

1. Introduction
This paper details the True Lifecycle Approach (“TLA”) towards governing artificial intelligence (“AI”) in health care. The TLA was first articulated in an earlier perspective paper, and this work builds on that proposal, providing greater details about what the TLA entails in practice for governing health care AI.Footnote 1 The TLA is defined as a comprehensive framework for governing AI in health care, whether the device is used in diagnostics, monitoring, health care administration, or other health care-related applications with a core emphasis on embedding patient protections derived from universal principles of medical law and bioethics.Footnote 2
Providing holistic governance of AI is necessary in health care because existing governance structures do not properly regulate AI.Footnote 3 Where regulations do exist, they largely ignore the patient and fundamental protections afforded to them under principles of medical law and bioethics.Footnote 4 AI is not merely passive, like a blood test kit. AI can be adaptive, autonomous, and be more accurate than practicing physicians.Footnote 5 AI is not imaginary; it is being used in health care systems throughout the world today, making significant impacts.Footnote 6
One survey of hospitals in the United States in 2023 found that sixty-five percent of hospitals used AI.Footnote 7 The United Kingdom’s (“UK”) National Health Service (“NHS”) uses AI to analyze X-ray images (such as mammograms) to support radiologists in making assessments, or helps clinicians read brain scans more quickly.Footnote 8 In China, projections suggested that up to ninety percent of hospitals would use some form of AI by the end of 2025.Footnote 9 In the European Union (“EU”), eighty percent of hospitals are experimenting with AI or attempting to scale the technology across the entire enterprise.Footnote 10 AI is used to support patients in “virtual wards” to receive care at home rather than a hospital.Footnote 11 AI is embedded in wearable devices and is used to monitor patients remotely.Footnote 12 A considerable number of emergency departments throughout England have rolled out AI to identify at-risk patients to provide care at an early stage.Footnote 13
Undoubtedly, there will be more pros than cons for use of AI in health care.Footnote 14 It will unleash exponentially greater benefits for patients. However, given the scale of its deployment, harms and hurdles will occur somewhere, and the law must be equipped to deal with those issues when they arise. Yet, governance frameworks have not been established nor calibrated to meet the scale of the challenge.Footnote 15 Since approximately 2020, a pathway from “AI to Law” has emerged.Footnote 16 Legal scholars have identified key legal issues (primarily arising in tort law) but have found the law to not be fully equipped to deal with the challenges of AI.Footnote 17 At the same time, soft laws have emerged in the form of guidelines underpinned by ethics “principles.” For example, the World Health Organization (“WHO”), the Organization for Economic Development (“OECD”), and others have produced guidelines.Footnote 18 Hard law has only recently emerged in the form of the EU’s AI Act, and the revised product liability directive.Footnote 19
This process of “AI to Law” has resulted in important developments but they miss the mark for health care, for reasons explored in this paper. Namely, existing frameworks do not capture best practices for research and development (“R&D”) of AI. It is unclear what standards (if any) R&D teams are following for devices that will ultimately be used on patients or in health care systems more broadly.Footnote 20 Market approvals processes for AI systems, like those under the Food & Drug Administration (“FDA”), leave out certain devices that make it to market without adequate checks.Footnote 21 Downstream, hospitals are deploying AI without any coherent or clear framework globally.Footnote 22 Most developers and hospitals do not have AI committees to assess the impact of technology internally, but some are making effort, such as in Singapore.Footnote 23 On the legal side, the EU’s AI Act deals with risk mitigation of AI systems, but the governance protections have little relevance to health care.Footnote 24
That is not to say that there have been no advances in scholarship regarding governance frameworks. There are important contributions examined below that certainly advance literature, and more are coming. The first part of this paper argues that where frameworks have been discussed or developed, they are not holistic, often focusing on certain aspects of the AI lifecycle rather than the broad and more complex care machinery that operates around patients. Indeed, another significant omission is the almost complete lack of regard for the most important person in all these considerations — the patient. Humans are at their most vulnerable when faced with a health crisis. A clear and robust system should operate in the background to protect the patient when AI is used, and that system should be communicated to the patient in a simple format, with clear expectations concerning their rights, duties, and redress pathways where things go wrong.
As such, the TLA was proposed as a governance model to bring together the entire AI lifecycle in a holistic manner, embedding health care law throughout and putting patients first.Footnote 25 The TLA consists of three phases of governance at the (1) R&D phase of AI; (2) market approval of AI; and (3) post-AI implementation in practice.Footnote 26 Part two of this paper examines the TLA in detail, and Part three critiques its limitations and highlights areas for future development. The TLA may not be the best proposal for AI governance in health care, but it is the first comprehensive proposal that considers the full lifecycle of AI from R&D to post-implementation deployment with health care law and ethics at its heart.Footnote 27 The TLA may ultimately be rejected as a proposal, but the purpose here is to stir debate and encourage discussion in an area where solutions feel stagnant and discussions repetitive.
It should be noted that the idea for the TLA first emerged when exploring governance models for AI in health care in Qatar, the UAE, and Saudi Arabia.Footnote 28 A separate analysis revealed that each country has had important developments in each area of the AI lifecycle, and it was observed that bringing those separate developments together would represent the TLA that could offer a global governance model for AI in health care.Footnote 29 As such, examples from those countries are primarily given in part two of this paper, whilst being situated within broader developments globally. Examples in the law are also drawn from the largest AI markets in the world (the United States, China, and Europe, particularly in part one of this paper).
2. Assessing Existing Frameworks for Health Care AI
2.1. The Universality of Medical Law & Ethics
Conceptualizing governance of health care-AI is complex. That process first requires some fundamental soul searching about issues that are hotly debated. Should AI even be regulated? The United States perspective might argue that we should only regulate AI to a limited extent and encourage the free market to flourish.Footnote 30 The EU has gone in another direction — creating a behemoth-like regulation via the AI Act.Footnote 31 China opts for another model, characterized by state-centric control linked to state security, social stability, and industrial policy.Footnote 32 There is no correct approach. Much depends on the economic, social, and political context.
Dig deeper into the application of AI to a specific sector like health care, though, and such distinctions in philosophy dissipate (or at least they should). Health care law is underpinned by ethical and legal principles that are largely universal. The interpretation and application of those principles varies but the core principles and approaches are similar. For example, patient autonomy should be respected and operationalized through informed consent.Footnote 33 Of course, autonomy manifests in diverse ways. Informed consent processes in the West are more individualistic and view the patient as a consumer.Footnote 34 In China and the Middle East, informed consent is more familial, with procedures directly involving or devolving authority to family members.Footnote 35 While the manifestation of informed consent processes differs, the core point is that informed consent is a universal principle.
So too are other principles in medical law and ethics. Non-maleficence and beneficence stipulate that providers should not harm patients and act in their best interests.Footnote 36 These principles manifest through the law via the duty of care and malpractice liability under torts.Footnote 37 There are “no-fault” liability schemes, such as in New Zealand and the Nordic countries but those are in the minority, and in any case, those schemes exist to compensate for wrongs even if no one is held to be liable.Footnote 38
The confidentiality of patients must be upheld to preserve trust in the doctor-patient relationship, and for many other important reasons. Such protections are found in a plethora of sources, such as laws, guidance documents, data protection standards, and more. For example, the General Data Protection Regulation (GDPR) treats health data as data of a “special category” with regulations from other countries also treating health data in a similar manner.Footnote 39 That designation gives such data extra legal protection.Footnote 40 Confidentiality is addressed in case law. For example, in England, the courts have long recognized that “there is an abiding obligation of confidentiality as between doctor and patient.”Footnote 41 In China, statute obligates that “medical and healthcare institutions, and medical and healthcare professionals shall … protect patient[]” privacy.Footnote 42
There are also obligations that apply internationally through treaties. The International Covenant on Economic, Social and Cultural Rights (“ICESCR”) recognizes the “right of everyone to the enjoyment of the highest attainable standard of physical and mental health.”Footnote 43 That right to health should be exercised “without discrimination of any kind as to race, colour, sex, language, religion, political or other opinion, national or social origin, property, birth or other status.”Footnote 44 In other words, health care should be delivered equitably and without discrimination.Footnote 45
Underpinning all the above is that there should be respect for human dignity, as recognized under the Universal Declaration on Bioethics and Human Rights (from UNESCO).Footnote 46 “The interests and welfare of the individual should have priority over the sole interest of science and society.”Footnote 47
Health care presents a universally consistent legal and ethical foundation for the protection of patients. The extent to which governments may seek to regulate AI will always run up against these expectations when AI systems are intended for use in health care; it is for this reason that any governance framework must encapsulate those universal protections and expectations.
