Soft tissue sarcomas represent a heterogeneous group of rare malignancies with an overall incidence of about 5/100,000/year. Incidence tends to vary according to age, ranging from approximately 2/100,000/year in the first two decades to 15–20/100,000/year in the elderly population. Soft tissue sarcomas can occur at any anatomic location; however, approximately half of all sarcomas occur in the limbs (wherein the thigh is by far the most common site), 30% occur intra-abdominally (including the retroperitoneum), and 15% arise in the trunk and in the head and neck region. As will be discussed in more detail, both incidence and site of occurrence are strongly influenced by the specific histotype. For example, alveolar rhabdomyosarcoma occurs most often in children, myxoid liposarcoma occurs most often in the thigh of adults in their third decade, dedifferentiated liposarcoma tends to occur in the retroperitoneum with a peak incidence in the fourth and fifth decades, and myxofibrosarcoma tends to occur in the superficial soft tissues of elderly patients.
Soft tissue sarcomas are aggressive neoplasms capable of local destructive growth, recurrence, and distant metastases, most often to lungs, liver, bone, soft tissue, and brain. Lymph node metastases are comparatively more rare, and tend to be associated with a relatively limited number of distinctive histologies, such as epithelioid sarcoma, clear cell sarcoma, alveolar rhabdomyosarcoma, and succinate dehydrogenase-deficient gastrointestinal stromal tumors (SDH-deficient GISTs). In approximately 20–30% of cases there is local recurrence, whereas about 30–50% of cases metastasize. Five-year overall survival varies between 55 and 65%, regardless of stage and histology.
Mesenchymal tumors have always been regarded as diagnostically challenging, rarity and morphologic heterogeneity representing the main factors affecting diagnostic accuracy. As a consequence, sufficient expertise can be achieved only through access to a large number of cases. To avoid major mistakes, careful evaluation of clinical presentation and integration of immunohistochemistry and molecular genetics whenever relevant are mandatory. As accurate classification increasingly correlates with the choice of specific treatments, every effort should be made to achieve diagnostic accuracy.
Soft tissue sarcomas are currently classified on the basis of the 2013 World Health Organization’s (WHO) classification of soft tissue tumors, which has further expanded and refined the concepts that were pioneered in the 2002 WHO classification, and which has collected and distilled all the major advances generated in the past 15 years. WHO classifies the different entities on the basis of histomorphology and includes all available immunophenotypic and genetic data. This perfectly matches a diagnostic approach that integrates sequentially the microscopic features of the lesion with its immunophenotype and its genetic profile. The changes that have occurred since publication of the latest WHO classification will be specifically addressed in the context of the discussion of the single tumor entities; however, it is useful at this stage to summarize the major changes introduced thus far. Soft tissue sarcomas and soft tissue tumors of intermediate malignancy currently recognized by the WHO 2013 classification are listed in Table 1.1.
Table 1.1 Intermediate (locally aggressive and/or rarely metastasizing) and malignant soft tissue tumors recognized by the 2013 WHO Classification of Soft Tissue Tumors
Adipocytic Tumors
One of the major conceptual shifts introduced after 2002 is the use of a stricter terminological definition of “well-differentiated liposarcoma,” which represents the most common liposarcoma subtypes. It has been clarified that the terms atypical lipomatous tumor and well-differentiated liposarcoma are synonyms, and that the latter term should be used only for lesions that occur in the retroperitoneum/mediastinum or in other anatomic sites where complete resectability is unachievable. The use of the term “atypical lipomatous tumors” for resectable lesions is justified by the fact they never recur and are most often cured by complete (even marginal) surgical excision. In 2002, it was recognized that in dedifferentiated liposarcoma (defined as morphologic progression from well-differentiated liposarcoma to high-grade non-lipogenic sarcoma), a low-grade dedifferentiation can also be observed. In 2013, the concept of homologous dedifferentiation (represented by the occurrence of lipogenic, high-grade morphology somewhat mimicking pleomorphic liposarcoma) was fully acknowledged. A major change also involved myxoid liposarcoma, which, until 2002, was kept separated from round cell liposarcoma. To reflect the fact that both lesions actually represent the ends of a morphologic spectrum of a genetically distinct histology, in 2002 myxoid and round cell liposarcoma merged into a single entity. In 2013 the term “round cell liposarcoma” was eliminated and replaced by high-grade myxoid liposarcoma to underscore the fact that clinical outcome depends on the amount of hypercellularity and not on the shape of neoplastic cells, which can be either rounded or spindled.
Fibroblastic/Myofibroblastic Tumors
An important conceptual change in 2002 was represented by the inclusion of hemangiopericytoma (HPC) within the WHO’s chapter on solitary fibrous tumors, because the borders between those lesions had become increasingly blurred. It was felt that the very concept of HPC was at risk of extinction, because it represented a collection of unrelated, benign as well as malignant, simple lesions sharing an HPC-like vascular network. Most cases (at any location) would currently be reclassified as solitary fibrous tumors, and the entity labeled as lipomatous HPC is considered a variant of solitary fibrous tumor. As a logical consequence of this conceptual evolution, in 2013 the label “hemangiopericytoma” (HPC) was completely abolished. Currently, the original (still valid) idea generated by Arthur Purdy Stout of the existence of lesions composed mainly of contractile cells organized in a perivascular pattern of growth survives within the label myopericytoma.
Fibrosarcoma also experienced a significant remodeling. Whereas it is currently recognized that most superficially located fibrosarcomas actually represent examples of fibrosarcomatous dermatofibrosarcoma protuberans (FS-DFSP), infantile fibrosarcoma is confirmed as a clinically, pathologically, and genetically distinct entity. However, new distinctive sarcoma subtypes featuring fibroblastic/myofibroblastic differentiation have been introduced. These are low-grade fibromyxoid sarcoma, myxoinflammatory fibroblastic sarcoma, sclerosing epithelioid fibrosarcoma, and low-grade myofibroblastic sarcoma.
So-Called Fibrohistiocytic Tumors
After reappraisal of malignant fibrous histiocytoma (MFH) and its variants, the label malignant fibrous histiocytoma was abolished in 2013. As discussed in depth in Chapter 7, pleomorphic MFH, once the most commonly diagnosed sarcoma, is now synonymous with high-grade undifferentiated pleomorphic sarcoma and it should not exceed approximately 5% of newly diagnosed sarcomas. Myxoid MFH is now included within the morphologic spectrum of myxofibrosarcoma. In addition, the so-called giant cell variant of MFH appears to be a heterogeneous collection of clinically as well as morphologically distinctive lesions – namely, giant cell tumor of soft tissue, extraskeletal osteosarcoma, and spindle cell sarcoma (most often leiomyosarcoma) featuring osteoclast-like giant cells. The inflammatory variant of MFH most often represents examples of inflammatory dedifferentiated liposarcoma. Angiomatoid MFH, the latest addition to the MFH family, is no longer considered a malignancy and has therefore been downgraded to the intermediate category. As its line of differentiation remains unknown, it has also been moved to the category of mesenchymal tumors of uncertain differentiation.
The existence of a broader category of undifferentiated sarcomas (pleomorphic, epithelioid, round cell, and spindle cell) is now fully acknowledged. Those round cell sarcomas harboring the CIC-DUX4 or the BCOR-CCNB3 translocation are temporarily classified under the heading “undifferentiated round cell sarcomas.” In consideration of the new data accumulated, these sarcomas are covered in Chapter 6 as separate entities.
Vascular Tumors
In the past two decades, several new entities have been characterized, particularly in the intermediate malignancy category, including kaposiform, retiform, and composite hemangioendotheliomas. Since the 2002 WHO classification, epithelioid hemangioendothelioma (EHE) has been reclassified as malignant because of its considerable metastatic rate that ranges between 15 and 30%. Endovascular papillary angioendothelioma (so-called Dabska tumor) has been renamed papillary intralymphatic angioendothelioma. Pseudomyogenic hemangioendothelioma, a novel, genetically distinct entity characterized by multifocality as well as relatively indolent clinical behavior, has been added to the group of vascular neoplasms of intermediate malignancy.
Tumors of Uncertain Differentiation
Tumors of uncertain differentiation is a category that contains tumors without a clear line of differentiation or without a normal cellular counterpart. Obviously, several new entities have been described since 1994, including myoepithelioma of soft tissue and PEComa. Because we now know more about divergent differentiation in various sarcoma subtypes, the category of malignant mesenchymoma is also losing ground, as it is currently acknowledged that heterologous differentiation may occur in the context of specific entities such as malignant peripheral nerve sheath tumors (MPNSTs) and dedifferentiated liposarcoma. The morphologically rather elusive category of intimal sarcoma was introduced as a new entity in this group.
Principles of Sarcomagenesis
The pathogenesis of the vast majority of soft tissue sarcomas is still unknown and most of them seem to arise de novo without an apparent causative factor. In rare cases, genetic and environmental factors such as radiation, lymphedema (secondary angiosarcoma of the breast), viral infections (human herpesvirus 8 infection is associated with Kaposi sarcoma), exposure to chemicals (vinyl chloride is linked to hepatic angiosarcoma), and immunodeficiency (Epstein-Barr virus infection in immunodeficient subjects is associated with the development of smooth muscle tumors) have been identified as risk factors. It is broadly accepted that trauma does not represent a predisposing factor and that, at best, it can simply draw attention to the presence of a pre-existing mass.
Genetic susceptibility plays a role in a minority of soft tissue sarcomas. Neurofibromatosis type 1 (NF1) and Li-Fraumeni syndromes represent two good examples. In NF1, up to 10% of patients will develop MPNSTs as well as multiple GISTs. The autosomal dominant Li-Fraumeni syndrome (wherein germline mutations of the TP53 gene occur) has been shown to predispose the development of malignant tumors, one-third of which are represented by bone and soft tissue sarcomas. Recent data have shown that approximately half of patients with sarcoma have putatively pathogenic monogenic and polygenic variation in known and novel cancer genes, among which are TP53, ATM, ATR, BRCA2, and ERCC2.
