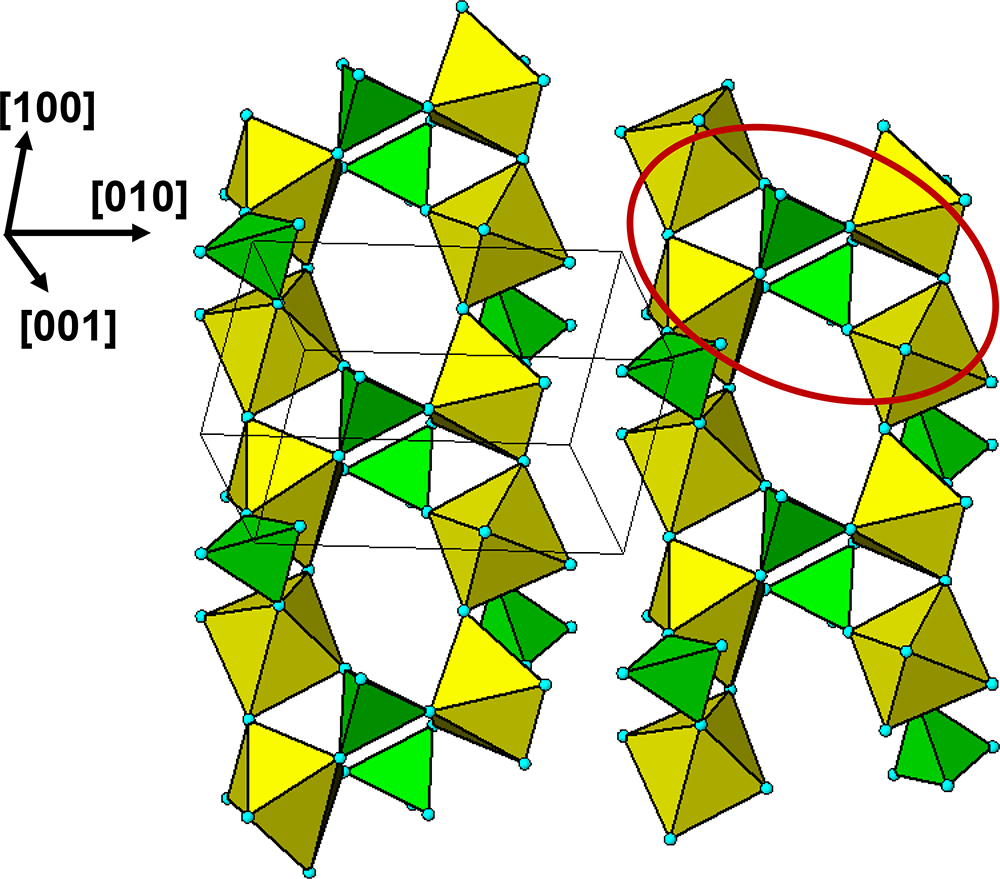
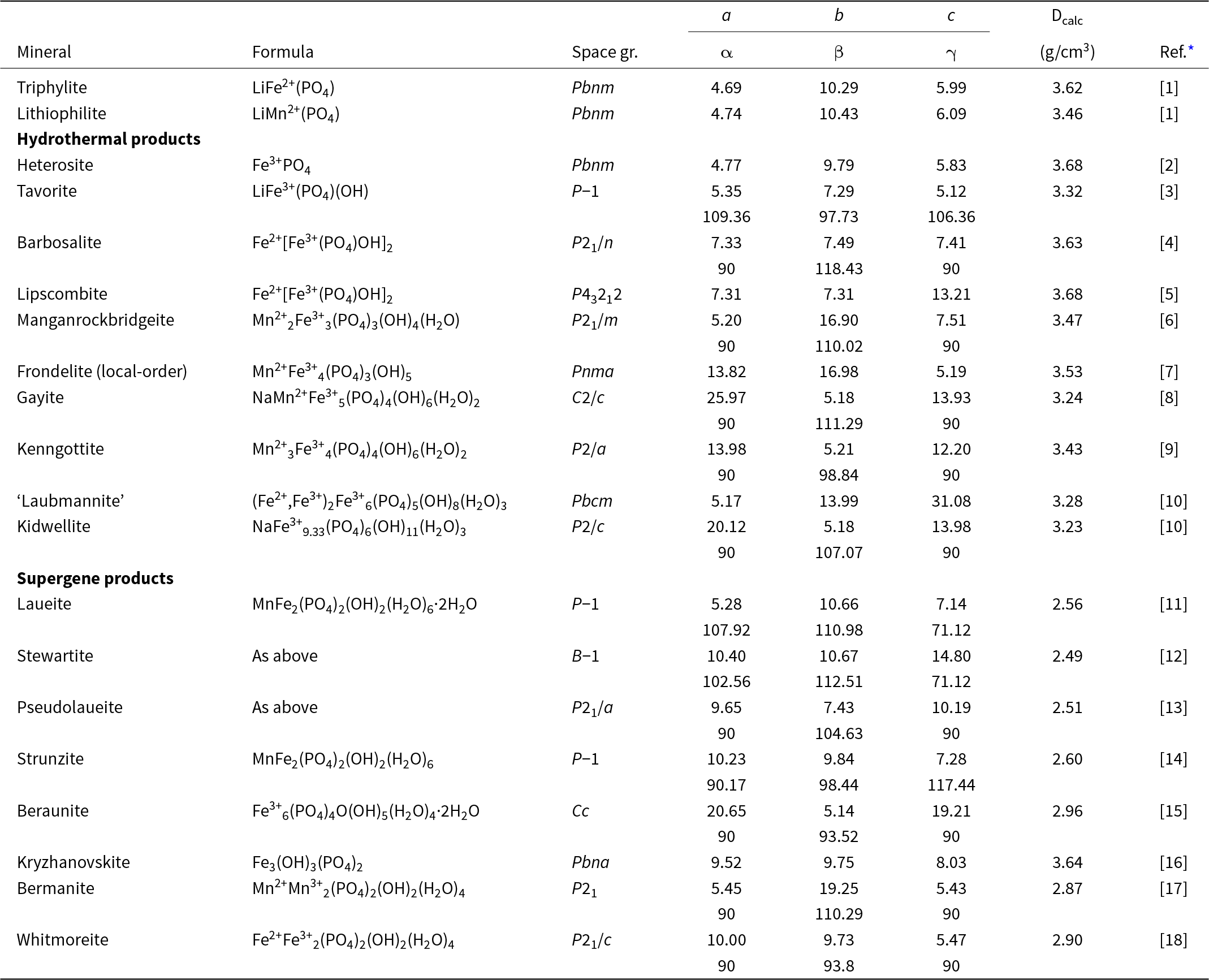