Another layer that is important is the existing national regulatory processes for medical device approvals to market. The FDA is the most well-known, but many such agencies exist, such as the Ministry of Food and Drug Safety (“MFDS”) in South Korea, or the Saudi Food and Drug Authority (“SFDA”), discussed later in this paper. These regulators have separate technical rules to determine whether medical devices can be brought to market.Footnote 48 The WHO has a global benchmarking tool (“GBT”) for evaluating national regulatory systems of medical devices, but very few countries meet the highest standards set by the tool, likely because the majority of countries do not have a ‘mature” infrastructure for medicine and vaccines.Footnote 49
2.2. Why AI Presents New Challenges in Health Care
Given that medical law and ethics principles are largely universal, and that there are at least some regulators, like the FDA, that already have processes for approving medical devices to market, it may be argued that the existing governance paradigm is sufficient (with some adaptations) when applied to AI devices used in health care. That proposition is true to an extent, but AI stretches the law further than its design.
AI devices can make decisions and interpret information without human input, which raises questions about legal accountability in medical decision-making.Footnote 50 AI devices have “adaptive” algorithms that learn and adapt over time to the data processed.Footnote 51 Medical device regulations have typically covered “locked” algorithms, meaning that the same result is produced from the same input every time it is used.Footnote 52 It is harder to regulate adaptive algorithms because the device may produce different outputs than expected at the time of regulatory approval.Footnote 53 The law has not been equipped to deal with that challenge.
Further, since adaptive algorithms can “drift” over time without any deliberate changes from the manufacturer,Footnote 54 the standard of care may become more contested where AI proves to outperform clinicians across a broad range of clinical practice. If that becomes the case, then the courts will be asked to decide whether the standard of care itself should be changed to align with AI outputs.Footnote 55 In other words, could we shift to a system where AI recommendations are the standard of care?
Explainability presents another challenge.Footnote 56 Informed consent requires that patients are able to understand all relevant information about diagnostic processes or proposed treatments for their care, and then voluntarily agree to that treatment.Footnote 57 To understand all relevant information, the health care professional should be able to provide an explanation to the patient in terms they can understand.Footnote 58 Challenges arise if AI is used to provide treatment recommendations, but AI cannot provide an explanation for why it made a specific recommendation.Footnote 59 Even if AI can provide an explanation, it cannot be trusted because AI often “hallucinates” and invents evidence to support its recommendations.Footnote 60 If patients rely on such hallucinations to make a decision, then the threshold for proper informed consent cannot be met because their decision may be based on a falsehood. This is not an imagined concern. AI has already made harmful recommendations that may have increased the risk of suicide in those following its advice.Footnote 61
Of course, there is case law about whether a particular medical interaction involved adequate informed consent, but those cases involve investigations about who said what, and one can ask the doctor why they made a particular recommendation.Footnote 62 For AI, even computer scientists can struggle to understand why AI made a particular recommendation.Footnote 63
Liability becomes further muddied in those waters. For example, there is the “learned intermediary” principle, where a manufacturer has a duty to warn a patient about the risks of a medical device, by providing adequate warnings to the prescribing doctor instead of to the patient.Footnote 64 Price has queried whether that doctrine must “bow to the recognition that doctors cannot understand all the technologies they use or the choices such technologies help them make when they are not provided the needed and/or necessary information?”Footnote 65 What if a clinician is overruled by AI? How is causation established in this arena of diffused responsibility when the traditional patient-doctor accountability loop is altered?
Data vulnerabilities present a double-edged legal challenge in AI. Health care data could be centralized (for example, within a hospital system), and AI could analyze data within that system only.Footnote 66 However, that single point of entry creates vulnerabilities for adversarial attacks.Footnote 67 At the same time, systems could be decentralized, with data stored in multiple locations across borders. AI processing could even be undertaken in the cloud. But this latter approach presents legal compliance issues.Footnote 68 Many companies are unclear about their obligations in this landscape.Footnote 69
Bias and inequity also present new challenges. Algorithms may be trained on inherently biased data, which could lead to useless or harmful outputs.Footnote 70 The law has not risen to this challenge. A major query is how bias and discrimination can be dealt with under equality laws.Footnote 71 This also invites the question about whether product liability laws or data protection laws are also applicable. There is also the potential for conflict between different areas of law. Manufacturers can refuse to disclose their algorithms for inspection under intellectual property law.Footnote 72 These tensions have played out in recent EU case law: the Court of Justice of the European Union (“CJEU”) held that the courts may have to balance trade secret protections against a data subject’s rights to access “meaningful information” about the logic involved in the automated decision of an algorithm.Footnote 73 Indeed, much remains unsettled in this space.
Finally, there is simply the issue of over-reliance on AI systems by medical professionals subordinating their own clinical expertise and decision-making.Footnote 74 AI models that are over-generalized may not be useful to a specific patient, but the clinician may nonetheless follow its recommendation.
Ultimately, it is inaccurate to argue that AI does not present new challenges for the patient. Indeed, AI presents new and pressing challenges that the law must deal with.
2.3. Are current governance frameworks sufficient?
There has been some important framing scholarship for identifying the legal issues noted above. For example, Gerke, Minssen, and Cohen highlighted legal and ethical challenges in the field in 2020.Footnote 75 They noted the ethical challenges of informed consent, safety and transparency, algorithmic fairness and bias, and data privacy.Footnote 76 Legal challenges highlighted were safety and effectiveness, liability, data protection and privacy, cybersecurity, and intellectual property.Footnote 77 Solaiman and Cohen’s book on “Health, AI and the Law,” included more detailed analyses of the legal issues, which they framed as algorithmic discrimination and health equity, data protection, data security, liability, informed consent, and intellectual property.Footnote 78 Solaiman and Cohen’s book also charts the governance developments globally, revealing that very little progress has been made on the underlying legal issues for health care.Footnote 79
However, there have been developments in three areas explored in the passages below that are consistently characterized or premised on a total lifecycle approach. First, at the global governance level through the WHO. Second, through the development of medical device regulations in some countries like the United States, Singapore, South Korea, China, and other countries. Third, through the passage of the EU’s AI Act. The main developments are outlined below and contrasted with the TLA proposed later in this paper.
2.3.1. The WHO and other bodies
In 2023, the WHO published Regulatory Considerations on Artificial Intelligence for Health. Footnote 80 The publication set out six broad topic areas: documentation and transparency, risk management, intended use and validation, data quality, privacy and data protection, and engagement and collaboration.Footnote 81 Central to the document was the Total Product Life Cycle (“TPLC”). The WHO emphasized:
A [TPLC] should be considered throughout all phases in the life of an AI system, namely: pre-market development management, post-market surveillance and change management. In addition, it is essential to consider a risk management approach that addresses risks associated with AI systems, such as cybersecurity threats and vulnerabilities, underfitting, algorithmic bias etc.Footnote 82
The WHO seeks to promote this approach with documentation and oversight that spans pre-market, deployment, and post-market phases of AI.Footnote 83 The focus is on holistic risk management through the use of a quality management system, development practices for AI systems, cybersecurity, performance evaluation, and more.Footnote 84 In this regard, the WHO’s recommendations are comprehensive, with a global outlook that aims to build shared regulatory expectations.