In the past two decades, molecular genetics has greatly contributed to the elucidation of some of the molecular mechanisms associated with the development of soft tissue sarcomas. Significant subsets of mesenchymal malignancies are associated with chromosome translocations, the presence of which is currently being exploited for diagnostic confirmation (Table 1.2). A smaller group of lesions is characterized by the presence of simple karyotypes associated with mutations. Good examples are represented by desmoid fibromatosis (the vast majority of which are associated with mutations of either the CTNNB1 or APC gene) and gastrointestinal stromal tumors (most often associated with mutations of the KIT and PDGFRA genes and far less often of the BRAF, SDH, and NF1 genes). A third (large) group of sarcomas exhibits variably complex karyotypes. In this context, particularly relevant is the occurrence of gene copy number alterations as observed in well-differentiated/dedifferentiated liposarcoma, wherein the amplification of the MDM2, CDK4, and HMGA2 genes represents the key driver genetic event.
Principles of Pathologic Diagnosis
Sarcomas are currently classified on the basis of their morphology, their immunophenotype, and their molecular status. The integration of conventional morphology with immunohistochemistry and molecular genetics represents the major contribution of the WHO classification since 2002 and this approach has been further confirmed in 2013. For practical reasons, the classification scheme follows a histogenetic approach, even though currently it is no longer believed that a given mesenchymal neoplasm actually originates from a mature normal counterpart. Interestingly, the list of lesions of unknown histogenesis (i.e., unknown line of differentiation) has increased in size, reflecting the uncertainties surrounding the mechanisms of sarcomagenesis.
Microscopic observation of hematoxylin- and eosin-stained slides obtained from formalin-fixed, paraffin-embedded material still represents the mainstay of sarcoma classification. The amount of information provided by this technically simple step is invaluable. Any other ancillary technique (immunohistochemistry and/or molecular pathology/genetics), even the most sophisticated, certainly represents an important complement to, but under no circumstances a replacement for, classic morphologic observation. It should be also noted that macroscopic observation also plays a fundamental role – first in providing accurate reporting of the status of surgical margins, and second in guiding proper sampling, and therefore acting as the milestone for correct classification. It is very important that any area showing a distinct gross appearance is sampled so that no relevant information is missed. It is also possible that in the near future, similar to what already occurs for osteosarcoma and Ewing sarcoma, the morphologic evaluation of tumor response to systemic treatment will gain significant clinical relevance.
Microscopic Examination of Soft Tissue Sarcomas
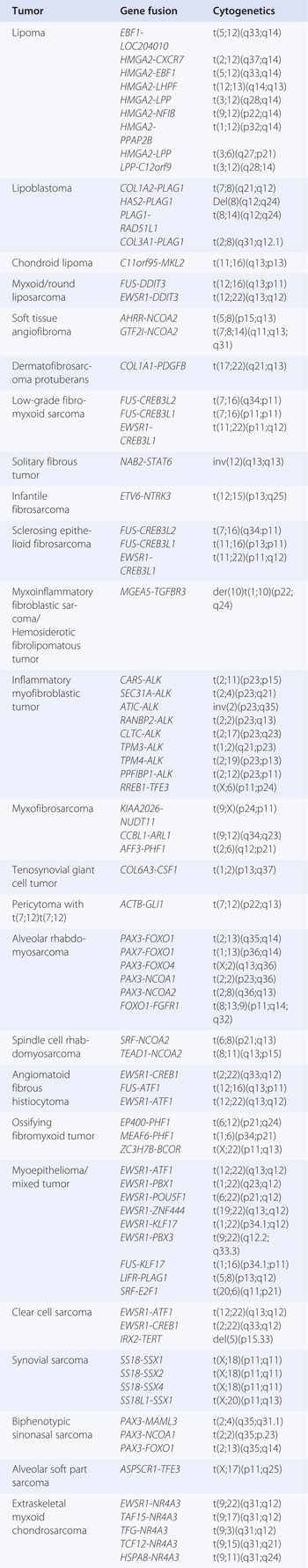
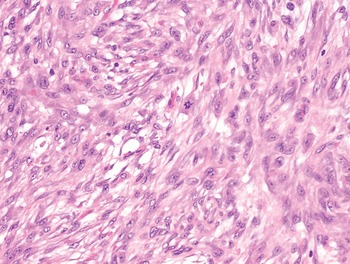
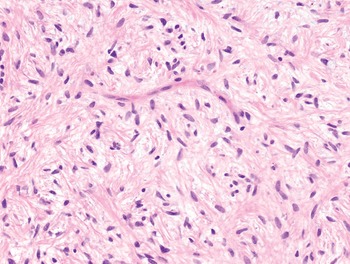
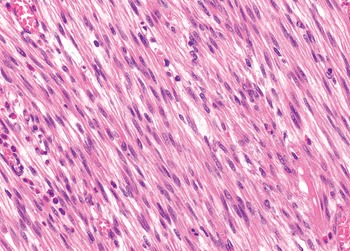
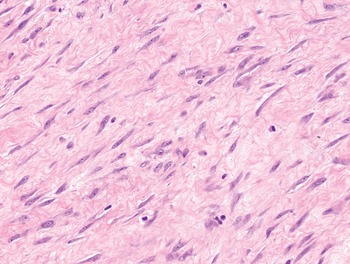
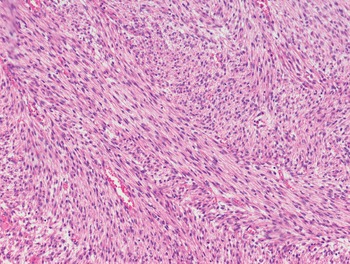
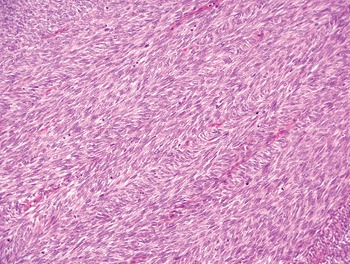
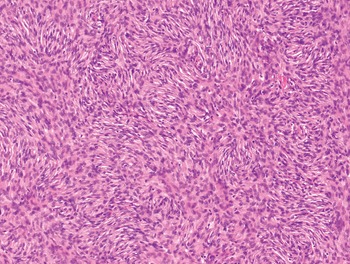
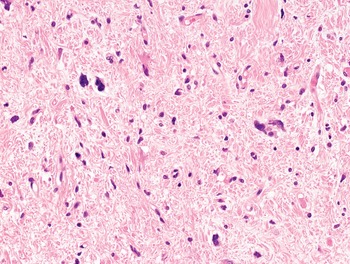
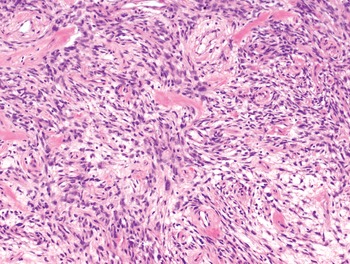
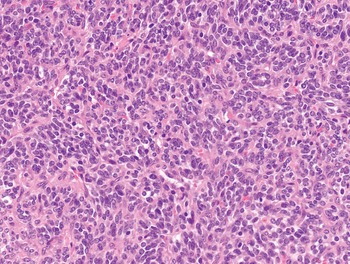
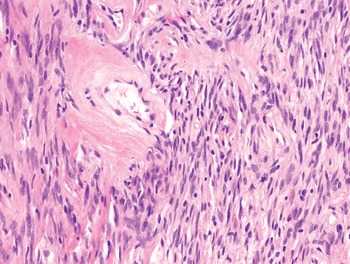
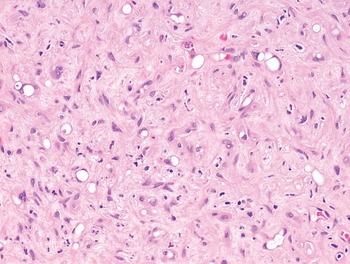
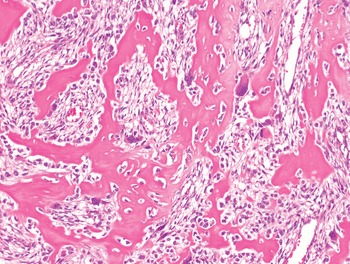
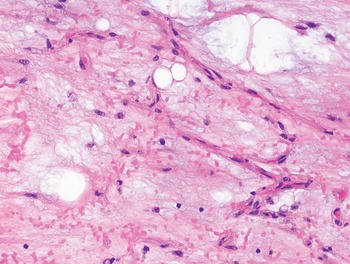
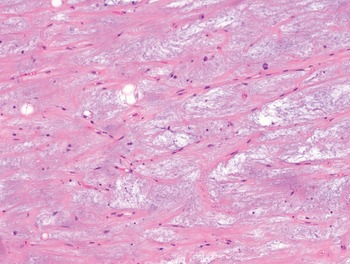
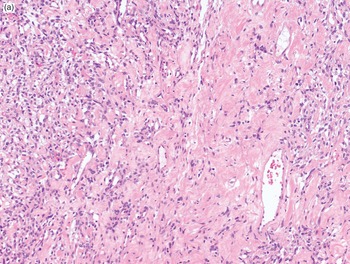
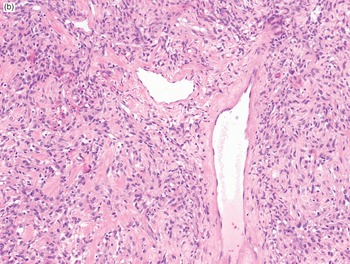
The diagnosis of mesenchymal malignancies represents a true challenge. This is largely owing to their rarity, a fact that hampers the chance to develop expert skills outside high-volume referral centers. Moreover, sarcomas relatively often exhibit a tendency to violate some of the common rules of malignancy that we routinely apply to non-mesenchymal cancers. Just imagine a lesion occurring in the forearm of a young adult that is clinically characterized by rapid growth, and that microscopically is composed of a spindle cell proliferation featuring both hypercellularity and high mitotic activity (Fig. 1-1). Understandably, in the absence of specific expertise, these morphologic (and clinical) features would all lead to a diagnosis of malignancy. However, those characteristics actually fit perfectly with the clinicopathologic presentation of nodular fasciitis, an entirely benign myofibroblastic proliferation that, in fact, is frequently mislabeled as a sarcoma. Several other examples of benign tumors mimicking malignant lesions are discussed in this book whenever appropriate (Table 1.3). At the opposite end, try to imagine a deep-seated mass featuring a hypocellular spindle cell proliferation with minimal atypia and irrelevant mitotic activity. The presence of cellular variation as well as of fibromyxoid background is of great help to the expert pathologist to suspect a low-grade fibromyxoid sarcoma (also known as Evans tumor). In less experienced hands, however, most of these cases are unrecognized and so diagnosed as benign (Fig. 1-2). Locally aggressive or malignant soft tissue lesions mimicking benign processes are listed in Table 1.4.