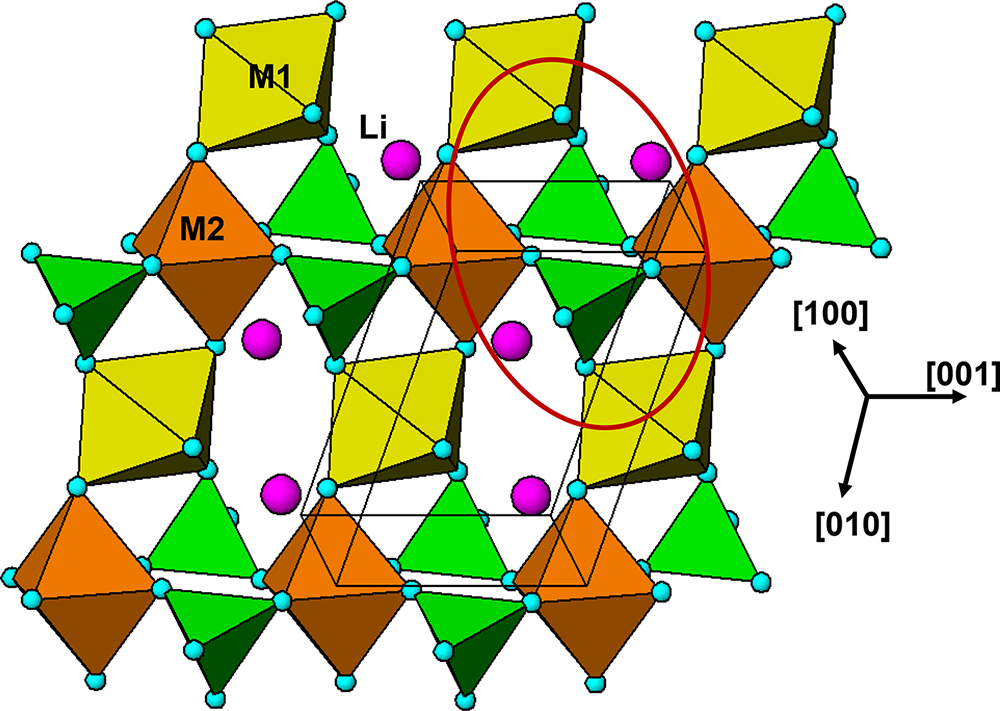
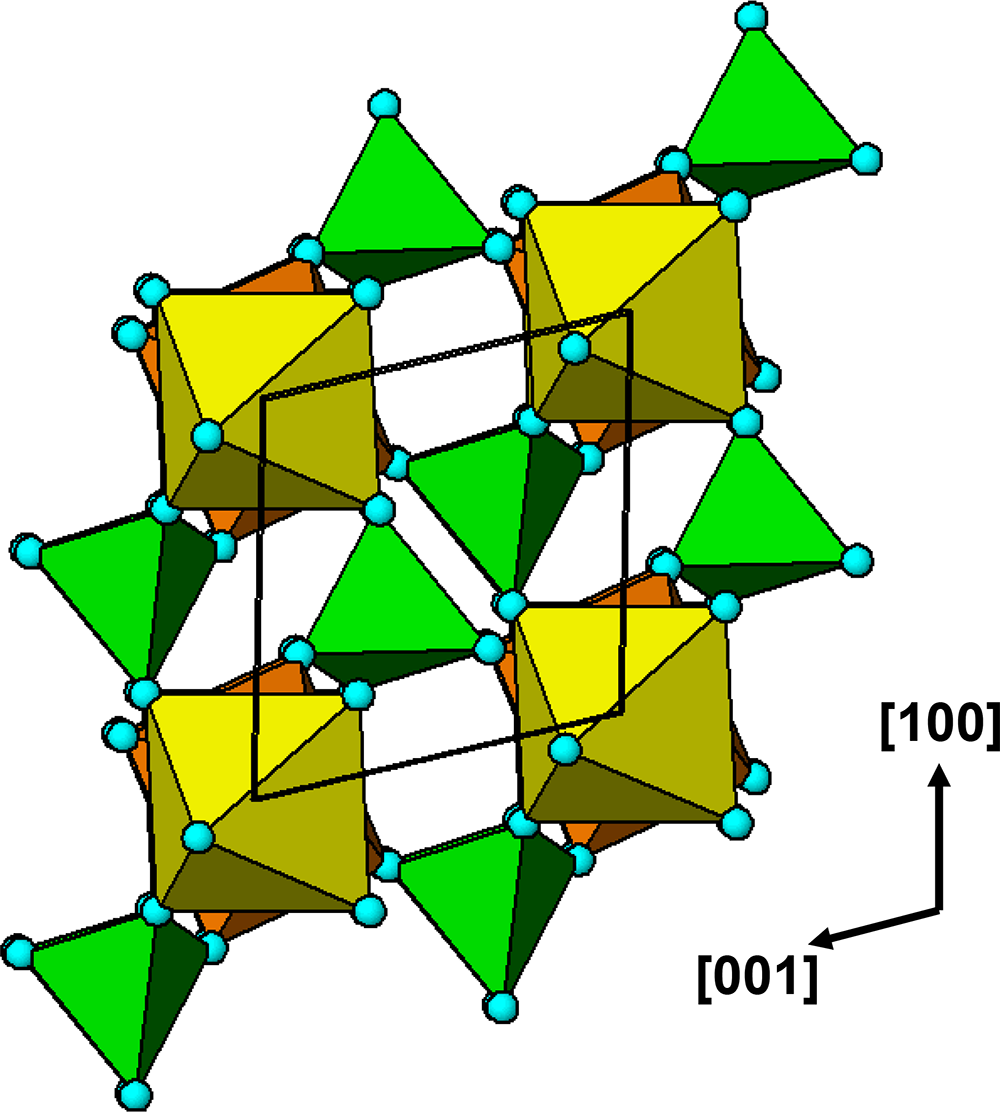
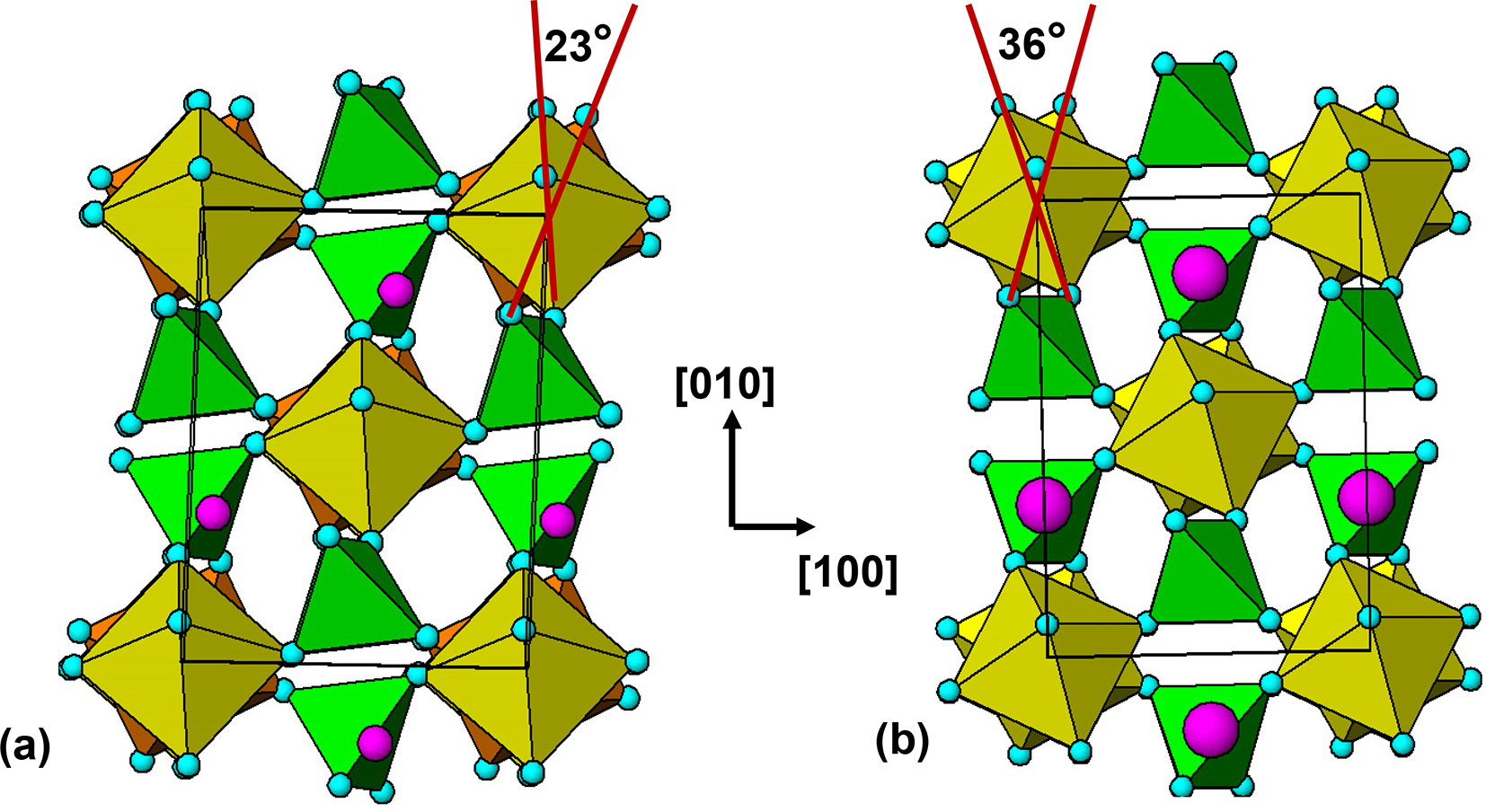