While this approach is important and detailed, it remains primarily technical and regulatory in nature. The recommendations do not embed accountability and redress mechanisms focused on patients, nor do they explicitly integrate medical law doctrines, or local and ethical considerations. By contrast, the TLA developed in this paper seeks to develop a more patient-centric governance framework.
2.3.2. The FDA’s Medical Device Regulations & Related Agencies
Aside from the WHO’s framework, medical device regulators have also developed rules to account for AI. The United States FDA is the most prominent example with its TPLC approach that is “focused on device oversight throughout the product’s life cycle — from device design and development to real-world use of the device.”Footnote 85 The TPLC seeks to promote transparency, efficiency, and agility of FDA oversight by making information sharing between regulators easier, compressing levels of review, and helping employees develop a deeper view of device safety.Footnote 86 It is emphasized that the suite of guidance documents produced by the FDA are of great import and there is some crossover in underlying philosophy between the TLA, but the focus is different for reasons unpacked below.
In recent years, the FDA has adapted the TPLC to AI-enabled devices. In 2025, it issued draft guidance on AI-enabled device software functions.Footnote 87 That guidance includes detailed recommendations on premarket submissions, with requirements on quality systems, risk assessments, data management, and performance validation.Footnote 88 It also discusses post market monitoring plans to detect data drift and to respond to safety issues once in use in the real world.Footnote 89 In 2021, the FDA also issued the Good Machine Learning Practice (“GMLP”) principles alongside Health Canada and the UK’s Medicines and Healthcare products Regulatory Agency (“MHRA”).Footnote 90 The GMLP emphasize the importance of leveraging multi-disciplinary expertise throughout the TPLC, implementing good software engineering and security practices, that clinical study participants and data sets are representative of the intended patient population, and that deployed models are monitored for performance, among others.Footnote 91
The focus of the GMLP on such issues squarely confronts the concern about bias and discrimination in the use of AI in health care. The FDA defines AI bias as follows:
AI bias is a potential tendency to produce incorrect results in a systematic, but sometimes unforeseeable way, which can impact safety and effectiveness of the device within all or a subset of the intended use population (e.g., different healthcare settings, different input devices, sex, age, etc.).Footnote 92
Thus, in monitoring a device post-deployment, appropriate controls should be in place to manage risks of overfitting, unintended biases, or degradation of the model that may impact the safety and performance of the model.Footnote 93 The FDA recommends that developers use “unbiased, representative training data for models” to avoid “perpetuating biases or idiosyncrasies from the data itself.”Footnote 94 While acknowledging that mitigating bias “may be difficult to eliminate completely” it recommends that, as a starting point, “validation data sufficiently represents the intended use (target) population of a medical device.”Footnote 95
In 2024, the same agencies also issued transparency principles for machine learning-enabled devices.Footnote 96 The principles are relevant to those who use the devices such as health care professionals, patients, and caregivers.Footnote 97 They note that “transparency is essential to patient-centered care and for the safety and effectiveness of a device” and that “logic and explainability are aspects of transparency.”Footnote 98 They recommend clear communication of the intended use, limitations, and performance to end users of AI devices.Footnote 99 The transparency principles allude to the informed consent process but do not address it directly. For example, they note that “effective transparency that helps parties make informed decisions can help control risks.”Footnote 100 However, this is never explicitly tied to principles of health care law and ethics, and the expectations and protections that patients may have in this regard.
Finally, in 2024, the same agencies developed quite innovative policies on Predetermined Change Control Plans (“PCCP”).Footnote 101 PCCPs are plans proposed by manufacturers of AI medical devices that specify planned modifications to devices, protocols for implementing and controlling those modifications, and include an assessment of the impact of modifications.Footnote 102 The PCCP principles are particularly focused on developing Principle 10 in the prior GMLP on monitoring for performance and managing re-training risks for deployed models.Footnote 103 Principle 10 stipulates that deployed models have the capability to be monitored in the “real world” with a focus on maintained or improved safety and performance.Footnote 104 In this regard, five guiding principles are developed.
First, any changes that a manufacturer intends to implement should be “focused and bounded.”Footnote 105 Changes should be limited to modifications within the intended use or intended purpose of the original device, and there should be plans in place to “safely modify the device within the bounds of the PCCP.”Footnote 106 Second, the intent, design, and implementation of a PCCP should be driven by a risk-based approach.Footnote 107 Doing so strengthens the value and reliability of a PCCP.Footnote 108 Third, PCCPs should be evidence-based, following scientifically and clinically justified methods and metrics used to measure device performance.Footnote 109 Fourth, PCCPs should have ongoing transparency to users and stakeholders. This helps those users to stay aware of the device’s performance before and after changes are implemented, which is crucial for monitoring, detection, and providing an appropriate response to deviations in device performance.Footnote 110 Fifth, the PCCP should be created from a TPLC perspective.
The guiding principles concerning PCCPs are by no means settled. The agencies intend that their principles lay a foundation for PCCPs and encourage international harmonization, and they encourage ongoing feedback in this space because PCCPs may be developed and implemented in different ways in different jurisdictions.Footnote 111
The collection of guidance documents released in recent years are reflected in the 2025 draft guidance noted above.Footnote 112 For example, that guidance states that “manufacturers should have a postmarket performance monitoring plan to help identify and respond to changes in performance in a postmarket setting.”Footnote 113 In doing so, manufacturers will “reduce uncertainty” and “support the FDA’s evaluation of risk controls.”Footnote 114 Such continuous monitoring is especially important because “models are highly dependent on the characteristics of data used to train them, and as such, their performance can be particularly sensitive to changes in data inputs.”Footnote 115 Existing obligations for monitoring devices remain, such as reporting serious injuries or malfunctions.Footnote 116 The distinction for AI is that the FDA emphasized continuous monitoring, which is an extension of the TPLC’s philosophy that governance does not end with device approval but is an ongoing process post-approval.Footnote 117 Embedded within that paradigm is a more concerted focus on issues that particularly matter in the AI space that the law has yet to properly grapple with, such as bias and discrimination.
Despite these aims, the FDA’s post-market surveillance framework for AI devices has been strongly criticized. The FDA has a repository for post-market surveillance reports called the Manufacturer and User Facility Device Experience (“MAUDE”) database.Footnote 118 This is part of the reactive element of the governance framework meant to ensure that adverse events and malfunctions are submitted by manufacturers and others. It has been argued that this system is “insufficient for properly assessing the safety and effectiveness of AI/ML devices.”Footnote 119 Scholars have assessed how well equipped the long-standing system is at “capturing the kinds of issues and problems that are especially likely to arise from AI/ML devices.”Footnote 120
For example, more than ninety percent of AI/ML device reports were labeled as “malfunctions” but the information given does little to reveal the true severity of the problems.Footnote 121 This, it was noted, was “emblematic of more general issues” as to why the reporting structure is “not fit for purpose for evaluating AI/ML devices.”Footnote 122 Those issues include the sheer extent of missing data that makes it difficult to study the safety of AI/ML devices, inadequate event classification which reveals a disconnect between the challenges arising in practice and the constraints of the categorization in the system, and the severity of the risk being unknown.Footnote 123
It is not only structural problems that are of concern. Conceptually, the FDA’s TPLC is designed to deal with approving medical devices, with a focus on monitoring safety and effectiveness throughout the device’s life cycle.Footnote 124 It will be seen below how the TLA is a broader and deeper framework that seeks to consistently embed patient protections arising from medical law and ethics.