Fig. 1-1. Nodular fasciitis. Hypercellularity and mitotic activity certainly represent worrisome morphologic features. However, they are the morphologic hallmark of this entirely benign mesenchymal neoplasm.

Fig. 1-2. Low-grade fibromyxoid sarcoma. The absence of nuclear atypia contrasts with the significant aggressiveness of this tumor entity.
Table 1.3 Clinically benign soft tissue lesions mimicking malignancy
| Nodular fasciitis |
| Proliferative fasciitis |
| Proliferative myositis |
| Ischemic fasciitis |
| Myositis ossificans |
| Pleomorphic angiectatic hyalinizing tumor |
| Pseudosarcomatous proliferation of urinary bladder |
| Cellular schwannoma |
| Atypical fibroxanthoma |
| PEComa |
| Pleomorphic lipoma |
Table 1.4 Intermediate and malignant soft tissue lesions mimicking benign tumors
| Desmoid fibromatosis |
| Low-grade fibromyxoid sarcoma |
| Low-grade myxofibrosarcoma |
| Low-grade myxoid liposarcoma |
| Epithelioid hemangioendothelioma |
| Epithelioid sarcoma, classical type |
| Low-grade malignant peripheral nerve sheath tumor |
Despite the intrinsic challenge of sarcoma diagnosis, it is still possible to achieve a correct classification in most instances, provided that cases are approached following a rigorous methodology. The diagnosis of sarcoma relies upon the evaluation as well as the integration of four main features:
1. Predominant shape of the neoplastic cells
2. Pattern of growth
3. Quality of the background
4. Architecture of the vascular network
This approach possesses the great merit of reducing dramatically the number of diagnostic options, also allowing a rational choice of ancillary immunohistochemical and molecular tests. Of course, this approach needs some degree of flexibility because numerous entities may at times exhibit a combination of different major morphologic features.
The Shape of Neoplastic Cells
Neoplastic cells can be classified on the basis of their shape into four main categories: spindle, epithelioid, round, and pleomorphic.
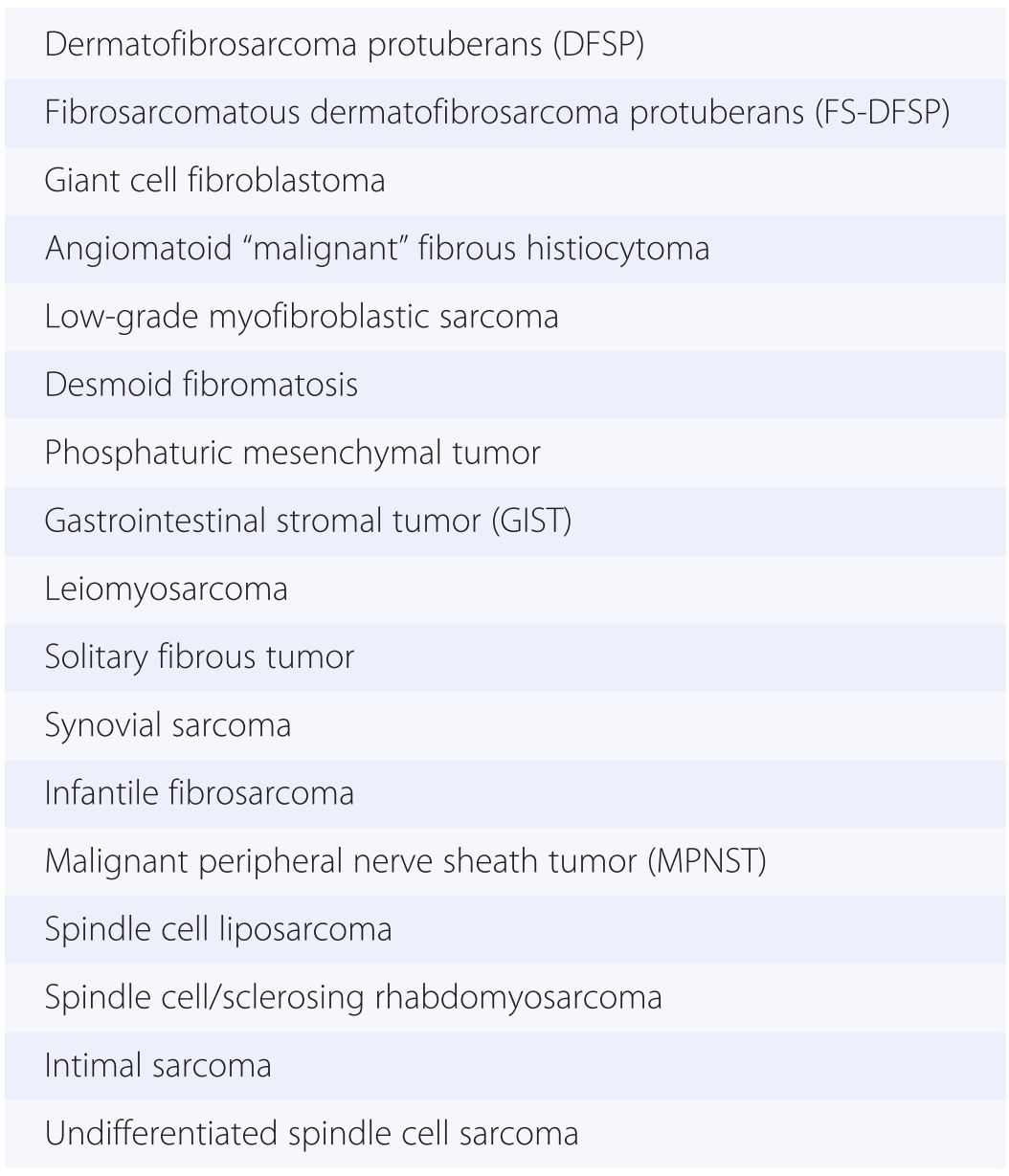
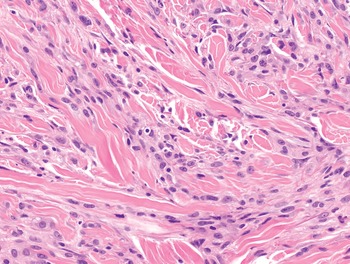
1. Spindle cells are defined by the presence of an elongated cytoplasm, harboring oval nuclei that can be blunt ended (as typically seen in smooth muscle tumors) (Fig. 1-3), tapering (as seen in myofibroblastic tumors) (Fig. 1-4), or pointed (as seen most often in neural neoplasms) (Fig. 1-5). Soft tissue malignancies featuring a predominantly spindle cell morphology are listed in Table 1.5 and described in Chapter 4.
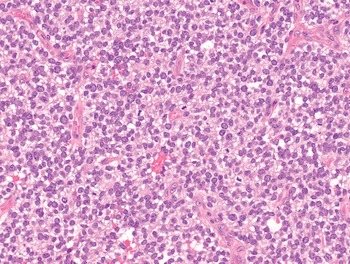
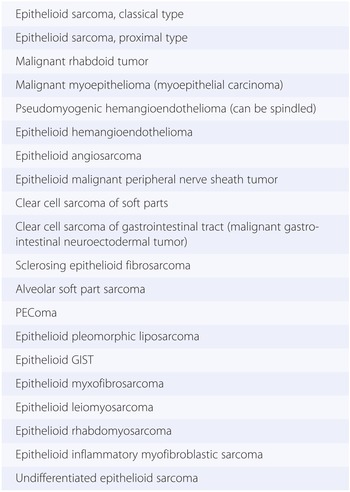
2. Epithelioid cells are defined by the presence of polygonal, abundant cytoplasm, most often harboring a round-shaped nucleus (Fig. 1-6). Soft tissue malignancies featuring predominantly epithelioid cell morphology are listed in Table 1.6 and described in Chapter 5.
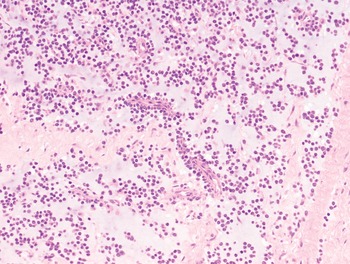
3. Round cells are defined by the presence of circular, scanty cytoplasm, harboring centrally located, round nuclei (Fig. 1-7). Soft tissue malignancies featuring predominantly round cell morphology are listed in Table 1.7 and described in Chapter 6.
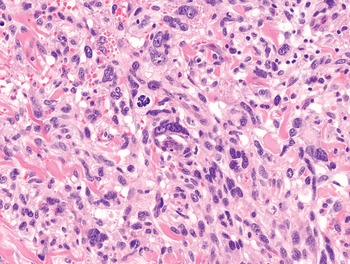
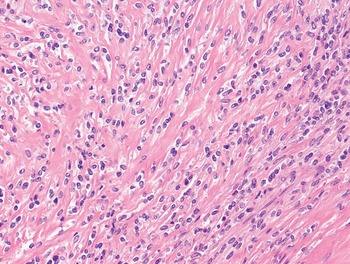
4. Pleomorphic cells are defined on the basis of marked nuclear atypia represented by extreme variation of nuclear size with or without macronucleolation and nuclear hyperchromasia (Fig. 1-8). Soft tissue malignancies featuring a predominantly pleomorphic morphology are listed in Table 1.8 and described in Chapter 7.



Fig. 1-5. Schwannoma. In neural neoplasms, nuclei tend to be irregularly shaped and often feature a pointed end.