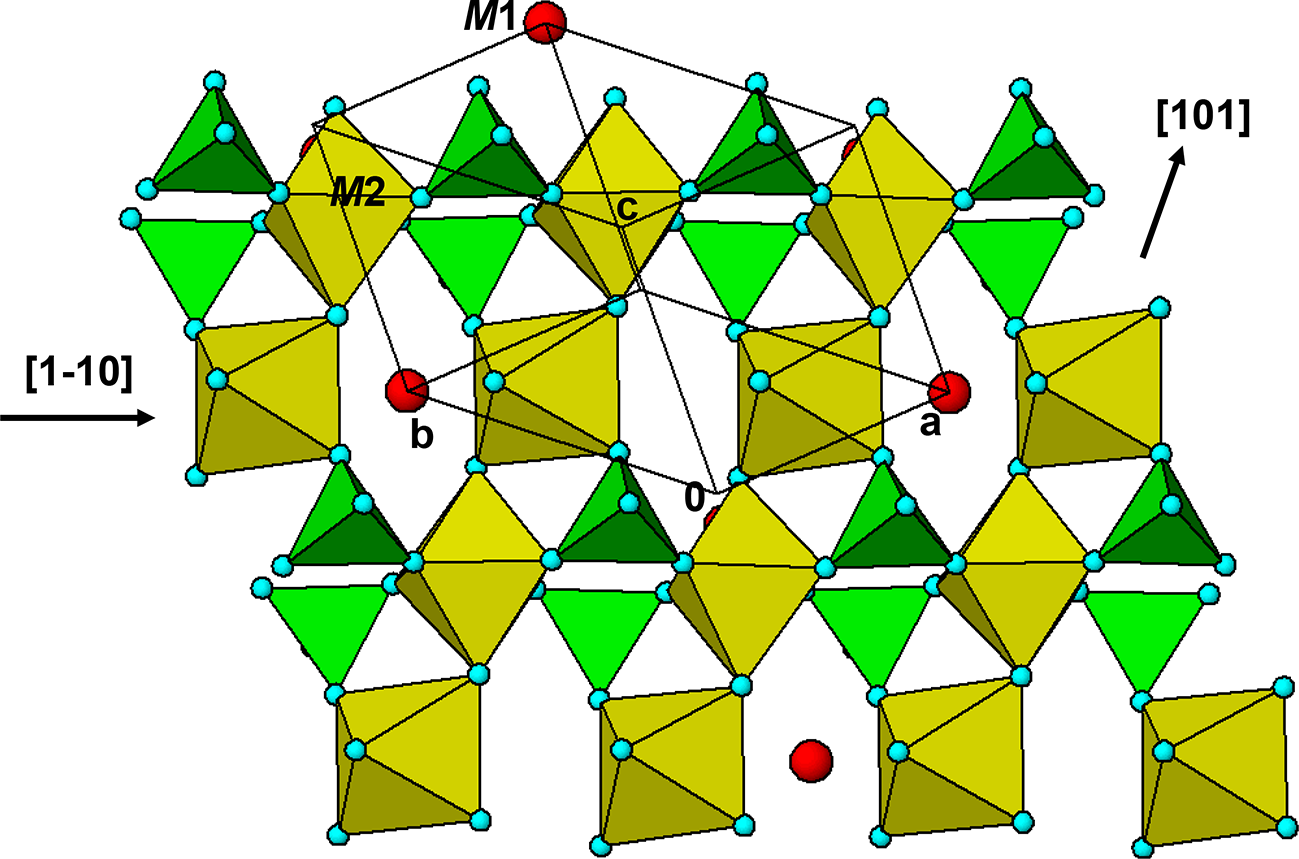

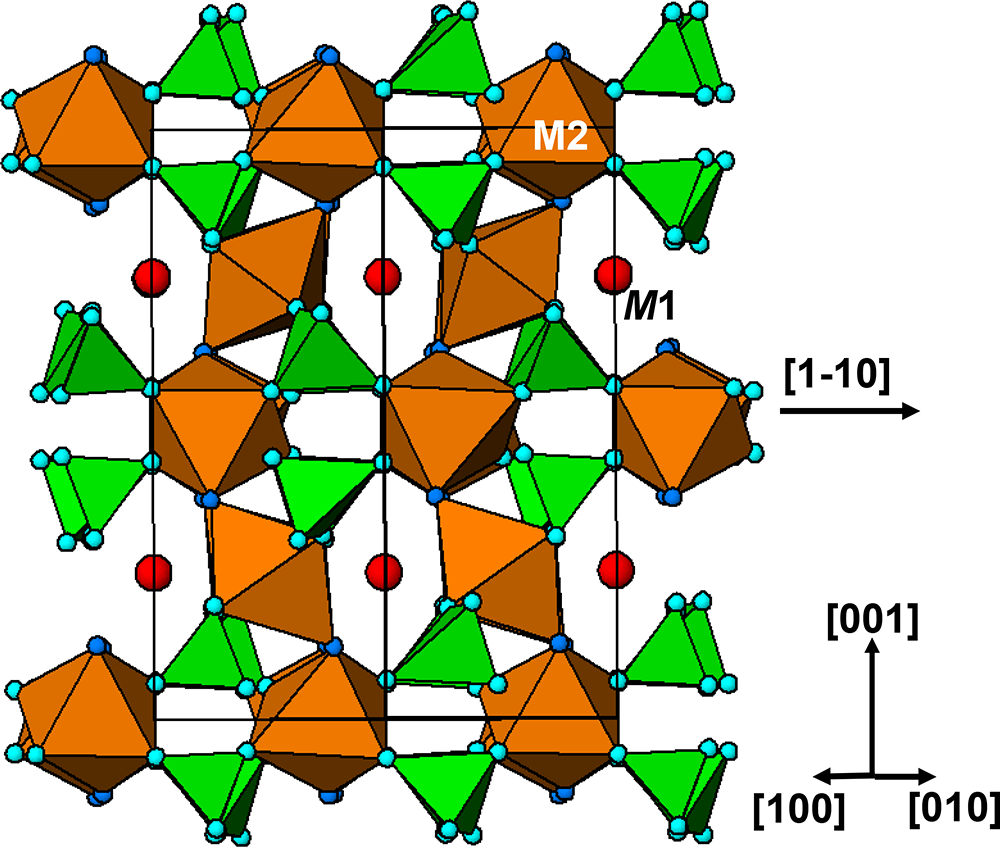