2.3.3. The EU’s AI Act
The AI Act should be understood in a quite different context to the FDA guidelines. The most significant difference is that it is binding “hard law,” as opposed to the FDA guidance documents which are technically non-binding, and remain under consultation.Footnote 125 The AI Act is also not focused on any sector but is a broad regulatory scheme premised on classifying the risk of AI systems and through that classification imposing certain requirements on manufacturers and others before permitting those devices to market.Footnote 126 Nevertheless, the AI Act is premised on promoting the uptake of AI while also ensuring a “high level of protection of health, safety, [and] fundamental rights as enshrined in the Charter of Fundamental Rights of the European Union.”Footnote 127 That articulation does broadly bring health within the explicit aims of the Act.
The Act classifies AI based on risk. AI devices such as social scoring systems are deemed to pose an unacceptable risk and should be prohibited from the market.Footnote 128 AI systems that pose a minimal risk, such as wellness apps on an app store are unlikely to be captured by the Regulation.Footnote 129 Most medical devices will fall under the “high risk” category and will be subject to certain checks before being permitted to market.Footnote 130 The Act incorporates the Medical Devices Regulation (“MDR”)Footnote 131 and the in vitro Diagnostic Medical Devices Regulation (“IVDR”)Footnote 132 into its annexes.Footnote 133 Manufacturers must comply with safety and other requirements of the MDR or IVDR, and also the specific requirements of the AI Act where AI is incorporated into the device.Footnote 134
There is a “conformity assessment” procedure for demonstrating whether the requirements for high-risk AI systems have been fulfilled.Footnote 135 Conformity assessments are rather convoluted processes involving a third party to obtain a “CE” mark.Footnote 136 The assessments are already required under the MDR/IVDR and have been criticized for providing weak protections for human health and safety.Footnote 137 The process ultimately amounts to self-certification, with some noting that “it is seriously questionable whether reliance on self-certification provides meaningful legal assurance that the requirements to obtain a CE mark in relation to high-risk AI systems are properly met.”Footnote 138
Aside from that specific conformity assessment procedure, the AI Act has some structural similarities to the FDA approach. It requires that a risk-management system should be implemented that is continuous, iterative, and runs “throughout the entire lifecycle of a high-risk AI system.”Footnote 139 It emphasizes post-market monitoring of devices to identify any need to immediately apply corrective or preventative actions.Footnote 140 A post-market monitoring system should actively and systematically collect, document, and analyze data to evaluate such compliance.Footnote 141
The AI Act also covers biases under its data requirements, requiring that training, validation, and data sets shall be subject to practices that include an “examination in view of possible biases that are likely to affect the health and safety of persons, have a negative impact on fundamental rights or lead to discrimination prohibited under Union law, especially where data outputs influence inputs for future operations.”Footnote 142
Appropriate measures must be taken to “detect, prevent, and mitigate” those possible biases.Footnote 143 In this manner, there are similarities to other lifecycle approaches but the explicit focus on health care is limited. Joint guidance issued by the EU’s Medical Device Coordination Group (“MDCG”) and the AI Board (an advisory body created by the AI Act), notes that the MDR/IVDR address general medical device software risks but “they do not explicitly address risks specific to AI systems. The AIA complements the MDR/IVDR by introducing requirements to address hazards and risks for health, safety[,] and fundamental rights specific to AI systems.”Footnote 144
The guidance emphasizes that the obligations under the AI Act complement the other regimes but are separate from them.Footnote 145 Compliance with one set of requirements does not equate to compliance with the other and both schemes must be complied with, but it is likely that compliance tasks will be integrated by developers in such a way to avoid duplicity of efforts, something that is explicitly encouraged.Footnote 146
Despite some areas of structural overlap between the AI Act and the FDA’s TPLC, they are ultimately different in design. The AI Act is a comprehensive binding governance model that defines rules, responsibilities, and creates new institutions with oversight. The FDA’s TPLC is more of a conceptual approach within existing legal frameworks. Importantly for the TLA proposed in this paper, both the AI Act and TPLC have a lifecycle approach and focus (to a certain extent) on ethical safeguards through their focus on data bias (for example), but both omit other patient-centric elements.Footnote 147 Neither system directly or explicitly incorporates the suite of health care law concerns identified in the scholarship in a comprehensive and consistent manner nor offer realistically actionable redress mechanisms for patients if things go wrong.
The AI Act has already been criticized elsewhere as being “flawed.”Footnote 148 The high-risk category has been labeled as “deficient” for patients because it limits their power of redress.Footnote 149 Individuals have the right to complain about infringements in the Regulation to the “market surveillance authority” but this ultimately bears little connection to patient’s rights in the health care system.Footnote 150 Indeed, Ebers argues that “one of the most crucial points” about the AI Act is that it provides “limited individual rights” beyond the ability to lodge a complaint or the right to an explanation of individual decision-making.Footnote 151
One of the fundamental aims of the AI Act was to build trust but it has been argued that it “falls short of connecting its premise on trust with its risk-based approach.”Footnote 152 Indeed, the rubrics discussed above are not sufficient for ensuring that patients are protected from potential AI harms.
2.3.4. Appraising the Existing Lifecycle Approaches
There is a growing body of research that underscores how those governance frameworks are fragmented and incomplete despite being a step in the right direction. Those gaps mean that patients may be exposed to harms, potentially undermining the very trust those frameworks seek to build.
Regarding the EU regime, Ebers notes that “neither the EU’s Medical Device Regulation nor its [AI] Act adequately address the risks to patient health and safety that arise in many situations where AI systems are used in the healthcare and nursing sector.”Footnote 153 The MDR only applies to tools that are explicitly marketed with a medical purpose, and it is concerning that labeling a product as general “wellness” would avoid oversight.Footnote 154 Ebers argues this is “problematic” given that there are no other rules to protect users from such products.Footnote 155 Further, even where the MDR applies, it only covers the manufacturer to ensure safety but omits health care professionals and others.Footnote 156 While the AI Act does create some obligations, it “does not establish duties to individuals affected by any AI system (patients, care recipients or others).”Footnote 157
The concerns are not limited to the lack of coverage for individual rights but extend to fundamental flaws in the regulatory scheme itself as intended. There are concerns from manufacturers that such regulations will place an undue burden financially and may pose an existential threat to their business.Footnote 158 There are already not enough notified bodies to undertake third party conformity assessments under the MDR, causing delays and backlogs for devices to come to market.Footnote 159 This will become worse under the AI Act given that more products will be captured by its scheme.
Scholars on all fronts have argued that patient-centered governance of AI should engage with fundamental rights more directly than existing schemes. Ho has praised the “adoption of a total device/product-specific lifecycle approach (rather than one that is point-in-time)” as being “more collaborative and participatory in nature, and anticipatory in character.”Footnote 160 However, he argues that even such lifecycle approaches should be grounded in an internationally recognized “human right to science” (HRS).Footnote 161 The HRS is a rights-based approach that obligates regulators to ensure that everyone can “participate in and enjoy the benefits of scientific and technological progress.”Footnote 162 In this manner, regulation should not merely be about risk mitigation but should also encompass public participation and equitable access to new innovations. Ho argues that by applying a HRS, patients can benefit from continuous improvements in AI devices.Footnote 163 A lifecycle regulatory model incorporating HRS would be both participatory (by involving stakeholders beyond manufacturers and regulators), and anticipatory by adapting governance as AI evolves.Footnote 164
Ho rightly notes that traditional static forms of medical device regulations fall short of HRS goals because “patients will not have the opportunity to benefit from” AI improvements.Footnote 165 Scientists will also “not be able to push forward AI science” because AI cannot be limited to trials undertaken in strictly “controlled conditions.”Footnote 166 The rights based approach he argues for is aimed at continuously balancing the interests of innovation with safeguarding patients.Footnote 167 Nevertheless, he also cautions against lifecycle governance approaches becoming overly bureaucratic and intrusive on scientific activities.Footnote 168 He notes that HRS provides “instructive guidance” in that the “enjoyment of the benefits of science” is contingent on protecting the freedom that is indispensable for research and the rights of developers, even where accountability is required.Footnote 169 Ultimately, Ho’s framework (while conceptual) goes further than the FDA’s risk based model, or the EU’s AI Act — with its abstract pledge to uphold fundamental rights and build trust in AI — by proposing to make such rights a fundamental part of the process.