| Dermatofibrosarcoma protuberans (DFSP) |
| Fibrosarcomatous dermatofibrosarcoma protuberans (FS-DFSP) |
| Giant cell fibroblastoma |
| Angiomatoid “malignant” fibrous histiocytoma |
| Low-grade myofibroblastic sarcoma |
| Desmoid fibromatosis |
| Phosphaturic mesenchymal tumor |
| Gastrointestinal stromal tumor (GIST) |
| Leiomyosarcoma |
| Solitary fibrous tumor |
| Synovial sarcoma |
| Infantile fibrosarcoma |
| Malignant peripheral nerve sheath tumor (MPNST) |
| Spindle cell liposarcoma |
| Spindle cell/sclerosing rhabdomyosarcoma |
| Intimal sarcoma |
| Undifferentiated spindle cell sarcoma |
| Epithelioid sarcoma, classical type |
| Epithelioid sarcoma, proximal type |
| Malignant rhabdoid tumor |
| Malignant myoepithelioma (myoepithelial carcinoma) |
| Pseudomyogenic hemangioendothelioma (can be spindled) |
| Epithelioid hemangioendothelioma |
| Epithelioid angiosarcoma |
| Epithelioid malignant peripheral nerve sheath tumor |
| Clear cell sarcoma of soft parts |
| Clear cell sarcoma of gastrointestinal tract (malignant gastrointestinal neuroectodermal tumor) |
| Sclerosing epithelioid fibrosarcoma |
| Alveolar soft part sarcoma |
| PEComa |
| Epithelioid pleomorphic liposarcoma |
| Epithelioid GIST |
| Epithelioid myxofibrosarcoma |
| Epithelioid leiomyosarcoma |
| Epithelioid rhabdomyosarcoma |
| Epithelioid inflammatory myofibroblastic sarcoma |
| Undifferentiated epithelioid sarcoma |
| Ewing sarcoma |
| CIC-DUX4-associated round cell sarcoma |
| BCOR-CCNB3-associated round cell sarcoma |
| Extraskeletal mesenchymal chondrosarcoma |
| Desmoplastic small round cell tumor |
| Alveolar rhabdomyosarcoma |
| Poorly differentiated round cell synovial sarcoma |
| High-grade myxoid (formerly, round cell) liposarcoma |
| Pleomorphic rhabdomyosarcoma |
| Pleomorphic liposarcoma |
| Dedifferentiated liposarcoma |
| Extraskeletal osteosarcoma |
| Pleomorphic high-grade myxofibrosarcoma |
| Pleomorphic leiomyosarcoma |
| Pleomorphic malignant peripheral nerve sheath tumor |
| Undifferentiated pleomorphic sarcoma |
The Patterns of Growth
The patterns of growth of the neoplastic cell population are extremely important, as they help to further refine the possible diagnostic options. Main growth patterns are as follows:
1. Fascicular: neoplastic cells are arranged in parallel to form fascicles of variable length. This pattern is typically observed in leiomyosarcoma and malignant peripheral nerve sheath tumors (Fig. 1-9).
2. Herringbone: neoplastic cells are arranged in long, intersecting fascicles. This pattern of growth is typically observed in the fibrosarcomatous variant of dermatofibrosarcoma protuberans (FS-DFSP) (Fig. 1-10).
3. Storiform: neoplastic cells are arranged in intersecting fascicles of variable length. This pattern is typically observed in DFSP but is also seen in undifferentiated pleomorphic sarcoma (Fig. 1-11).
4. Alveolar: neoplastic cells form roundish aggregates often surrounded by vascularized stroma. Loss of cohesion at the center of the neoplastic aggregate may occur, somewhat reminiscent of a lung alveolus. This pattern of growth is typically observed in alveolar soft part sarcoma and alveolar rhabdomyosarcoma (Fig. 1-12).
5. Solid: neoplastic cells are arranged in solid sheets of variable dimension (Fig. 1-13).
6. Biphasic: neoplastic cells are organized in two (rarely more) patterns. The prototype is represented by biphasic synovial sarcoma wherein a spindle cell component is organized in fascicles, whereas an epithelioid cell component forms glands or gland-like structures (Fig. 1-14). A biphasic pattern is typically observed in mesenchymal chondrosarcoma (cartilage plus high-grade round/spindle cell proliferation) but it can also be observed in dedifferentiated liposarcoma and in malignant peripheral nerve sheath tumors, wherein several types of heterologous differentiation may occur.






The Quality of the Background
The quality of the background refers to the characteristics of the extracellular stroma in which the neoplastic cell population is embedded.
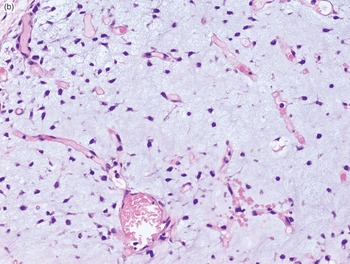
1. Fibrous: neoplastic cells are set in a variably collagenized stroma. Collagen fibers can be of variable thickness, from thin and fibrillary to coarse. A fibrillary background is typically observed in the sclerosing subtype of well-differentiated liposarcoma (Fig. 1-15). Collagen fibers are typically coarser in solitary fibrous tumor (Fig. 1-16). The distribution of the collagen is also of great help in recognizing specific entities. The presence of pericellular collagen is, for example, associated with synovial sarcoma (Fig. 1-17), whereas the presence of perivascular collagen is most often observed in solitary fibrous tumor (Fig. 1-18) and in benign schwannoma (Fig. 1-19).
2. Sclerotic: neoplastic cells are set in a heavily collagenized stroma that acquires a solid, somewhat glassy appearance of an osteogenic-mimicking matrix. A sclerotic background is typically observed in sclerosing epithelioid fibrosarcoma (Fig. 1-20), and sclerosing rhabdomyosarcoma (Fig. 1-21).
3. Myxoid: neoplastic cells are set in a stroma rich in mucin. Myxoid degeneration of the stroma is rarely observed in virtually all soft tissue tumors. However, as the presence of a myxoid background characterizes distinctive subtypes of soft tissue malignancies such as myxoid liposarcoma, myxofibrosarcoma, and extraskeletal myxoid chondrosarcoma (Fig. 1-22; Table 1.9), they are discussed separately in Chapter 8.
4. Myxochondroid: neoplastic cells are set in a stroma rich in mucin. In addition, the stroma assume a more condensed texture, somewhat similar to the chondrogenic matrix. This type of stroma can be observed in epithelioid hemangioendothelioma (Fig. 1-23).
5. Osteogenic: neoplastic cells are surrounded by a dense eosinophilic matrix most often organized in a lace-like configuration. The presence of an osteogenic matrix represents a prerequisite to the diagnosis of extraskeletal osteosarcoma (Fig. 1-24).










The Architecture of the Vascular Network
Blood vessels can variably organize to form a plexiform, archiform, or HPC-like architecture. These features can be extremely helpful in recognizing specific tumor entities, at least to address the differential diagnosis within a limited number of options.
1. Plexiform architecture: blood vessels are capillary sized and organized to form a richly anastomosed network. This pattern has been variably labeled as “chicken wire” or “crow’s feet,” depending on the imagination of the pathologist. This architecture is typically observed in myxoid liposarcoma (Figs. 1-25 and 1-26).
2. Archiform architecture: blood vessels are capillary sized and exhibit an archiform shape. This architecture is typically observed in myxofibrosarcoma (Figs. 1-27 and 1-28).
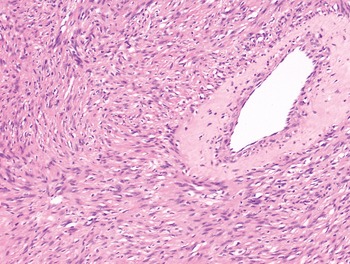
3. Hemangiopericytoma-like architecture: blood vessels are branching, dilated, and thin walled, generating a “staghorn” configuration. This architecture is observed in several (from benign to malignant) tumor entities but most often in solitary fibrous tumor and synovial sarcoma (Fig. 1-29). The lesions associated with an HPC-like vascular network are listed in Table 1.10.


Fig. 1-26. Myxoid liposarcoma. Anastomosing, thin-walled blood vessels may form the so-called chicken-wire pattern.


Fig. 1-28. High-grade myxofibrosarcoma. The presence of archiform, capillary-sized blood vessels is also retained in high-grade forms of myxofibrosarcoma.

| Benign soft tissue neoplasms featuring an HPC-like vascular network |
| Myofibroma/myofibromatosis |
| Myopericytoma |
| Deep-seated benign fibrous histiocytoma |
| Malignant soft tissue neoplasms featuring an HPC-like vascular network |
| Solitary fibrous tumor |
| Phosphaturic mesenchymal tumor |
| Synovial sarcoma |
| Extraskeletal mesenchymal chondrosarcoma |
| Malignant peripheral nerve sheath tumor |
| Infantile fibrosarcoma |
The integration of the aforementioned morphologic features reduces significantly the number of diagnostic options to the extent that, in some instances, ancillary technique may play a rather limited role. As an example, a hypocellular spindle cell proliferation set in a myxoid background and associated with a plexiform vascular network has the greatest chance to represent an example of myxoid liposarcoma (Fig. 1-30A–B). Following the same approach, a spindle cell proliferation set in a fibrous background, exhibiting variation in cellularity, and featuring an HPC-like vascularization most often represents an example of solitary fibrous tumors (Fig. 1-31A–B). However, in most situations an accurate diagnosis may require a second step represented by the application of a variable (ideally relatively limited) number of immunohistochemical stains.

Fig. 1-30A. Myxoid liposarcoma. A hypocellular spindle cell proliferation is seen at low power.

Fig. 1-30B. Myxoid liposarcoma. At high power, the distinctive plexiform vascular network is appreciated. This combination of morphologic features identifies almost unequivocally a myxoid liposarcoma.

Fig. 1-31A. Solitary fibrous tumor. At low power, a spindle cell proliferation set in a fibrous background and featuring variation of cellularity is seen.

Fig. 1-31B. Solitary fibrous tumor. The association of the morphologic features described in Fig. 1-31A with the presence of a hemangiopericytoma-like vascular network almost certainly identifies a solitary fibrous tumor.