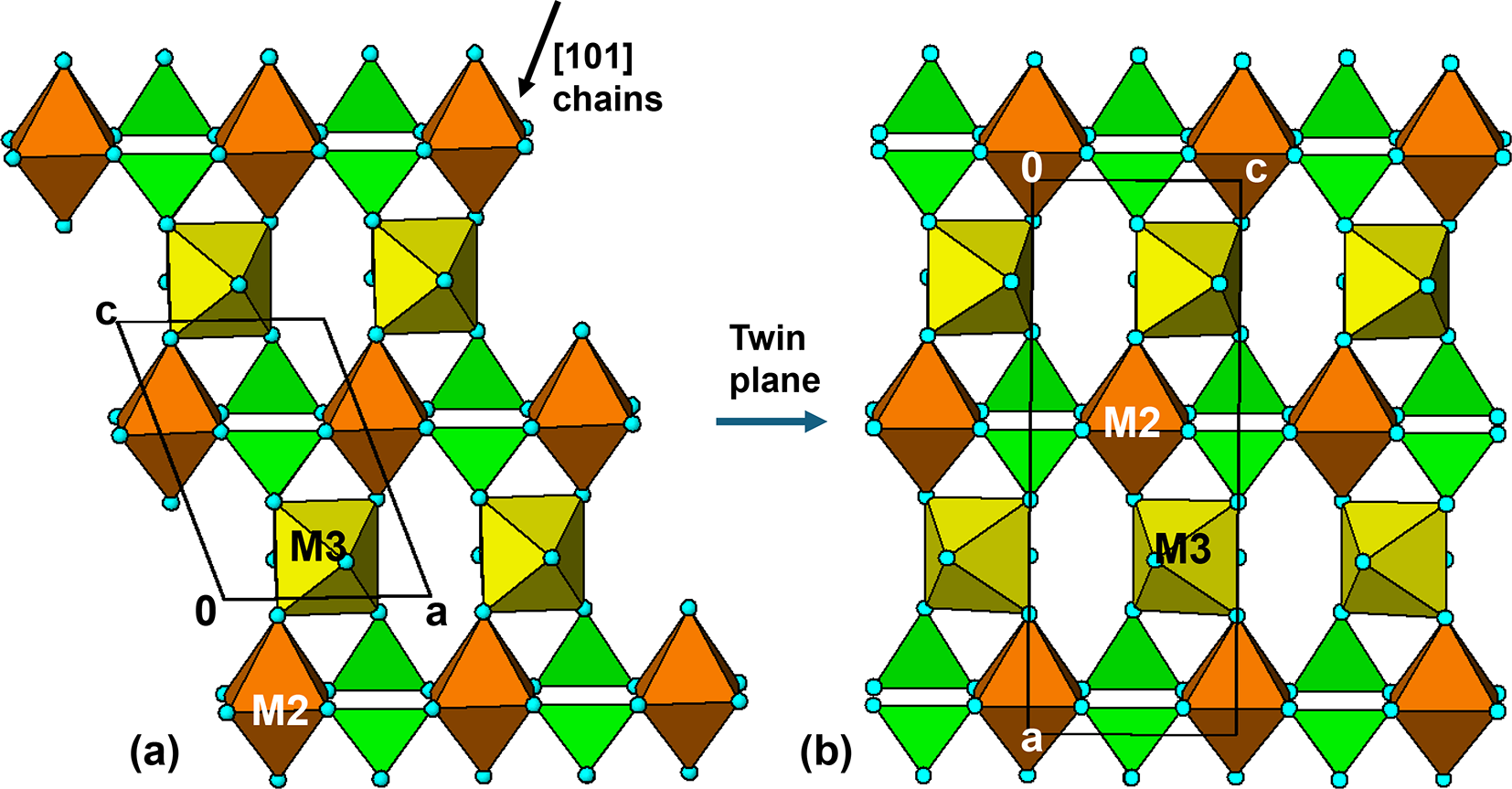
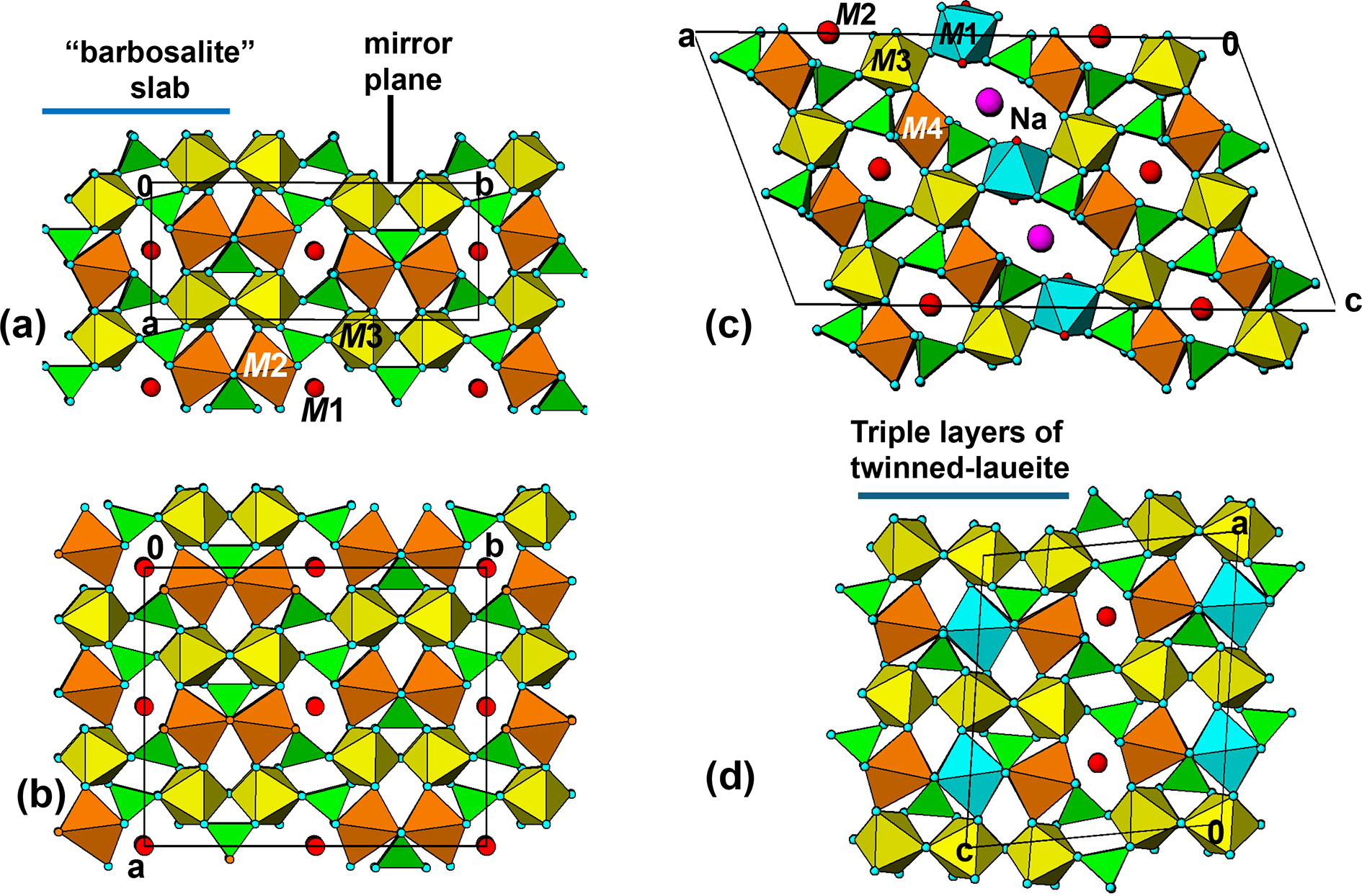