Elsewhere, Cohen et al. have argued that patients should be empowered through the creation of an appeals process for AI decisions.Footnote 170 Looking to legal appeals processes, they suggest that there should be a structured way to appeal an important AI decision, such as the prioritization of care for one patient over another, or a diagnosis that the patient or doctor doubts.Footnote 171 Human expert judges could be included in those appeals processes to review AI decisions. A human reviewer could consider case specific information and provide more nuanced clinical, moral, or legal reasoning — serving as an error correction check on AI.Footnote 172 This approach resonates with care that is patient-centered because patients could seek a human review of an AI decision. While the EU’s AI Act requires human oversight, that does not translate into a specific right for patients to have a review.
The call to follow a true lifecycle mindset is echoed in other scholarship going as far back as 2020. For example, Gerke et al. argued that regulators like the FDA need to “widen their scope” for assessing systems.Footnote 173 The authors noted that the shift in perspective from a product to a system view is necessary to maximize the safety and efficacy of AI in health care.Footnote 174 To achieve this through transitional ‘first steps’ (rather than a full system model) they proposed examining how healthcare professionals react to AI and require training.Footnote 175 The ‘full system approval’ could be expanded to include workflow integrations, hospital authorizations (specific to the particular hospital), liability considerations, insurance, and the ongoing reevaluation of algorithms.Footnote 176 Whilst the FDA does not regulate the practice of medicine, the authors argued that it could require AI developers to set up training programs for their product.Footnote 177 They also argued for ongoing system monitoring, periodic retraining, software and usage inspections, and reviews of aggregate usage statistics (suggestions that are beginning to form part of the post-market monitoring thinking today).Footnote 178 Thus, even in 2020, these ideas illustrated how a regulator like the FDA might operationalize a lifecycle approach by going beyond the pre-market evaluation of devices and towards the ongoing interactions between AI and its users post-market. Those recommendations align with Ho’s more recent HRS arguments.Footnote 179 They converge on the idea that regulating health care AI requires continuous governance.
Hassan et al. have also argued for an “adoption-centered governance framework” that “covers the entire cycle of an AI system, from concept through to sustainability.”Footnote 180 That framework is premised on the creation of a governance committee, which applies “gated mechanisms” at various stages of AI development.Footnote 181 This approach incorporates an analysis of concerns surrounding “bias, equity, transparency, ethics (of AI), explainability, data handling, and safety.”Footnote 182 While their research does not discuss the FDA or the law, the approach somewhat reflects the FDA’s TPLC, but appears to include more ethical checks.Footnote 183
Ultimately, the literature is coalescing around a lifecycle approach towards governing AI that focuses on the patient, the continuous evaluation of AI systems, and the adaptability of design following market deployment. The TLA broadly captures these aims as will be seen below. Like Ho’s HRS framework, the TLA requires ongoing engagement and participation from all stakeholders, especially the patient.Footnote 184 And, like Gerke et al.’s system approach, the TLA emphasizes the monitoring of AI in real world clinical settings while updating that oversight as the systems evolve.Footnote 185 However, it also differs from those frameworks. Thus, Ho’s framework focuses on high-level human rights obligations, but the TLA translates similar values into specific requirements and checkpoints spanning the AI lifecycle.Footnote 186
The next section details how the TLA can build on these insights in practice. This proposal may not solve every problem, but if it at least spurs fresh discussions, we can come a step closer to resolving the health care AI governance puzzle.
3. The TLA
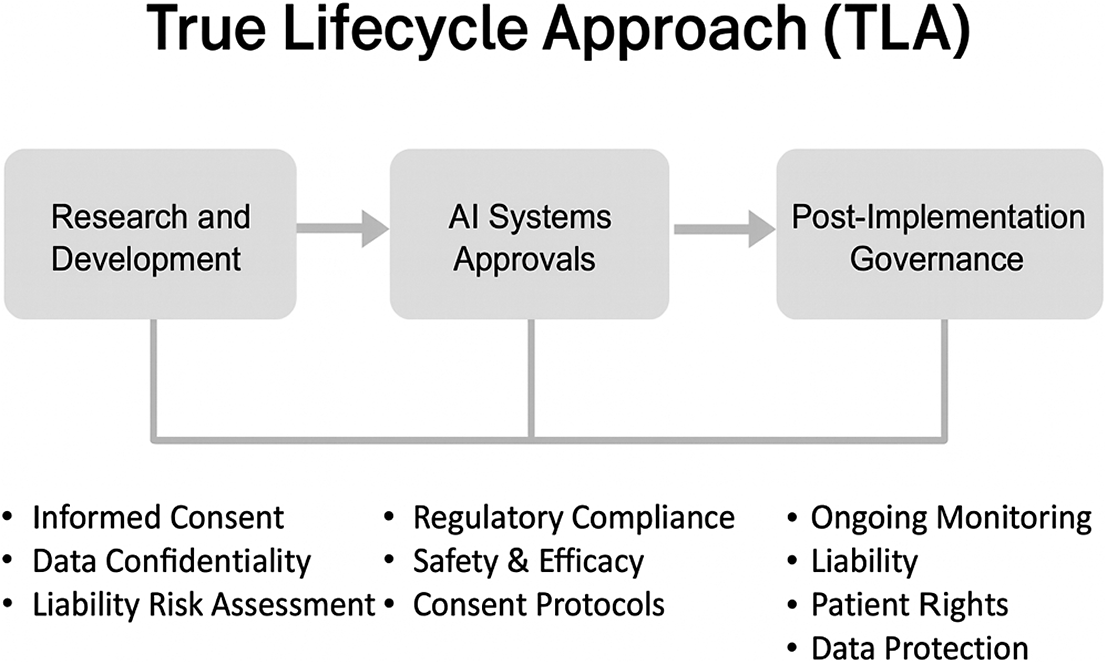
The concept of the TLA builds on existing research. In 2024, Solaiman, Bashir, and Dieng outlined how health care AI is governed in Qatar, Saudi Arabia, and the UAE (with a focus on Dubai and Abu Dhabi).Footnote 187 Their research revealed how AI is governed in those countries and highlighted that each jurisdiction was governing different aspects of AI.Footnote 188 In Qatar, there was the creation of the Research Guidelines for Healthcare AI Development by Solaiman et al. with advisory support of the Qatar Ministry of Public Heath, which were finally published in 2025.Footnote 189 Those non-binding guidelines cover the research and development stage of health care AI.Footnote 190 In 2024, Solaiman also undertook a detailed analysis of the Saudi Food and Drug Authority’s Guidance on Artificial Intelligence (AI) and Machine Learning (“ML”) technologies based Medical Devices.”Footnote 191 That guidance covers the approval of medical AI devices to market, and goes beyond efforts made by other regulators.Footnote 192 Finally, in 2025, Solaiman et al. proposed the TLA for governing health care AI that synthesized earlier research.Footnote 193 Footnote This section provides are more detailed examination of the TLA, building on previous research. The term True in “True Life Cycle Approach” is used because other frameworks do not fully capture the AI lifecycle, which focus instead on certain stages of its use or development. The TLA is designed to cover all stages of AI as can be seen in Figure 1 below:
The TLA Footnote 194

3.1. R&D & the Qatar Example
Qatar’s Research Guidelines for Healthcare AI Development arose from a multidisciplinary three-year research grant funded by Hamad Bin Khalifa University (“HBKU”) led by the author.Footnote 195 They are a pioneering attempt to regulate AI at the research and development stage, which has often been overlooked by other frameworks.Footnote 196 To ensure a seamless discussion with stakeholders, Qatar’s Ministry of Public Health (“MOPH”) were included as official advisers to the grant and involved at all major stages.Footnote 197 The team consisted of scholars and experts from HBKU’s College of Law, College of Health, the Qatar Biomedical Research Institute, the College of Science and Engineering, the College of Islamic Studies, and the Qatar Genome Program (now, the Qatar Precision Health Institute).Footnote 198 Stakeholders external to the grant but located in Qatar were also consulted in-depth at expert roundtables. The aim was to develop guidelines grounded in the experiences and expertise of those in health, law and bioethics with a focus on local norms. The guidelines were, therefore, adapted in places to the local context but the overarching framework was designed to be applicable in other countries with adaptations.Footnote 199
The guidelines provide non-binding best practices for researchers creating AI systems for use in the health care sector broadly defined.Footnote 200 They embed legal and ethical principles from the very outset of AI innovation across three stages: (1) development; (2) external validation; (3) deployment. Across all stages, the principles of fairness, accountability, and transparency are emphasized but adapted to the local Qatar context. For example, “fairness” in the guidelines is broadly a concern about the underrepresentation of data from the Middle East used to train AI systems.Footnote 201 The principle of accountability encompasses Islamic Bioethics perspectives that emphasize human rather than machine responsibility.Footnote 202 This approach seeks to support existing priorities at the national level concerning health care research that should empower patients, empower researchers and medical professionals, advance public health goals, facilitate doctor-patient relations, and develop digital tools that are ethically sound.Footnote 203 Examining the national context is crucial when developing such guidelines to ensure that priorities are aligned across existing ecosystems.