Immunohistochemical Characterization of Soft Tissue Tumors
Immunohistochemical characterization plays a key role in the diagnostic workup of soft tissue sarcomas. However, a blind application of a broad range of immunophenotypic markers unsupervised by morphology most often leads to diagnostic errors. The determination of the line of differentiation is crucial in order not only to ensure proper classification but also to provide prognostic and/or predictive information. The number of potential diagnostic markers has grown exponentially through the years; in consideration of the natural evolution of the field, however, some markers have lost their role, while others have gained diagnostic relevance. It has to be underlined that, with some exceptions that will be discussed, the majority of classic differentiation markers tends to show good sensitivity, although associated with rather limited specificity. This may not represent a problem if interpretation is strictly handled in context with morphology.
We will herein focus on those differentiation markers showing major diagnostic as well as clinical relevance. Of course, more details will be given while discussing the specific tumor entities. The use of a panel of immunostains driven by morphology is felt to represent the most efficient approach. We will briefly describe the application of time-honored (however, still valid) differentiation markers as well as of newly reported ones, focusing on those having a consolidated diagnostic utility.
Myogenic Differentiation Markers
Demonstration of myogenic (both smooth muscle and striated) differentiation is clinically relevant in the following situations:
1. To differentiate between rhabdomyosarcoma and non-rhabdomyosarcoma pediatric soft tissue tumors (those two broad groups definitely undergo distinct systemic treatments).
2. To recognize adult myogenic sarcomas in general. As is discussed in depth in Chapter 7, the separation of both pleomorphic leiomyosarcoma and rhabdomyosarcoma from the undifferentiated pleomorphic sarcoma category is also relevant, as they represent prognostically unfavorable histologic subtypes.
3. To identify rhabdomyoblastic differentiation in malignant peripheral nerve sheath tumors, as it is associated with worse prognosis.
4. To identify myogenic differentiation in dedifferentiated liposarcoma, wherein it also seems to associate with poorer outcome.
Classic myogenic markers are basically represented by smooth muscle actin, muscle specific actin, desmin, and h-caldesmon.
Smooth muscle actin immunopositivity is observed in most smooth muscle tumors and in myofibroblastic, myoepithelial, and pericytic neoplasms. Muscle specific actin (HHF-35) also stains smooth muscle lesions; however, it also represents a very sensitive marker of striated muscle differentiation, staining up to 90% of rhabdomyosarcomas of all subtypes (embryonal, alveolar, and pleomorphic). Desmin belongs to the category of intermediate filaments and represents a valid marker for both smooth muscle and striated muscle lineage. A subset of myofibroblastic neoplasms may also express desmin. Expression of desmin is also observed in non-myogenic lesions such as desmoplastic small round cell tumor and angiomatoid fibrous histiocytoma, wherein it represents a great diagnostic help. h-Caldesmon immunoreactivity is observed in most smooth muscle neoplasms. In contrast to other smooth muscle markers, it tends to be negative in myofibroblastic lesions as well as in rhabdomyoblastic neoplasms. h-Caldesmon positivity is consistently observed in GIST and glomus tumors.
The most specific and sensitive markers to demonstrate rhabdomyoblastic differentiation remains myogenin (MYF4), a lineage-restricted nuclear transcription factor involved in striated muscle differentiation. Recently, it was shown that MyoD1 (MYF3), an alternative nuclear transcription factor involved in the development of striated muscle, represents the most sensitive marker for the spindle cell variant of rhabdomyosarcoma, wherein MyoD1 gene homozygous mutations have been shown to occur.
1. Focal smooth muscle actin positivity can be observed in a broad variety of spindle cell mesenchymal and even non-mesenchymal neoplasms (i.e., sarcomatoid carcinoma and sarcomatoid melanoma).
2. Desmin is consistently expressed in non-myogenic neoplasms such as desmoplastic small round cell tumor and angiomatoid fibrous histiocytoma.
3. Myogenin expression in pleomorphic rhabdomyosarcoma can be limited to a few cells and therefore can be easily overlooked.
4. Myogenin expression is retained by infiltrated and degenerated normal striated muscle fibers, and therefore it can be misinterpreted as evidence of rhabdomyoblastic differentiation.
5. Rhabdomyoblastic differentiation does not equate to rhabdomyosarcoma as it can be seen as a heterologous component in MPNSTs (so-called malignant triton tumor), Wilms’ tumor, dedifferentiated liposarcoma, mullerian adenosarcoma, malignant mullerian mixed tumor of the genital tract (so-called carcinosarcoma), biphenotypic sinonasal sarcoma, and, in malignant phyllodes, tumor of the breast.
Neural Differentiation Markers
Paradoxically, the best use of the prototypic schwannian differentiation markers, namely, S100 protein (named after its 100% solubility in ammonium sulphate), is achieved out of context of recognition of malignant peripheral nerve sheath tumors (MPNSTs). In fact, approximately only 30% of MPNSTs exhibit S100 positivity, which is usually limited to less than 30% of neoplastic cells. Epithelioid MPNST represents an important exception, as S100 usually decorates most neoplastic cells. It has to be stressed that S100 exhibits a distinctive multispecificity (Table 1.11) to the extent that its evaluation needs to be strictly performed in context with morphology.
| Benign neoplasms featuring S100 immunopositivity |
| Melanocytic nevi |
| Benign neural neoplasms |
| Benign adipocytic neoplasms |
| Granular cell tumor |
| Langerhans cell histiocytosis |
| Rosai-Dorfman disease |
| Malignant neoplasms featuring S100 immunopositivity |
| Malignant melanomas |
| Clear cell sarcomas (both soft tissue and GI tract) |
| Alveolar rhabdomyosarcoma |
| Epithelioid malignant peripheral nerve sheath tumor (100%) |
| Synovial sarcoma (30%) |
| Ossifying fibromyxoid tumor |
| Myoepithelial carcinoma |
| Extraskeletal myxoid chondrosarcoma (20%) |
| Malignant peripheral nerve sheath tumors (30%) |
| Biphenotypic sinonasal sarcoma |
| Myxoid liposarcoma |
| Langerhans cell sarcoma |
| Interdigitating reticulum cell sarcoma |
The best use of S100 immunostains is as follows:
1. Recognition of benign neural neoplasms.
2. Support in the diagnosis of cellular schwannoma (that in contrast to MPNSTs expresses S100 diffusely).
3. Recognition of metastatic sarcomatoid melanoma (wherein commonly used melanocytic differentiation markers such as HMB45 and Melan-A can be lost).
4. Support (in association with expression of epithelial differentiation markers) in the recognition of myoepithelial differentiation.
5. Identification (in association with the expression of melanocytic markers) of clear cell sarcoma of soft tissue, with the important caveat that clear cell sarcoma of the GI tract most often lacks expression of melanocytic markers.
6. Recognition (along with loss of nuclear expression of INI1) of epithelioid MPNST.
7. Support in the recognition of interdigitating dendritic cell sarcoma.
8. Recognition of cartilaginous differentiation (particularly useful in identifying the focal heterologous component in MPNST and dedifferentiated liposarcoma).
Another member of the group of the intermediate filaments, glial fibrillary acidic protein (GFAP) is also detectable in Schwann cells. When dealing with soft tissue neoplasms, it may stain a minority of MPNSTs; however, its sensitivity does not exceed that of S100. GFAP immunopositivity can also be observed in myoepithelial neoplasm.
SOX10 and H3K27me3 (histone 3K27 trimethylation) represent more recently introduced markers that can be used in the diagnosis of neural soft tissue neoplasm. SOX10 represents a transcription factor belonging to the SRY-related HMG-box family. SOX10 is involved in melanogenesis and schwannian differentiation, and has been proposed as a valid immunohistochemical marker for both melanocytic and neural neoplasms. SOX10 tends to immunostain the vast majority of benign neural mesenchymal lesions, whereas when dealing with MPNSTs its sensitivity is much lower, with approximately 20% of cases showing positivity in a minority of neoplastic cells. Importantly, SOX10 immunopositivity is also observed in myoepithelial neoplasms, astrocytic tumors, and metaplastic and basal-like breast carcinomas. H3K27me3 expression tends to be lost in approximately half of MPNSTs as a consequence of homozygous inactivation of the polycomb repressive complex 2 (PRC2). H3K27me3 seems to show relatively good specificity but it does not offer greater sensitivity than other neural markers.
1. Approximately 30% of monophasic synovial sarcomas may also exhibit S100 immunopositivity, causing significant immunophenotypic overlap with MPNSTs.
2. S100 immunopositivity in a cutaneous spindle cell “sarcomatous” tumor supports metastatic sarcomatoid melanoma rather than MPNST that virtually never occurs as a primary cutaneous neoplasm.
3. S100 immunopositivity is associated with expression of melanocytic markers HMB45 and Melan-A in clear cell sarcoma (CCS) of soft tissue; however, the same association seems to be absent in most (but not all) examples of CCSs of the gastrointestinal tract.
4. SOX10 immunoreactivity is also observed in melanocytic as well as myoepithelial neoplasms.
Epithelial Differentiation Markers
Any soft tissue neoplasm featuring true epithelial differentiation or epithelioid morphology is characterized by variable expression of epithelial differentiation markers, namely, cytokeratin and epithelial membrane antigen (EMA). Cytokeratin expression is observed in up to 80% of classic synovial sarcomas and in about half of those that are poorly differentiated. Cytokeratins actually represent a family of molecules classically catalogued by Moll into 20 types. The most widely used is the multispecific cytokeratin cocktail AE1/AE3 that recognizes keratins 9–17 (AE1) and keratins 1–8 (AE3). Importantly, AE1/AE3 does not recognize keratin 18. Cytokeratin AE1/AE3 decorates virtually all examples of epithelioid sarcomas, desmoplastic small round cell tumors, and pseudomyogenic hemangioendotheliomas. It is also observed in half of epithelioid angiosarcomas and, less frequently, in epithelioid hemangioendotheliomas (EHEs). Importantly, expression of cytokeratin is relatively commonly observed in histotypes wherein it is somewhat unexpected, such as leiomyosarcoma, retroperitoneal schwannomas, inflammatory myofibroblastic sarcoma, alveolar rhabdomyosarcoma, and Ewing sarcoma. Epithelial membrane antigen also stains most epithelioid sarcomas, and 90% of synovial sarcomas (including those that are poorly differentiated), therefore representing the most sensitive marker of epithelial differentiation in this context. By contrast, in sarcomatoid carcinoma cytokeratin expression can be very limited and therefore easily overlooked. EMA is also expressed consistently in meningothelial cells, perineurial cells, and subsets of plasma cells.