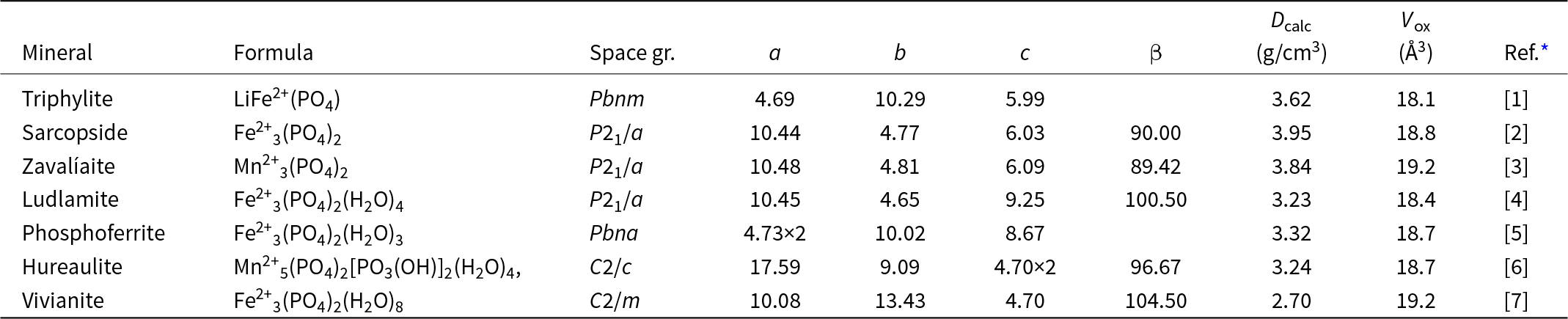
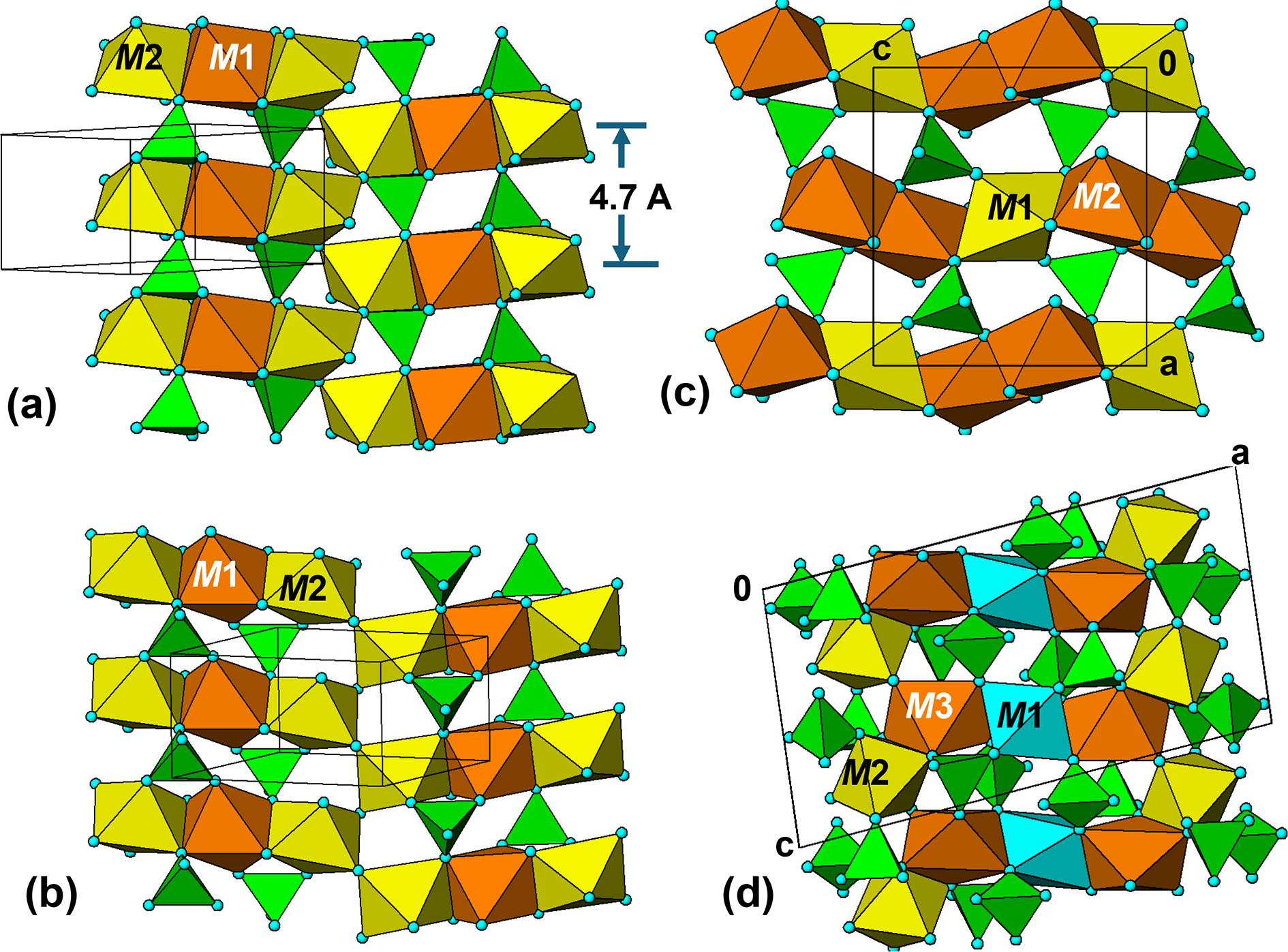
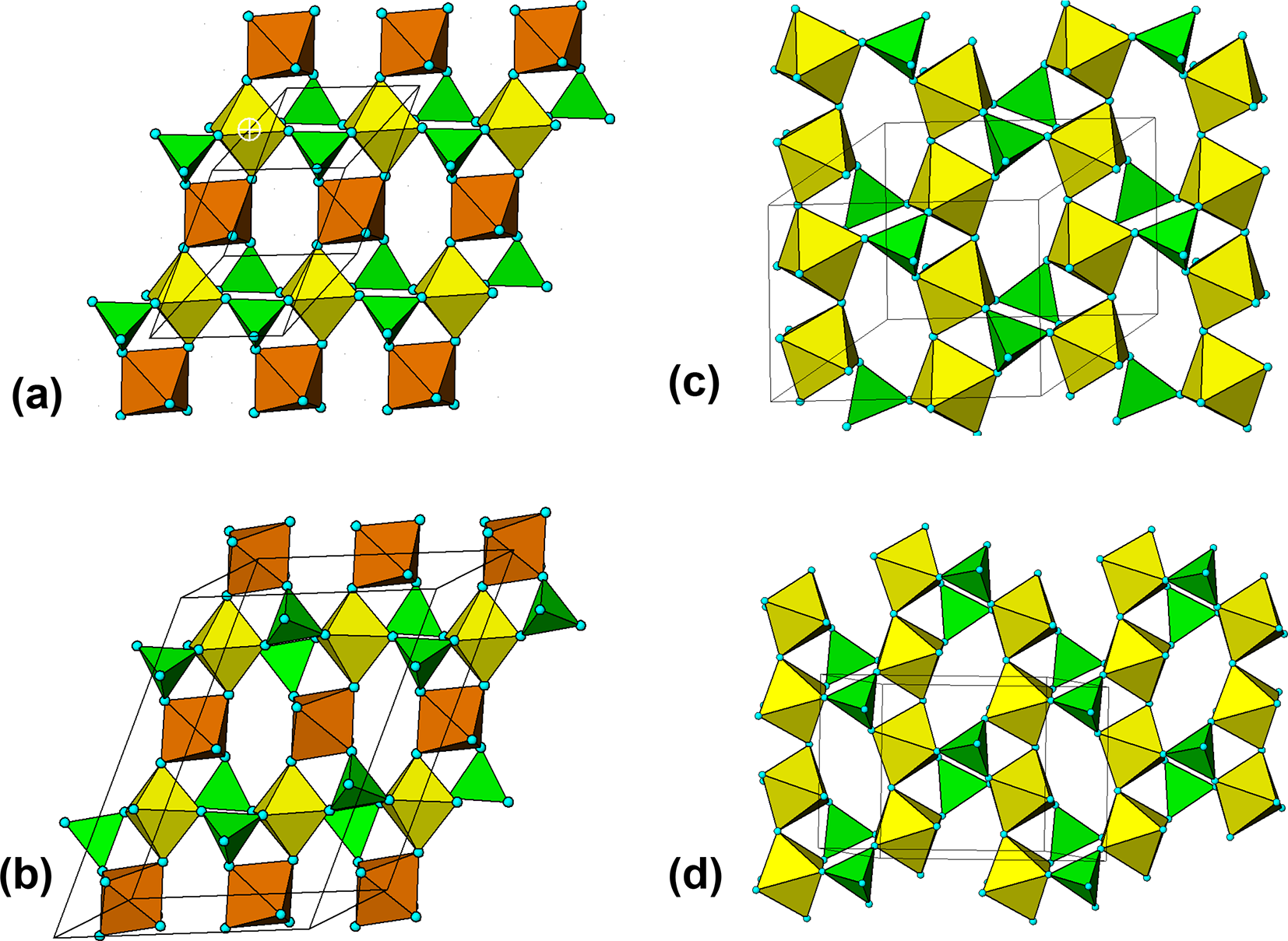