The principles and underlying aims carry through into the guidelines themselves that serve as a detailed technical checklist for researchers to complete to have a detailed record of following best practices.Footnote 204 They explicitly address Qatar’s regulatory and cultural context. For example, one section of the guidelines asks researchers to address data limitations and biases by recording the following information in Table 1.
Extract From Qatar Research Guidelines for Health Care AI Development: Footnote 205

In this regard, Qatar’s diverse population is considered at the outset because AI researchers must account for under-represented groups in datasets (for example, non-Arabic or English speakers) to prevent bias. By asking researchers to consider issues such as ethnicity, citizenship, and language, the aim is to preempt AI biases before the device has been deployed for use. This aligns the national strategies noted above that encourage the empowerment of patients and developing digital tools that are ethically sound.
Elsewhere, the guidelines state that researchers should state “whether data processing used in the research complies with data protection laws in Qatar”.Footnote Footnote 206 In particular, compliance with Law No. 13 of 2016 on Privacy and Protection of Personal Data and accompanying guidelines for regulated entities.Footnote 207 In essence, the rules are encouraging legal and ethical best practices during development rather than trying to resolve issues after the fact once system has been deployed. The guidelines encourage researchers to keep comprehensive documentation of how the AI model was developed, its intended use on patient populations, and the chosen accuracy thresholds for the algorithm, and much more across its approximately fifty pages.Footnote 208
Researchers do not have to satisfy every requirement, but this level of operational detail would certainly help to address any legal compliance questions later. One provision even encourages the researchers to state “any liaison with the Ministry of Public Health or any other relevant regulatory entity on any aspect of the algorithm’s development and deployment.”Footnote 209 These are not merely abstract principles but are concrete tasks that teams can incorporate into their project management and research protocols.
In this regard, these guidelines can help fill a critical gap in existing governance approaches. Current approaches, such as the FDA or EU address device approvals or post-market monitoring but offer little at the research stage where design choices will be most consequential. By integrating such guidelines into a TLA governance framework, this approach could offer a baseline for best practices for ethical and legal AI design with the local context in mind.
A key question that remains outstanding is how to formally integrate the guidelines into research oversight. The grant project envisaged a certification process.Footnote 210 Researchers can complete the checklist, submit it to a committee to review the AI system for compliance, and receive certification as a mark of credibility for the project.Footnote 211 A mock certification website was developed for researchers to complete the checklists and keep a record.Footnote 212 This worked from a technical standpoint but did not resolve the overarching question of whether a certification process is the best method. An alternative method would be to integrate the guidelines somewhere into Institutional Review Board (IRB) processes. For example, an IRB might require researchers to append the completed AI guideline checklist to their application, demonstrating due diligence. However, ultimately, it was determined that the guidelines should exist to complement IRB processes rather than replace them, a prudent approach since the optimal way to integrate these AI guidelines is still unresolved.Footnote 213
Ideally, a separate expert committee should exist through a Ministry approved body, or department within the Ministry of Public Health that can review submissions and provide certification. The committee would be analogous to the IRB but with a specific focus on AI ethics and safety. While not binding, the MOPH could recognize such certification and provide certain legal safeguards for purchasers of certified AI systems in health. For example, certification could act as a defense against liability claims for harms caused by the AI system — perhaps a no-fault scheme could operate where a certified AI system that followed existing best practices causes harm. Another approach might be for the MOPH to require certification as a pre-requisite for government funded health AI projects or for access to national health data.
Regardless of the path chosen, the underlying point is that health care AI reviews could be institutionalized with a mandate for researchers to undergo such assessments. It would require finding experts and forming such a committee (no easy task). But it could “support smoother transitions into the approval and post-implementation stages of the TLA.”Footnote 214 As noted in earlier research, in addressing “these considerations early, researchers reduce the risk of delays or rejections during approval while ensuring systems are better positioned to meet the requirements of medical device regulators.”Footnote 215
Overall, Qatar’s guidelines demonstrate a strong proof of concept for the first phase of the TLA. Early-stage governance at the research phase can be both comprehensive and practical, covering a plethora of legal and ethical issues. The challenge ahead lies in formalizing these practices. Qatar’s early-stage AI guidelines illustrate that rigorous R&D governance is feasible in practice and adaptable beyond the Qatari context. The core design — embedding law and ethics into technical checklists — can be localized to different legal systems, including those without the same centralized health care oversight.
3.2. Market Approval & the Saudi Example
An AI medical device should require some form of regulatory approval before going to market in a country. The FDA is clearly at the forefront of discussions in the AI space but not all countries have the equivalent of the FDA. Another pioneer is Saudi Arabia through the Saudi Food & Drug Authority (“SFDA”). In 2022, the SFDA issued the Guidance on Artificial Intelligence (AI) and Machine Learning (ML) technologies based Medical Devices (“MDS-G010”).Footnote 216
In a prior detailed analysis, the MDS-G010 was characterized as a “patchwork of existing international best practices concerning AI regulation [that] incorporates adapted forms of non-AI-based guidelines, and builds on existing legal requirements in the SFDA’s existing regulatory architecture.”Footnote 217 The guidance has both binding and non-binding components that integrate and build on global standards from the United States FDA, the International Medical Device Regulators Forum (“IMDRF”), and the WHO, adapting them to the Saudi context.Footnote 218 The MDS-G010 is, therefore, congruent with leading approaches, but also goes beyond them by incorporating additional best practices. Indeed, some requirements were incorporated before the FDA.Footnote 219 Notedly, some elements of the SFDA’s architecture are binding whereas the FDA leans into soft law.Footnote 220
The MDS-G010 is novel in several ways. It establishes requirements for manufacturers to obtain Medical Device Marketing Authorization (“MDMA”).Footnote 221 The SFDA adopts principles related to “Software as a Medical Device” (“SaMD”) as agreed by the IMDRF, and uses those principles as a basis for evaluating AI devices.Footnote 222 AI devices are ones that “diagnose, manage or predict diseases by analyzing medical data.”Footnote 223 Within that scheme, the SFDA notes that there are no internationally agreed standard for clinical evaluations.Footnote 224 To close that gap, it partially adapts standards from the WHO that other regulators have not.Footnote 225 Several provisions are worth restating here:
The manufacturer should assess whether the promised medical benefit is achieved is consistent with the state of the art.
Manufacturers should provide assurance that metrics of effectiveness and safety include outcomes that are meaningful to patients and clinical outcome, i.e. measures of improvement in patient outcomes, clinical process or time efficiency, measures of acceptable unintended consequences, and absence of harm to patients.