1. Focal expression of cytokeratin can be observed in a variety of unrelated neoplasms featuring sarcomatoid morphology such as melanoma and carcinoma.
2. Expression of epithelial differentiation markers in sarcomatoid carcinoma can be limited to a few neoplastic cells and therefore easily overlooked.
3. Poorly differentiated synovial sarcoma is often keratin negative, whereas EMA expression is retained in the vast majority of cases.
4. EMA positivity within plasma cells can be misinterpreted as expression in neoplastic cells.
Endothelial Differentiation Markers
Demonstration of endothelial differentiation appears crucial when dealing with poorly differentiated vascular neoplasms, in particular epithelioid angiosarcoma that may often lack overt vasoformative morphology. CD34, CD31, and FlI1 represent time-honored but still valid markers. The use of factor VIII-RA, despite its high specificity, is strongly limited by a relatively low sensitivity. CD34 is a sialomucin expressed in the precursors of hematopoiesis, endothelial cells, and various subsets of fibroblasts. When dealing with vascular neoplasms, CD34 is very sensitive, but it has to be stressed that it is also expressed in half of epithelioid sarcomas in addition to an endless list of non-vascular, spindle cell neoplasms (Table 1.12). Much more specific and extremely sensitive is the transmembrane glycoprotein CD31 (also known as platelet-endothelial cell adhesion molecule-1 [PECAM-1]); however, it is consistently expressed by histiocytes, megakaryocytes, and subsets of myeloblasts. FLI1 represents an ETS-related transcription factor that shows high sensitivity in detecting normal as well as neoplastic endothelium. Importantly, FLI1 immunopositivity is observed in most Ewing sarcomas, lymphoblastic lymphomas, and, more rarely, in melanomas and carcinomas. ERG (another member of the ETS-related family of nuclear transcription factors) represents a recently developed, sensitive endothelial marker. ERG specificity is also reasonably good, with a major caveat represented by the fact that ERG also stains prostatic adenocarcinomas, a small subset of Ewing sarcomas carrying the EWSR1-ERG translocation, and approximately 40% of epithelioid sarcomas.
Table 1.12 Non-vascular soft tissue neoplasms exhibiting CD34 immunopositivity
Important diagnostic adjuncts in the context of the diagnosis of vascular lesions are represented by nuclear detection of human herpesvirus 8 (HHV-8) in Kaposi sarcoma that appears to be very specific and of c-MYC in post-radiation cutaneous angiosarcoma.
In recent years, as a byproduct of new insights in the molecular pathogenesis of vascular neoplasms, a number of new diagnostic immunohistochemical markers have been made available. The recognition of EHE is currently greatly helped by the detection of the nuclear expression of CAMTA1 that is observed in those cases harboring a WWTR1-CAMTA1 gene fusion. TFE3 nuclear expression is observed in a subset of EHEs carrying the YAP1-TFE3 gene fusion. FOSB expression is consistently observed in pseudomyogenic hemangioendothelioma (associated with a FOSB-SERPIN1 gene fusion) as well as in a subset of epithelioid hemangioma (associated with a ZFP36-FOSB gene fusion).
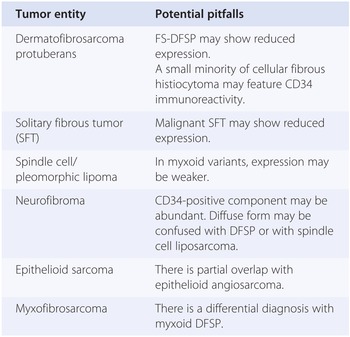
1. CD34 is expressed in dermal fibroblasts and in a broad variety of spindle cell neoplasms. By contrast, it can be negative in a variety of vascular neoplasms, particularly in epithelioid angiosarcomas.
2. CD34 expression is observed in approximately 50% of epithelioid sarcomas, leading to potential diagnostic confusion with epithelioid angiosarcomas.
3. CD31 is consistently expressed in intra-tumoral histiocytes, potentially leading to overdiagnosis of vascular malignancies.
4. CD31 is expressed in platelets where the antigen can be absorbed on the outer surface of neoplastic cells.
5. ERG is consistently expressed in prostatic adenocarcinomas as well as in up to 40% of epithelioid sarcomas.
Melanocytic Differentiation Markers
Despite total lack of specificity, S100 remains the most sensitive marker of melanocytic differentiation. HMB45, Melan-A (MART1), and MITF1 all represent sensitive melanocytic differentiation; however, their sensitivity drops significantly when dealing with sarcomatoid variants of malignant melanomas, wherein S100 is most often the only expressed diagnostic marker. Negativity of S100 in “bona fide” examples of metastatic melanomas may occur, but it represents a much more rare event. They not only play an obvious role in the differential diagnosis of malignant melanoma, but they have also proved extremely helpful in recognizing the members of the clinically and morphologically heterogeneous family of PEComas, as well as being helpful in the proper classification of clear cell sarcomas.
1. Most melanocytic differentiation markers such as Melan-A and HMB45 tend to be negative in sarcomatoid melanoma.
2. Melanocytic markers are strongly expressed in clear cell sarcoma of soft tissue but much less in those lesions arising in the GI tract.
Other Useful Immunohistochemical Markers
There exists an increasing number of immunohistochemical markers that despite variable specificity play a major role in the differential diagnosis of soft tissue sarcomas.
ALK (Anaplastic Lymphoma Kinase)
Cytoplasmic expression of the tyrosine kinase ALK plays a key role in supporting the diagnosis of inflammatory myofibroblastic tumors (IMTs), including the aggressive epithelioid subtypes. The pattern of staining (cytoplasmic, perinuclear, membrane) may predict the underlying genetic aberration. Its presence is also potentially relevant from a therapeutic standpoint to justify its use in the metastatic setting of ALK inhibitors. Unfortunately, approximately half of IMTs lack ALK expression. In addition to anaplastic large cell lymphomas and to a small subset of lung adenocarcinomas, ALK expression is also observed in alveolar rhabdomyosarcoma and in epithelioid benign fibrous histiocytoma.
Beta-catenin
Nuclear expression of beta-catenin (a 92 KDa protein that normally localizes in the cytoplasm wherein it acts both as an intracellular signal transducer in the Wnt signaling pathway and in cadherin-mediated cell adhesion) represents an extremely valuable confirmatory finding in the diagnosis of desmoid fibromatosis. Nuclear accumulation is due to mutation of the CTNNB1 gene (found in up to 90% of sporadic desmoid fibromatosis) or, alternatively, of the APC gene (in the context of Gardner syndrome). A major pitfall is represented by the fact that beta-catenin expression can be observed in approximately one-third of solitary fibrous tumors, in synovial sarcoma, and in low-grade myofibroblastic sarcoma. Interestingly, nuclear expression of beta-catenin (associated with CTNNB1 gene mutations) occurs in sinonasal hemangiopericytoma.
Brachyury (T)
Brachyury is a protein acting as a transcriptional activator of notochordal development. Nuclear expression of brachyury represents a key diagnostic feature in the recognition of chordoma and is very helpful in the differential diagnosis with chondrosarcoma, metastatic carcinoma, and myoepithelial neoplasms.
CD63
The most relevant diagnostic application of CD63 is in the recognition of cellular neurothekeoma. CD63 is not specific because it is also commonly seen in melanocytic lesions.
CD68
CD68 (a 110 kDa glycoprotein) represents a lysosome-specific marker and therefore is generally used as a histiocytic differentiation marker. However, CD68 tends to be positive in several lysosome-rich lesions such as granular cell tumor and neural and melanocytic neoplasms.
CD163
CD163 represents a specific as well as sensitive histiocytic marker that at variance with CD68 exhibits very limited expression in non-histiocytic lesions.
CD99
When dealing with the differential diagnosis of small round cell sarcomas, CD99 (a cell surface glycoprotein normally expressed on thymic T cells) represents a powerful diagnostic marker. In fact, virtually all Ewing sarcomas exhibit strong CD99 immunopositivity, to the extent that such a diagnosis should be regarded as doubtful in case of CD99 negativity. However, it has to be underlined that CD99 is expressed rather broadly, and among potential mimics of Ewing sarcoma, CD99 is also detectable in 90% of synovial sarcomas (including the poorly differentiated round cell variant), in 40% of Merkel cell carcinomas, in lymphoblastic lymphomas, and in mesenchymal chondrosarcoma. Importantly, immunopositivity in all these lesions tends to be more diffuse, rarely matching the thick membrane staining typically observed in Ewing sarcoma.
Cyclin-B3
Expression of the cell cycle regulator cyclin-B3 seems to be useful in the recognition of a small subset of poorly differentiated round cell sarcomas associated with a BCOR-CCNB3 gene fusion.
DOG1
DOG1 (discovered on GIST 1, also known as anoctamin 1 [ANO1] and TMEM16A) represents a useful and sensitive marker for diagnosis of GIST. DOG1 is a calcium-activated chloride channel and is expressed in the interstitial cells of Cajal. DOG1 is extremely helpful in rescuing at least half of KIT-negative GIST cases. Importantly, DOG1 immunopositivity can be observed in smooth muscle tumors of the retroperitoneum as well as in synovial sarcoma.
INI1 (SMARCB1)
INI1 (integrase interactor 1, also known as SMARCB1) is a member of the BAF molecular complex that contributes to the process of chromatin remodeling and plays a key role in regulating transcription of DNA. Loss of INI1 nuclear expression is consistently observed in malignant rhabdoid tumors (including atypical teratoid rhabdoid tumors of the central nervous system) as a consequence of INI1 biallelic inactivation. Loss of INI1 is also observed in up to 95% of both classical and proximal variants of epithelioid sarcoma wherein it tends to be associated with homozygous deletion of the INI1 locus. INI1 negativity has been described in up to 70% of epithelioid MPNSTs, in 10 to 35% of myoepithelial carcinomas, and in almost all renal medullary carcinomas.