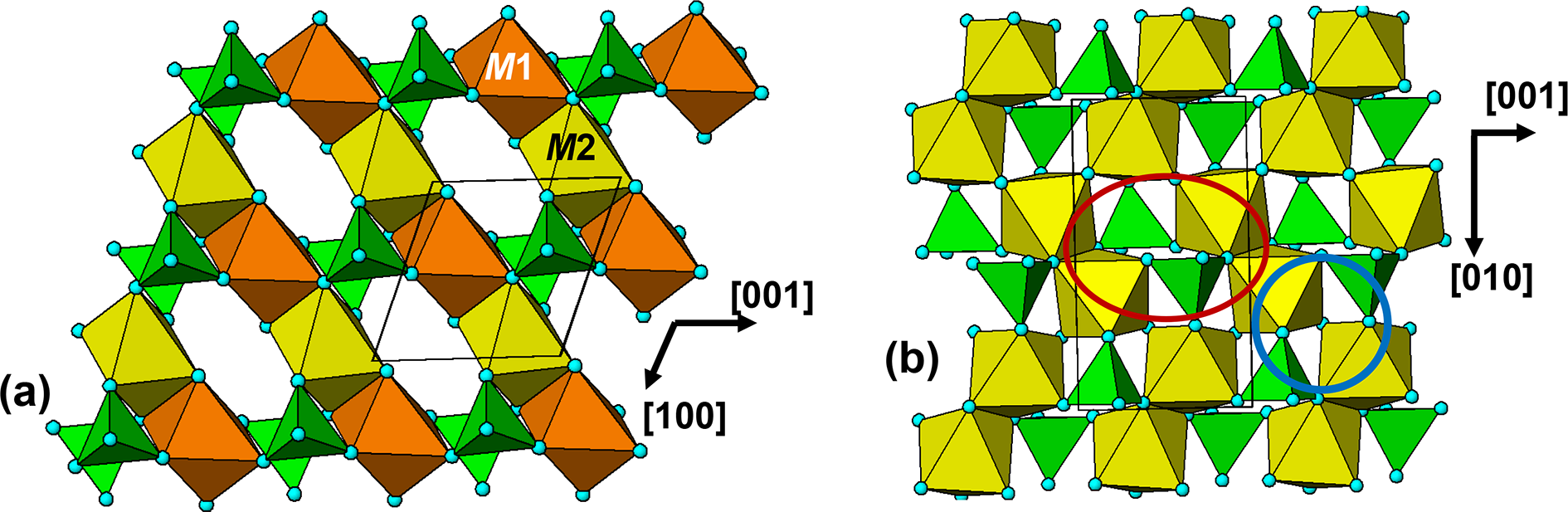
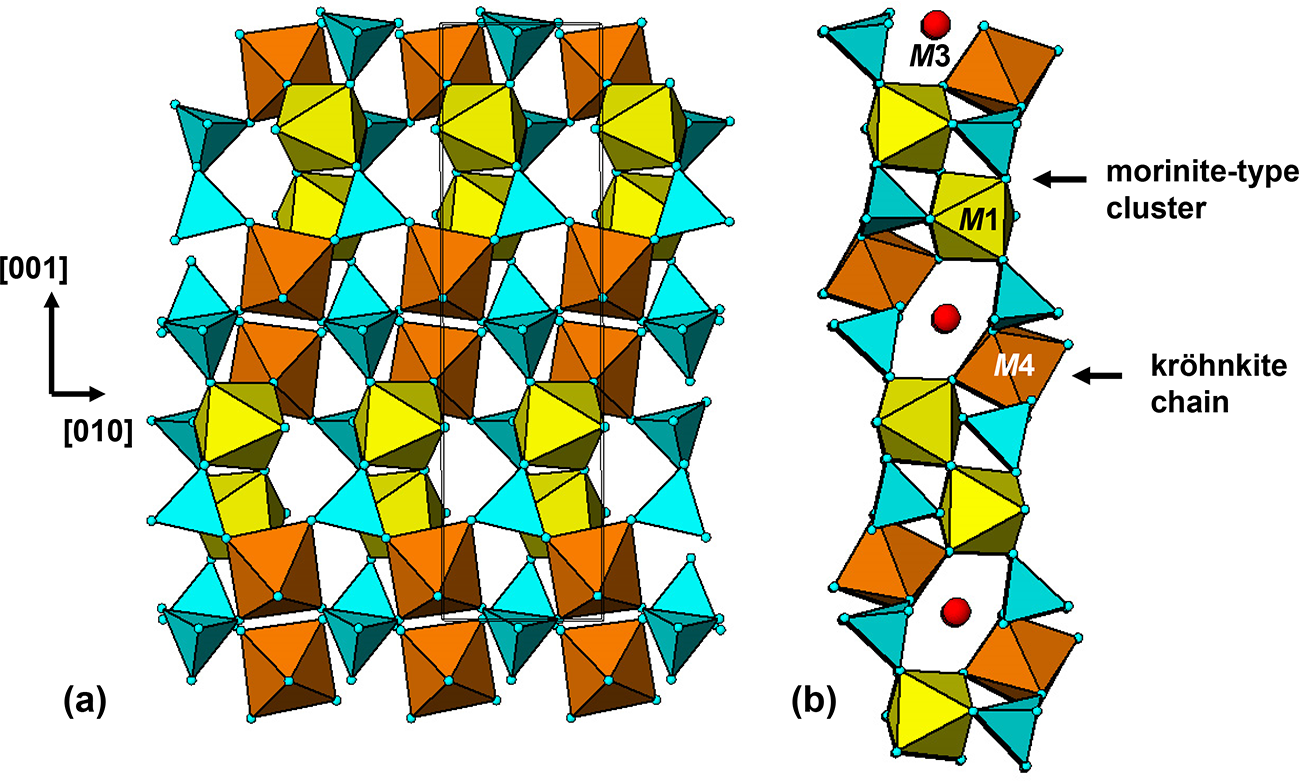