The manufacturer should generate evidence on device performance that can be generalized to the entire intended population, demonstrating that performance will not deteriorate across populations and sites.
The effects of AI/ML-based medical devices should be evaluated in clinically relevant conditions, i.e. this requires integration into the existing clinical workflow
Manufacturers in their study design should consider proactively the effects that their studies may have on healthcare organizations and potentially explore the possibility of prospective real-world studies in order to minimize selection bias, have more control over variables and data collection, and examine multiple outcomes.Footnote 226
These articles are non-binding components of the MDS-G010 because the phrases “should” and “advise” repeatedly occur.Footnote 227 Nevertheless, the inclusion of adapted WHO guidance represents an innovation by the SFDA through the combination of hard law and soft law to close gaps where they exist. Additionally, some provisions in the MDS-G010 have a patient-centric focus despite its technical nature. The SFDA notably states that AI “may present risks that could jeopardize patient health and safety, increase inequalities and inefficiencies, undermine trust in healthcare, and adversely impact the management of healthcare.’Footnote 228 This is a rare acknowledgement among regulators that patient trust and welfare are as crucial as technical safety.
This patient-centric emphasis is central to the TLA and should be encouraged by all regulators. Indeed, the SFDA also effectively brings the FDA’s TPLC into binding practice through its requirement that manufacturers institute “post-market continuous monitoring of safety, effectiveness, and performance” for AI devices operating in the real world.Footnote 229 On risk management, manufacturers are also required to “demonstrate that their medical devices do not pose unacceptable risks, and that the benefits of their intended use outweigh the overall residual risk.”Footnote 230
The FDA has to date cleared AI devices through existing pathways like 510(k) and de novo.Footnote 231 This has led to concerns about “predicate creep” whereby new AI devices get cleared based on comparisons with older non-AI devices, which raises safety concerns.Footnote 232 The SFDA has been more proactive and decisive by incorporating bespoke standards for AI.Footnote 233 This approach clearly goes further than the EU because the AI Act does “not address matters of health directly relevant to AI-based medical devices.”Footnote 234 Nevertheless, the unresolved binding nature of the MDS-G010 is noteworthy. There are binding “components” in the MDS-G010 that arise from underlying medical device law,Footnote 235 but there are also non-binding international best practices.Footnote 236 This hybrid approach is innovative but may blur lines of accountability, creating challenges in delineating stakeholders’ responsibilities. For example, determining whether a lapse is a legal violation or a best practice gap.
The key point to emphasize is that the SFDA’s approach is novel in certain aspects and takes on a patient-centric focus. That is important for the TLA which seeks to weave a consistent focus on individual rights. The requirements for market approvals will be necessarily more technical in nature but the language and philosophy of approach are also important. Market approval should not merely be a tick box technical exercise concerning risk but should feed into a broader narrative of patient safety. The SFDA’s approach is also congruent with the TLA because it also stipulates post-market monitoring and a focus on the continuous learning of AI systems.Footnote 237 Embedding the SFDA’s approach within the TLA’s device approval phase represents a forward-thinking patient-centric approach. The Saudi example demonstrates that AI device approvals can integrate global best practices while being tailored to national context. Its hybrid binding structure – though imperfect – might also be an approach for low- and middle-income countries to follow, to leapfrog older models by adopting select innovations from multiple jurisdictions.
3.3. Post Implementation and the UAE Example
The final part of the TLA applies once AI is deployed and integrated into health care practice. Abu DhabiFootnote 238 and DubaiFootnote 239 have addressed such governance through two “policies” which were among the first in the world on post implementation in health care. These exist within a much broader framework of developments for AI governance in the UAE.Footnote 240 While termed as policies, they contain binding requirements with penalties for lack of compliance.Footnote 241 As such, these are not voluntary best practices. The policies are similar in focus, but Dubai’s offers more depth and will therefore be primarily outlined below.
Both policies are broad in scope and contain clear requirements to protect patients and ensure accountability for the use of AI. Both policies apply to all health care providers, pharmaceutical manufacturers, insurers, researchers, and AI developers using data from local health care systems.Footnote 242 Dubai’s policy requires that “all AI solutions for healthcare” must conform to the relevant local and international laws and regulations, “with respect to values, patient autonomy, people rights, and acceptable ethics.”Footnote 243 AI must be free of biases, and accountability for AI outputs in health care “must be agreed between designer, researcher, developer, operators/ commissioners, and end users.”Footnote 244 AI must also have in-built appeals processes for users to challenge significant decisions.Footnote 245 Abu Dhabi has similar provisions that seek to “minimize any potential risks to patient safety.”Footnote 246
There are important requirements on transparency, such as disclosing which data sets were used, what the role of the health care professional is in making the final decision, an ethical examination of how the data is used, and how the AI solution must be integrated into health care provision.Footnote 247 The emphasis on patients is robust, with the recognition that AI solutions for health care “may directly impact people’s lives in a significant way” and so “must be designed with utmost care.”Footnote 248 The policy imposes “minimum acceptable requirements for AI tools” that encompass compliance with a plethora of laws, namely, federal laws and regulatory requirements pertaining to telehealth, data protection, and cybersecurity, as well as, an ICT health law, a medical liability law, human research laws, health insurance law, and more.Footnote 249 In this manner, the policy makes post-market AI governance an extension of the legal frameworks governing health care. It moves beyond other frameworks by giving regulatory oversight to the Dubai Health Authority (“DHA”).Footnote 250 End users must report incidents, deficiencies, and issues arising from the implementation of AI to the DHA.Footnote 251 It is also the responsibility of the DHA to create a regulatory framework that governs AI in health care, to monitor compliance with the policy through reporting, audits, and inspections, and to impose sanctions for breaches.Footnote 252
If these policies become a model for other countries to follow, then hospitals and other stakeholders will need internal processes to ensure compliance with continuous monitoring and reporting requirements. The requirements on patient redress mechanisms or appeals procedures will require some thought to ensure consistency in application of those mechanisms across institutions to ensure proper compliance with the law. Perhaps Cohen et al.’s proposal for an appeals process (noted supra) could indicate the path forward here.Footnote 253
Another approach could be to create an AI Bill of Rights that connects patient protections clearly and holistically to the ethical and legal obligations that surround stakeholders.Footnote 254 Some jurisdictions have a patient bill of rights, including Qatar,Footnote 255 Saudi Arabia,Footnote 256 the UAE,Footnote 257 the UK,Footnote 258 the United States,Footnote 259 and many other countries. These existing patient charters could be updated with an AI Bill of Rights, with provisions on informed consent, the right to an explanation for the use of AI, rights concerning data use with AI and transparency, and a straightforward right to redress for patients to pursue (such as complaining to an AI committee in a hospital that can resolve or escalate matters on behalf of patients through appropriate ethics or legal channels).Footnote 260 Patients could be given the right to opt out of AI being used in their care where appropriate human alternatives exist. These could be formulated in a manner that is congruent with the expectations of regulators such as the authorities in Dubai and Abu Dhabi, with their focus on protecting patients.
By institutionalizing rights in this manner, the gap can be closed between high level principles and complex legal systems to the on-the-ground patient experience. A Bill of Rights supports the TLA’s requirement that law, and ethics must be central considerations from research and development through to deployment. Ultimately, the UAE’s post-implementation framework illustrates how binding patient protection measures can be embedded in national AI governance, and how such provisions could be adapted in other jurisdictions to make lifecycle approaches tangible for patients.
4. The Future of the TLA
While the TLA offers a pathway for governing health care AI, it is not without its limitations. Highlighting those limitations candidly is essential to refining the framework and encouraging constructive debate. As such, this paper closes by critiquing the potential shortcomings of the TLA before synthesizing its strengths to demonstrate how it exemplifies a comprehensive and interconnected holistic governance model.