KIT (CD117)
KIT represents a tyrosine kinase receptor that is involved in the development of mast cells, melanocytes, and interstitial cells of Cajal. KIT has become one of the most clinically relevant phenotypic markers. In fact, its expression in GIST permits the accurate recognition of this once orphan tumor, offering proper selection of patients for treatment with tyrosine kinase inhibitors. KIT expression does not predict response to tyrosine kinase inhibitors and can be observed in unrelated neoplasms such as seminoma, thymic carcinoma, melanocytic neoplasm, and mast cell disorders.
MDM2
MDM2 (murine double minutes) is the product of the MDM2 proto-oncogene, the main function of which is to promote cell proliferation via inhibition of TP53. Both well-differentiated liposarcoma and dedifferentiated liposarcoma are characterized by strong nuclear overexpression of MDM2 as a consequence of MDM2 gene amplification. MDM2 immunopositivity is most often associated with CDK4 overexpression. A possible pitfall is represented by the fact that MDM2 multifocal immunopositivity (unrelated to gene amplification) can be observed in MPNST, rhabdomyosarcoma, and myxofibrosarcoma. Among mesenchymal malignancies, MDM2 overexpression associated with gene amplification is also observed in intimal sarcoma and in low-grade central osteosarcoma of bone.
MUC4
MUC4 (mucin 4) is a high molecular weight transmembrane glycoprotein normally expressed in epithelial cells. The major diagnostic role of MUC4 is represented by diagnostic confirmation of both low-grade fibromyxoid sarcoma and sclerosing epithelioid fibrosarcoma, wherein gene expression profiling has identified its upregulation. MUC4 expression is not observed in spindle cell lesions that enter the differential diagnosis with LGFMS (Low Grade Fibromyxoid Sarcoma), perineurioma, low-grade myxofibrosarcoma, and desmoid fibromatosis; nor in other tumors associated with prominent sclerotic background such as sclerosing epithelioid rhabdomyosarcoma. Expression of MUC4 is, however, observed in the epithelial component of biphasic synovial sarcoma. MUC4 immunostaining (along with SATB2) is also helpful in the differential diagnosis with extraskeletal osteosarcoma, a lesion in which distinguishing osteogenic matrix from sclerotic collagen is not always an easy task.
RB
RB (retinoblastoma gene product) is the product of the RB tumor suppressor gene that acts as a potent negative regulator of the cell cycle. From a diagnostic standpoint, loss of nuclear expression of RB is observed in spindle cell and pleomorphic lipomas. The same phenomenon is also observed in atypical spindle cell lipomatous tumor.
SATB2
Nuclear expression of SATB2 (special AT-rich sequence-binding protein 2) is observed in neoplasm featuring osteogenic differentiation. Normal function of SATB2 is to enable osteoblast lineage commitment. Importantly, SATB2 expression is shared by both normal and neoplastic osteoblasts and therefore plays no role in the differential diagnosis between benign and malignant osteogenic neoplasms. SATB2 is best used whenever hematoxylin and eosin (H&E) stain does not discriminate unequivocally between osteoid and sclerotic collagen, or when osteoid is either minimally represented or not represented in the sample.
STAT6
Nuclear expression of STAT6 (signal transducer and activator of transcription-6) is consistently expressed in all variants of solitary fibrous tumors. Nuclear relocation of STAT6 nuclear expression is determined by the occurrence of the NAB2-STAT6 gene fusion.
TFE3
TFE3 (transcription factor E3) decorates the neoplastic cell population of alveolar soft part sarcoma (as a consequence of the presence of the ASPSCR1-TFE3 gene fusion that leads to the overexpression of the TFE3 protein) but it is not an entirely specific marker. In fact, it is also expressed in Xp11 translocated renal cell carcinomas, in a small subset (approximately 5%) of perivascular epithelioid cell tumors (PEComa), and, as already discussed, in a subset of epithelioid hemangioendothelioma.
TLE1
TLE1 (transducing-like enhancer of split 1) is a transcriptional co-repressor that inhibits Wnt signaling. Gene profiling studies have demonstrated high levels of TLE1 in synovial sarcoma that can be detected immunohistochemically. An important potential pitfall is represented by the fact that TLE1 has limited as well as weak immunopositivity may also be observed in unrelated sarcomas such as MPNSTs or solitary fibrous tumors.
WT1
Nuclear expression of the c-terminus WT1 (Wilms’ tumor 1) can be used as a confirmatory tool in the diagnosis of desmoplastic small round cell tumor.
Molecular Characterization of Soft Tissue Tumors
The marriage of molecular genetics and soft tissue tumor pathology certainly represents one of the most fruitful events of the past decade. The close relationship between morphology (including immunohistochemistry) and molecular genetics was certified by the 2002 WHO classifications of bone and soft tissue tumors and further expanded in the 2013 update.
This integration has had an impact on several aspects of pathology:
1. Created a more accurate definition of disease entities and validation of classification schemes.
2. Improved diagnostic accuracy.
3. Identified molecular predictive and prognostic markers.
4. Discovered and validated therapeutic molecular targets.
Definition of Disease Entities and Validation of Classification Scheme
Molecular genetics has contributed greatly to a more robust definition of histologic subtypes. This is particularly true whenever new entities are described and also applies to the revision of classification schemes. As an example, the fact that the morphologic description of pseudomyogenic hemangioendothelioma has been followed by the identification of a “disease-specific” FOSB-Serpine1 gene fusion has greatly contributed to the credibility of this newly identified entity. In addition, molecular genetics has contributed greatly to refining and modifying histologic classification. The merging of myxoid liposarcoma and round cell liposarcoma into a single entity (first generated by morphologic observations) has been greatly supported by the detection in both lesions of the same rearrangement involving the CHOP gene. Similarly, identification of MDM2 gene amplification in so-called inflammatory MFH has represented a key finding, allowing reclassification of those malignancies as an inflammatory variant of dedifferentiated liposarcoma.
Improvement of Diagnostic Accuracy
From a diagnostic standpoint, during the past two decades it has become clear that molecular testing may add diagnostic accuracy in important subsets of challenging soft tissue tumors. In fact, many of these lesions are known to harbor a variety of relatively specific point mutations, gene amplifications, and chromosome translocations. Gene fusion seems to be particularly frequent in soft tissue neoplasms, the most common of which are listed in Table 1.2. Their occurrence can be routinely assessed for diagnostic purposes via fluorescence in situ hybridization (FISH) and/or polymerase chain reaction (PCR) techniques. It can be foreseen that next-generation sequencing (NGS) technology will greatly contribute to a comprehensive genetic evolution of mesenchymal malignancies.
The diagnostic utility of molecular genetics can be best exemplified in the following situations:
1. Distinguishing specific subtypes of sarcomas.
2. Supporting diagnosis in non-canonical clinical presentations.
3. Distinguishing sarcomas from benign mimickers.
Distinguishing Specific Subtypes of Sarcomas
The distinction of specific sarcoma subtypes is becoming increasingly important as more specific local as well as systemic treatments are being developed. Molecular genetics/pathology has proved diagnostically useful in all morphologic groups of mesenchymal malignancies. Round cell sarcomas represent the perfect example to underscore how clinically relevant molecular diagnostics has become. Round cell sarcomas include Ewing sarcoma, desmoplastic small round cell tumor (DSRCT), alveolar rhabdomyosarcoma, poorly differentiated round cell synovial sarcoma (PDSS), CIC-rearranged and BCOR-rearranged undifferentiated round cell sarcomas, and a minority of cases of high-grade liposarcomas featuring a round cell morphology. They all represent aggressive neoplasms, and their distinction is crucial because therapeutic approaches may differ significantly. As morphologic overlap may be at times extreme, to the extent that even immunohistochemical characterization cannot help in achieving a definitive classification (i.e., keratin positive Ewing sarcoma vs. poorly differentiated round cell synovial sarcoma), molecular genetics may play a key diagnostic role. As shown in Table 1.2, all these entities harbor relatively specific genetic aberrations that can be routinely assessed, contributing to increased diagnostic accuracy. The demonstration by FISH of EWSR1, SS18, and FOXO1 rearrangements in Ewing sarcoma, rhabdomyosarcoma, and PDSS, respectively, or, alternatively, of specific chimeric transcripts by PCR-based techniques or via an NGS-based approach, is of great help in achieving a correct diagnosis. As far as FISH analysis of the EWSR1 gene is concerned, a major caveat is represented by the fact that its rearrangement can occur in a variety of unrelated lesions (Table 1.2). As a consequence, any result needs to be mandatorily interpreted in context with morphology and immunohistochemical findings.
The detection of translocations is not the only diagnostically relevant genetic event. The identification of gene copy number variations has also proved extremely helpful. The perfect example is represented by dedifferentiated liposarcoma (DDLPS), a pleomorphic, usually (but not exclusively) retroperitoneal adipocytic malignancy that, in contrast to other sarcomas such as leiomyosarcoma, exhibits the tendency to recur locally with a comparatively lower rate of metastatic spread. As a consequence, the surgical treatment of retroperitoneal dedifferentiated liposarcoma is currently based on multivisceral resection aimed at prolonging the time period until a local destructive recurrence. The recognition of dedifferentiated liposarcoma is generally based on the identification of a well-differentiated (WD) lipogenic component associated with a high-grade, most often non-lipogenic, sarcoma. In consideration of the increasing tendency to use core biopsies for diagnostic purposes, the well-differentiated lipogenic component is often not made available. In this context, the detection of MDM2 amplification by FISH or quantitative RT-PCR certainly represents a useful diagnostic adjunct. MDM2 testing is also potentially useful in distinguishing between myxoid liposarcoma (consistently MDM2 negative and most often instead characterized by a DDIT3/CHOP gene rearrangement) and WD/DDPLS with myxoid change, as well as to separate homologous dedifferentiated liposarcoma from pleomorphic liposarcoma. In most of these situations, it must be stressed that MDM2 immunohistochemistry (under corrrect laboratory conditions) matches both in terms of sensitivity and specificity of molecular techniques.
Supporting Diagnosis in Non-canonical Clinical Presentations
As a result of the widespread use of molecular pathology as a confirmatory diagnostic tool, the range of clinical presentations of many entities has broadened. In fact, the combination of morphologic criteria and genetics validates the recognition of rare diseases even when arising at non-canonical anatomic locations. This is particularly true for referral centers wherein challenging cases tend to concentrate. Molecular genetics has undoubtedly greatly contributed, for instance, to the identification of primary Ewing sarcoma of the skin, kidney, and meninges, as well as of synovial sarcomas occurring at visceral sites such as the lungs and the gastrointestinal tract.