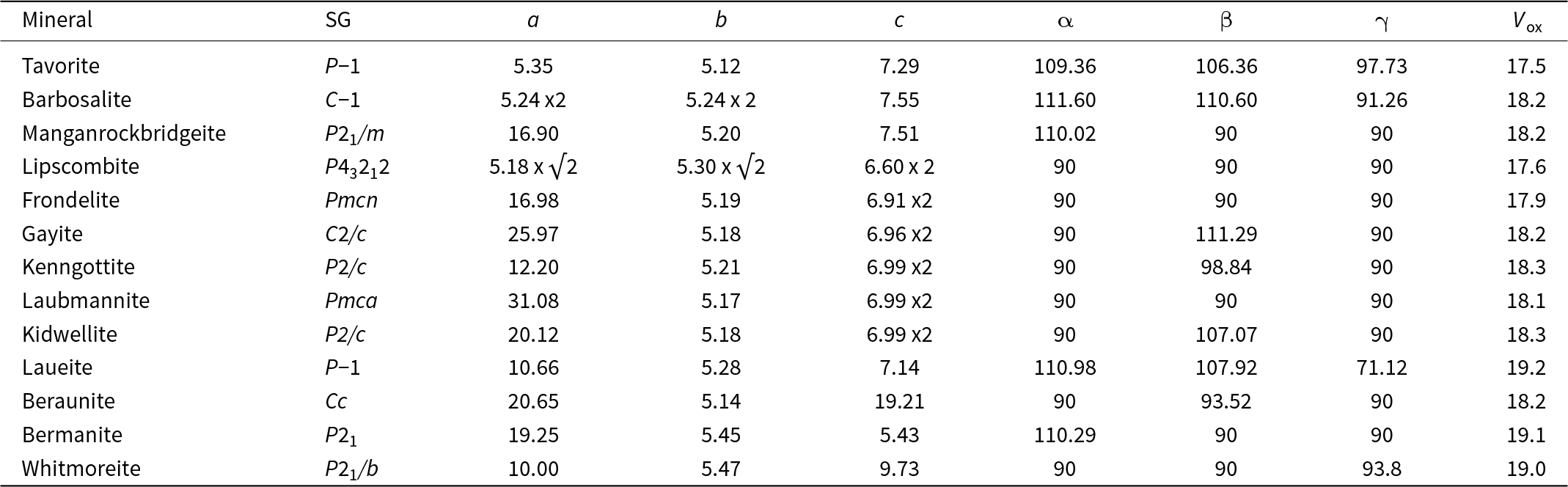
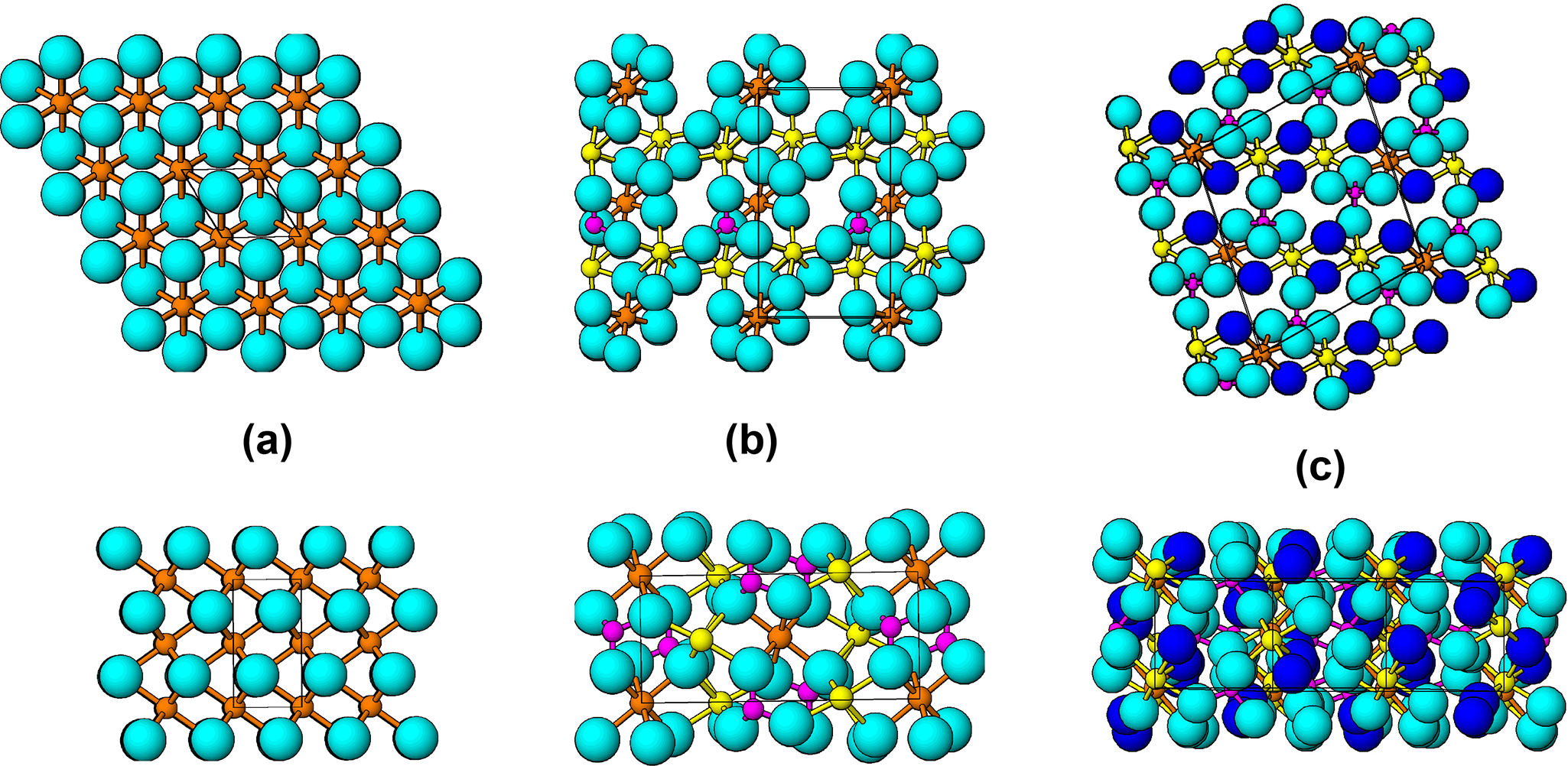