One of the main concerns noted above relates to where certain phases of the TLA fit into the overall governance architecture. For example, the first phase of research and development might risk replicating IRB functions or unnecessarily overburdening them. IRBs already evaluate matters of ethics, informed consent, data privacy, and so on.Footnote 261 Also, even if an AI committee is created, one then must deal with the obvious challenge in finding relevant experts for such committees. Many regions will not have many experts spanning AI, ethics, law, and health care to evaluate relevant systems.Footnote 262 Even in well-resourced Gulf countries, finding such talent is difficult, but in low resource settings, creating more governance could exacerbate inequities.Footnote 263 This risk underscores the need for proportionate implementation. The TLA’s phases can be embedded into existing review or audit processes rather than creating standalone bureaucracies, allowing for context-sensitive adoption.
The examples of Qatar, Saudi Arabia, and the UAE work, in part, because they have compact health care ecosystems and there can be strong centralized government coordination (although, that does not always translate to greater efficiency). In larger dynamic economies like the United States and the EU, there are more diverse stakeholders, siloed oversight, and rapid innovation cycles. For example, the first phase of the TLA on research and development would be difficult to integrate into the FDA’s TPLC because of existing layers of bureaucracy. The EU’s AI Act has already introduced heavy regulatory demands on stakeholdersFootnote 264 – injecting TLA governance could further increase costs and complexity. Proportionality also means recognizing when certain elements can be scaled back or merged with existing requirements. For instance, in jurisdictions already operating robust post-market monitoring, the TLA’s post-implementation checks could be incorporated as targeted enhancements rather than wholesale additions.
Along those lines, it may also simply be unnecessary to have TLA governance, creating more red tape and overcomplicated governance. There are already lifecycle frameworks noted above that emphasize post market monitoring.Footnote 265 Implementing the TLA could lead to fragmented oversight over the different phases and may burden manufacturers and those subject to regulations, duplicating documentation without enhancing patient safety. It could be argued that the SFDA’s MDS G010 already incorporates adapted forms of best practices from the WHO and others. Ultimately, proportionate integration and adaptive implementation are key to ensuring that the TLA does not duplicate or overburden but instead strengthens patient protections without stifling innovation.
Despite these limitations, the TLA can move beyond them. This framework should cause us to think about how we can create a coherent and unified approach that puts patients first and closes the gaps that exist in the existing piecemeal approaches. AI brings with it specific challenges in health care and patients should not be ignored at any stage of the AI lifecycle. Some frameworks ignore them,Footnote 266 while others only address their concerns in passing.Footnote 267 In an earlier paper, it was noted how the examples from Qatar, Saudi Arabia, and the UAE exemplify the “interconnectedness of the [TLA].”Footnote 268 By creating a patient-centered approach that emphasizes law and ethics “each regulatory phase reinforces and builds upon the others.”Footnote 269
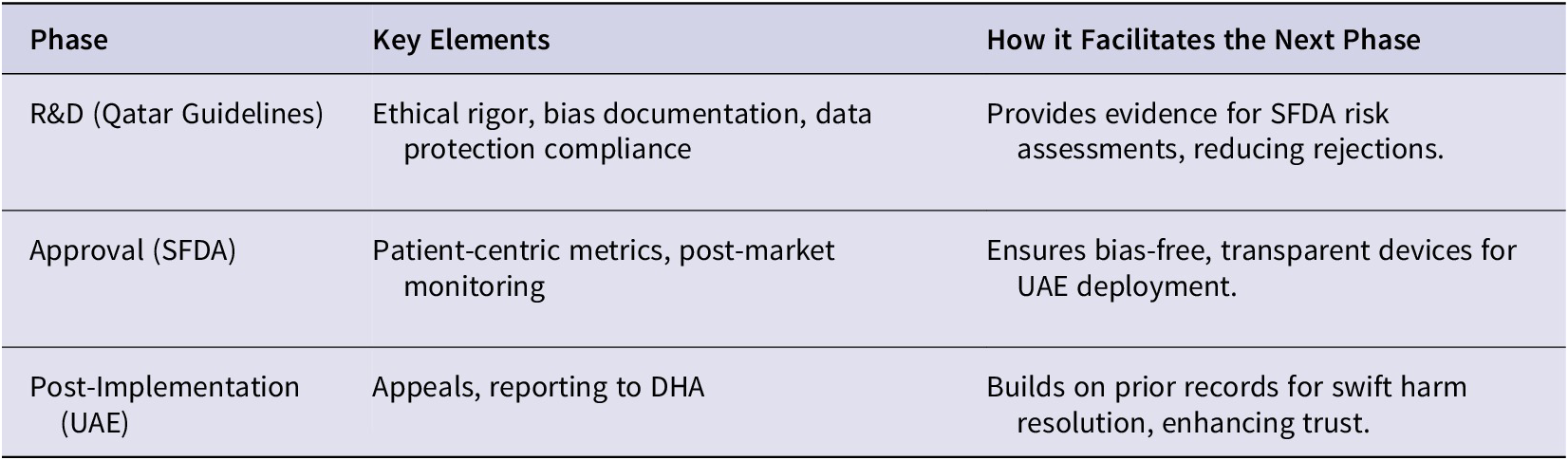
Thus, compliance with Qatar’s guidelines during research and development with its rigorous documentation on matters such as data biases, and compliance with laws such as those on data protection, would prepare developers for obtaining market approval later. For example, the SFDA requires that AI devices work on their intended population, that biases are mitigated, and that performance is evaluated in clinical practice.Footnote 270 Obtaining certification through Qatar-style guidelines with its record keeping on ethical justifications for the AI system and data limitations, could provide the evidence required for SFDA risk assessments without the need for duplication, and may reduce the risk of devices being rejected given that best practices would have been followed. The emphasis on transparency in the Qatar guidelines would also support the post-market monitoring requirements under the SFDA architecture. Addressing biases early would avoid making costly adjustments later when an SFDA audit reveals concerns.
The same is true for post-implementation requirements that move beyond mere market approval of devices. The UAE example highlights how more governance can be expected beyond FDA-style regulators, with regulators potentially having far broader powers to monitor the use of AI in practice. For example, the UAE policies require that AI is free from biases, transparent, and compliant with medical liability laws.Footnote 271 An AI system that has navigated Qatar-style guidelines at the research stage and SFDA style regulations on device approval will be prepared for downstream regulatory enquiries. If documentation on explainability is in place, and there are accountability mechanisms built in to facilitate an appeals process, then this would support the requirements of the DHA. Consider an AI system used in a hospital that hallucinates, makes an incorrect recommendation, and ultimately harms the patient. There will be foundational R&D records in place to ensure efficient reporting to the DHA which would assist with determining corrective actions. In this manner, rigorous ethical R&D at the outset would help streamline approvals and support post-market trust and redress. This interconnectedness can be seen in Table 2.
Interconnectedness of TLA Phases

At this stage then, the TLA may be thought of as a dynamic and evolving framework that weaves patient centric principles through its three phases from the lab to clinic. By focusing on patients in this systematic way, it goes beyond what FDA and EU frameworks can do in health care. Actionable rights are fundamental considerations throughout, such as opt outs and appeals, and this complements the WHO guidance by encouraging practical tools that apply at the national level. By synthesizing these pioneering approaches, the TLA offers a template that can be adapted globally to protect patients wherever AI is used in health care. Through Qatar’s approach to research and development, Saudi Arabia’s forward-thinking medical device approvals frameworks, and the UAE’s comprehensive obligations post-deployment, the TLA offers a model to remedy existing gaps in governance that focus on protecting patients. The TLA is not perfect, but the aim is to stir debate about health care AI governance, urge global collaboration, inspire similar national-level initiatives, and demonstrate that models trialed in the Gulf can inform AI governance in health care worldwide.
Acknowledgements
I would like to thank the editorial team at the AJLM for the significant time and effort for helping with reviewing and copy-editing this paper.


 Open access
Open access