Distinguishing True Sarcomas from Benign Mimics
As mentioned, the morphologic appearance of mesenchymal lesions does not always reflects the clinical behavior. The distinction of sarcomas from benign mimics most often relies on morphologic criteria; however, in a minority of cases molecular genetics may also prove diagnostically helpful. This is particularly true when dealing with low-grade fibromyxoid sarcoma (LGFMS), a deceptively bland-looking spindle cell mesenchymal malignancy characterized by an aggressive clinical behavior on a long-term basis. The differential diagnosis of LGFMS includes benign lesions such as perineurioma, neurofibroma, cellular myxoma, and nodular fasciitis, as well as locally aggressive neoplasms such as desmoid fibromatosis. Even if MUC4 expression is currently regarded as a key diagnostic feature, the identification of FUS rearrangement via interphase FISH or the identification of either FUS-CREB3L2 or FUS-CREB3L2 transcripts represents an extremely useful diagnostic tool. As mentioned, desmoid fibromatosis enters the differential diagnosis, and it should be therefore noted that in addition to immunohistochemical detection of nuclear accumulation of β-catenin, mutational analysis of the CTNNB1 gene may also represent a valuable diagnostic tool.
Identification of Molecular Predictive and Prognostic Markers
During the past decade, several attempts have been made to determine the prognostic value of molecular genetic findings. Most analyses have focused on Ewing sarcoma, alveolar rhabdomyosarcoma, and synovial sarcoma. Following initial enthusiasm, we have to admit that subsequent results have been contradictory and at this point no meaningful molecular prognostic stratification can be foreseen. The attempt to correlate molecular status and clinical behavior in desmoid fibromatosis has proved controversial. A potential exception is represented by a recently published molecular signature, complexity index in sarcomas (CINSARC), which reportedly allows better separation of grade 2 sarcomas. Nonetheless, this attempt was based on the use of a relatively complex technique (comparative genomic hybridization [CGH]-array) and requires availability of fresh material. Both factors may unfortunately hamper a large-scale clinical application of CINSARC.
An extremely important, clinically relevant exception is represented by gastrointestinal stromal tumors (GISTs), wherein the types of mutations involving both the KIT and PDGFRA genes are associated with distinctive outcomes. As examples, it is now well known that deletions occurring at exon 11 of the KIT gene are associated with more aggressive disease, whereas mutations of exon 18 of the PDGFRA gene generally identify a more indolent clinical course. Again, GIST represents the best example of successful prediction of response to treatment in sarcomas. Distinct mutation types reflect different objective response rates (greater for the KIT exon 11 mutation and much lower for so-called wild-type GIST) as well as different progression-free survival and overall survival. The presence of specific mutations of exon 18 of the PDGFRA gene (D842V) predicts primary resistance to tyrosine kinase inhibitors. Considering that imatinib is currently administered as an adjuvant treatment, the molecular assessment of GIST assumes a central role in clinical decision making. Mutational analysis in GIST also has an impact on dose selection; in fact, the progression-free survival of GIST patients with KIT exon 9 mutations is known to be significantly better in patients treated with 800 mg as compared with 400 mg per day.
More recent examples are represented by the use of crizotinib in inflammatory myofibroblastic tumors wherein assessment of ALK gene status may represent an important diagnostic confirmatory finding as well as a key biomarker of prediction.
It must be stressed how molecular pathology/genetics represents the most valuable tool used to identify and validate new therapeutic targets. Good examples are represented by MDM2 and CDK4 in dedifferentiated liposarcoma, the mTOR pathway in malignant PEComa and lymphangioleiomyomatosis, PDGFB in DFSP, KDR in angiosarcoma, NTRK3 in GIST, and CSF1 in giant cell tumor of tendon sheath.
The Grading of Soft Tissue Sarcomas
Soft tissue sarcomas tend to behave aggressively and metastasize in a large percentage of cases. Tumor size, location, depth, and histologic type are all prognostic factors in terms of metastatic risk and overall survival.
With many exceptions, histologic typing does not provide sufficient information for predicting the clinical course of the disease and, therefore, grading systems based on histologic parameters were introduced to provide a more accurate estimation of the degree of malignancy of tumors.
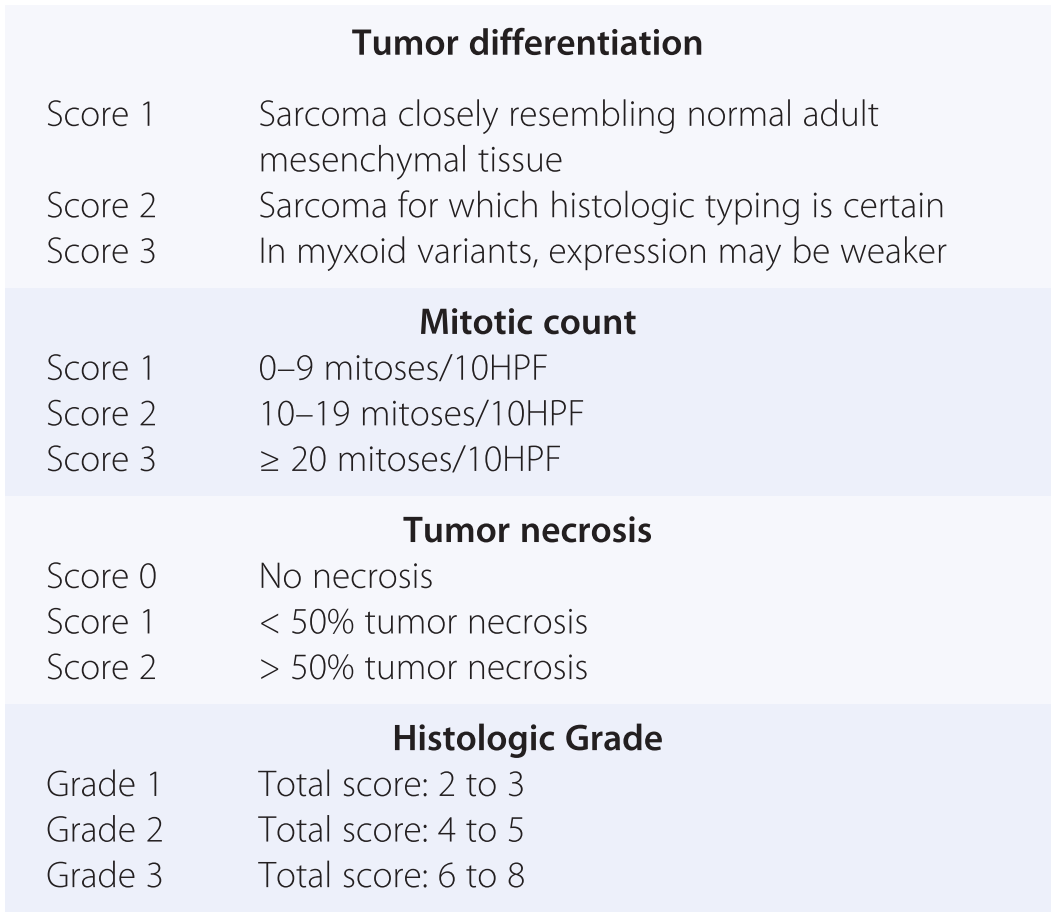
Several different grading systems have been developed. The most successful have been the National Cancer Institute (NCI) and the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) (French Federation of Cancer Centers Sarcoma Group) systems. Both are three-tiered systems. The FNCLCC system, created in 1984 and updated in 1997, was chosen for use in European Organisation for Research and Treatment of Cancer (EORTC) trials and advocated in both the 2002 and 2013 versions of the WHO classification. It offers slightly better discrimination between low- and high-grade sarcomas, the intermediate group being smaller. The FNCLCC grading system is based on the evaluation of three main parameters: (1) Differentiation; (2) Mitotic count per high-power fields (HPF); (3) Presence of necrosis. As summarized in Table 1.13, each parameter generates a score, the sum of which determines a final score that assigns the tumor to one of the three groups.
| Tumor differentiation | |
| Score 1 | Sarcoma closely resembling normal adult mesenchymal tissue |
| Score 2 | Sarcoma for which histologic typing is certain |
| Score 3 | In myxoid variants, expression may be weaker |
| Mitotic count | |
| Score 1 | 0–9 mitoses/10HPF |
| Score 2 | 10–19 mitoses/10HPF |
| Score 3 | ≥ 20 mitoses/10HPF |
| Tumor necrosis | |
| Score 0 | No necrosis |
| Score 1 | < 50% tumor necrosis |
| Score 2 | > 50% tumor necrosis |
| Histologic Grade | |
| Grade 1 | Total score: 2 to 3 |
| Grade 2 | Total score: 4 to 5 |
| Grade 3 | Total score: 6 to 8 |
The main limitations of the FNCLCC system (actually, of any grading system) are as follows:
1. Many sarcoma subtypes are ungradable. A typical example is alveolar soft part sarcoma, an aggressive lesion that by applying the FNCLCC system would be rated as G1.
2. The concept of differentiation is rather subjective and therefore hampered by poor reproducibility.
3. The FNCLCC system has been devised for surgical specimens from untreated patients. In real life, pathologic diagnosis is increasingly based on core biopsy, wherein no grading system has yet been validated. The same concept applies to surgical specimens from patients treated with neoadjuvant systemic therapy, wherein the presence of necrosis may actually reflect the effect of chemotherapy instead of an intrinsic quality of the tumor itself.
4. The FNCLCC system is not applicable to metastasis. The rationale is rather simple and based on the assumption that a metastatic tumor has already shown its potential for aggressiveness. Relatively often, however, metastases exhibit a morphology that does not match with that of the primary lesion and actually clearly indicates morphologic progression. Interestingly, clinicians tend to show increased interest in the morphologic features of metastatic lesions as a parameter for modulating systemic treatments.
Prognostic monograms that recently have incorporated the revised WHO classification of soft tissue tumors represent an additional, valuable clinical tool. From one of these monograms, there is now a freely downloadable application for portable devices called Sarculator.