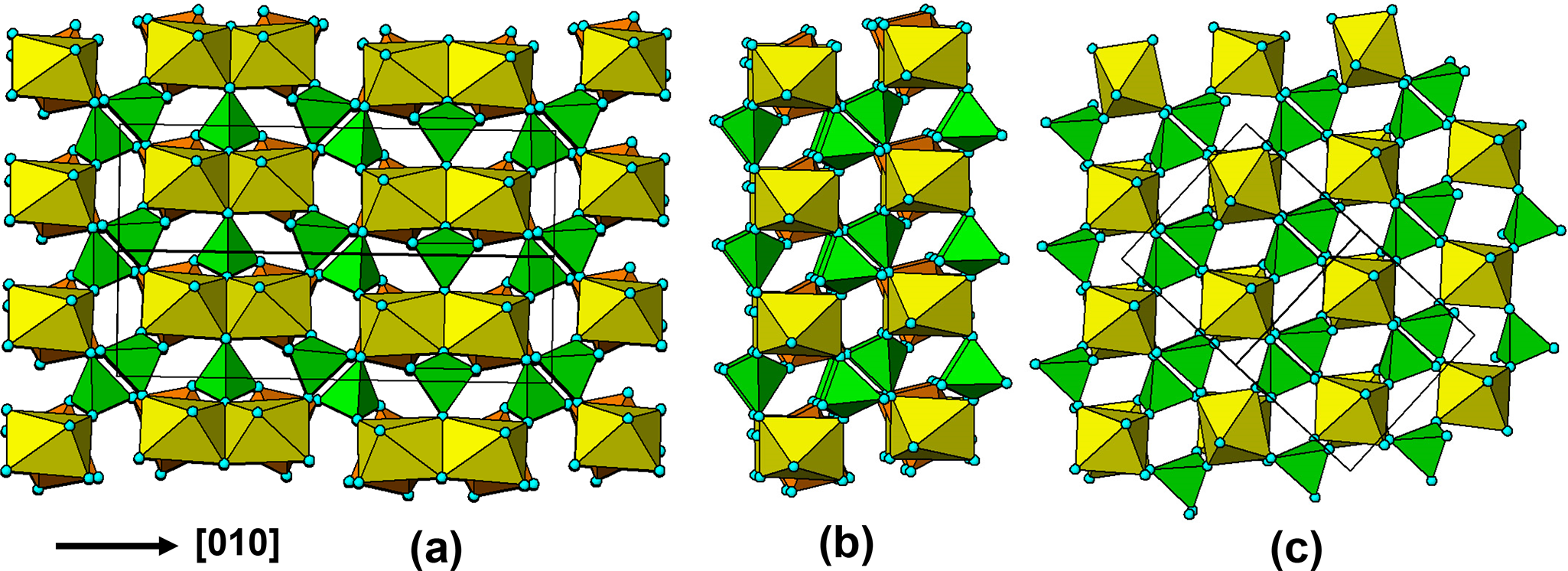
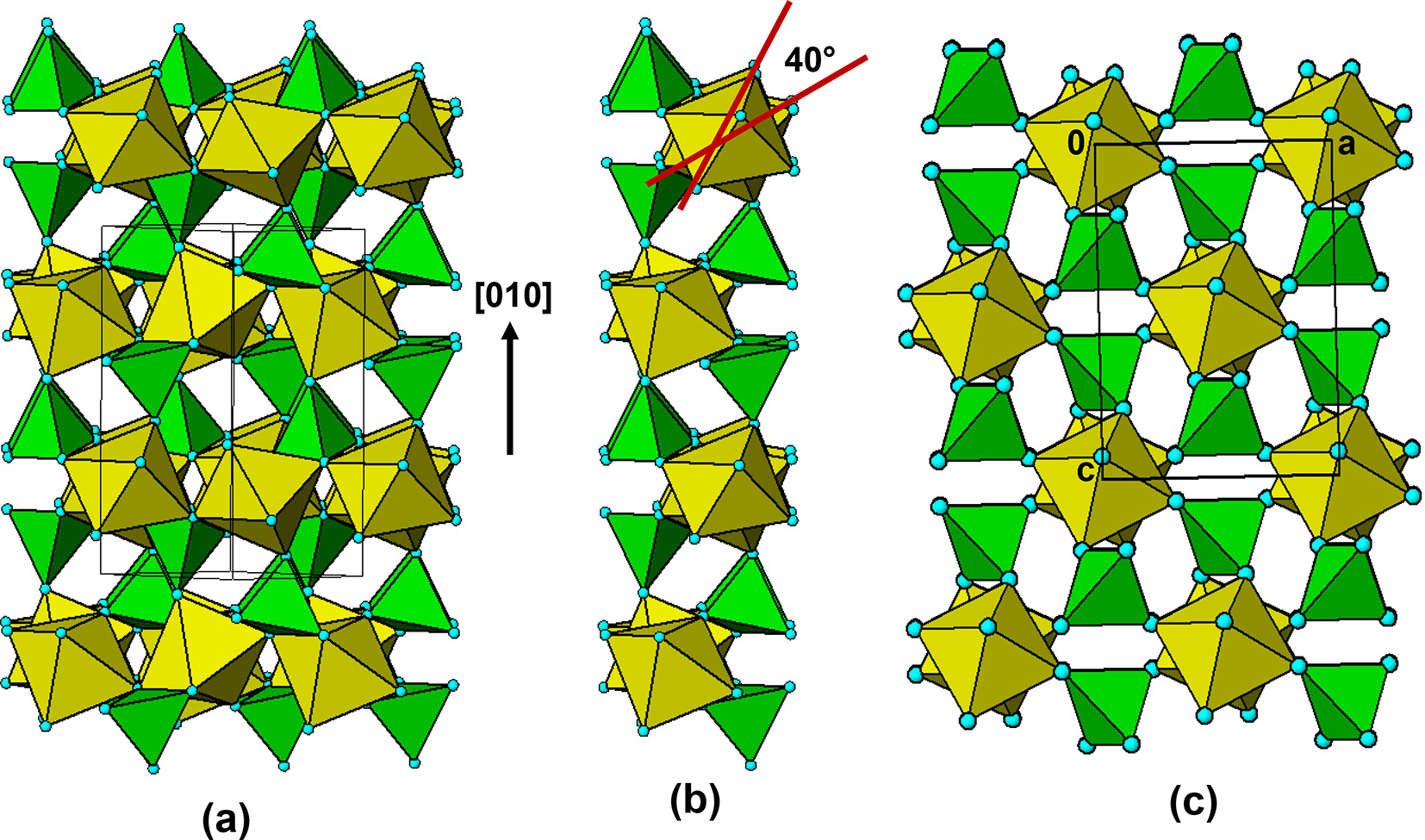


 Open access
Open access