


Introduction
The seed microbiome is the primary microbial inoculum for the plant microbiota (Barret et al., Reference Barret, Bonneau and Préveaux2015). These plant-associated microbes are acquired horizontally from the environment or vertically across generations (Shade et al., Reference Shade, Jacques and Barret2017; Abdelfattah et al., Reference Abdelfattah, Wisniewski, Schena and Tack2021). Some seed microbes were found to colonize the emerging seedling and adult plants efficiently, and exert lasting beneficial impacts on plant fitness by promoting seed germination and root symbiosis, producing phytohormones and essential nutrients, and increasing plant tolerance to biotic and abiotic stress (Bergna et al., Reference Bergna, Cernava, Rändler, Grosch, Zachow and Berg2018; Rodríguez et al., Reference Rodríguez, Antonielli, Mitter, Trognitz and Sessitsch2020; Matsumoto et al., Reference Matsumoto, Fan, Wang, Kusstatscher, Duan, Wu, Chen, Qiao, Wang, Ma and Zhu2021; Walsh et al., Reference Walsh, Becker-Uncapher, Carlson and Fierer2021). Therefore, seed-borne microbes have been identified as key organisms for biotechnological applications, including their use as biostimulants or biocontrol agents (Berg and Raaijmakers, Reference Berg and Raaijmakers2018; Matsumoto et al., Reference Matsumoto, Fan, Wang, Kusstatscher, Duan, Wu, Chen, Qiao, Wang, Ma and Zhu2021; Lobato et al., Reference Lobato, de Freitas, Habich, Kögl, Berg and Cernava2024). Recent studies suggested their consideration in microbiome-assisted breeding strategies (Adam et al. Reference Adam, Bernhart, Müller, Winkler and Berg2018; Wassermann et al., Reference Wassermann, Adam, Cernava, Berg, Verma and White2019). Healthy seeds containing a beneficial microbiome are important for sustainable plant production to ensure global food security. Therefore, it is important to understand the main drivers impacting the seed microbiome composition. A variety of factors have been identified, e.g., the plant genotype (Rybakova et al., Reference Rybakova, Mancinelli, Wikström, Birch-Jensen, Postma, Ehlers, Goertz and Berg2017; Chen et al., Reference Chen, Krug, Yang, Li, Yang, Berg and Cernava2020; Bziuk et al., Reference Bziuk, Maccario, Straube, Wehner, Sørensen, Schikora and Smalla2021), microbial inheritance from parental breeding lines (Fort et al., Reference Fort, Pauvert, Zanne, Ovaskainen, Caignard, Barret, Compant, Hampe, Delzon and Vacher2021; Wassermann et al., Reference Wassermann, Abdelfattah, Wicaksono, Kusstatscher, Müller, Cernava, Goertz, Rietz, Abbadi and Berg2022), seed origin (Lobato et al., Reference Lobato, de Freitas, Habich, Kögl, Berg and Cernava2024), agricultural management (Christian et al., Reference Christian, Sullivan, Visser and Clay2016; Wassermann et al., Reference Wassermann, Cernava, Goertz, Zur, Rietz, Kögl, Abbadi and Berg2023), and storage conditions (Chandel et al., Reference Chandel, Mann, Kaur, Norton, Edwards, Spangenberg and Sawbridge2021). Further, vertically transmitted endophytes, as persistent and prevalent components, can successfully colonize plant tissues (Sanz-Puente et al., Reference Sanz-Puente, Redondo-Salvo, Torres-Cortés, de Toro, Fernandes, Börner, Lorenzo, de la Cruz F and Robledo2025) and most likely play a significant role in influencing host phenotype and immunity (Matsumoto et al., Reference Matsumoto, Fan, Wang, Kusstatscher, Duan, Wu, Chen, Qiao, Wang, Ma and Zhu2021; Tariq et al., Reference Tariq, Tanvir, Barasarathi, Alsohim, Mastinu, Sayyed and Nazir2025). However, the impact of seed aging on the seed microbiome is still poorly understood.
Seed aging involves deterioration processes and loss of protection from damage, such as inactivation of antioxidant enzymes, elevated presence of reactive oxygen species, and membrane and genetic damage (Yin et al., Reference Yin, Xin, Song, Chen, Zhang, Wu, Li, Liu and Lu2014; Ebone et al., Reference Ebone, Caverzan and Chavarria2019). Therefore, if seeds have to be stored for long periods, i.e., in breeding companies and seed banks, they are kept under controlled dry and cold conditions to conserve seed vigour. Nonetheless, storage conditions affect aging mechanisms and hence seed vigour, resulting, amongst others, in lower total germination and higher abnormal germination (Finch-Savage and Bassel, Reference Finch-Savage and Bassel2016). Artificial aging treatments are used to investigate seed vigour and storability, which are critical to ex situ seed conservation, e.g., in genebanks (Hay and Whitehouse, Reference Hay and Whitehouse2017). Different artificial aging tests are used, such as controlled deterioration (Powell and Matthews, Reference Powell and Matthews2005) or accelerated aging. The artificial aging tests mimic seed aging by short-term exposure to high temperature and moisture (Yin et al., Reference Yin, He, Gupta and Yang2015; Fenollosa et al., Reference Fenollosa, Jené and Munné-Bosch2020; Malviya et al., Reference Malviya, Dey, Pandey and Gayen2023). They are conducted to estimate the storability of seed lots, where seed lots with a high percentage of germination are considered as high quality, and those with a low germination percentage are considered as low quality (Delouche and Baskin, Reference Delouche and Baskin2021). Artificial aging treatments were reported to result in symptoms typical exhibited by naturally aged seeds (Yin et al., Reference Yin, He, Gupta and Yang2015; Finch-Savage and Bassel, Reference Finch-Savage and Bassel2016), where natural seed aging refers to the gradual, irreversible loss of viability and vigour caused by cumulative physiological and molecular damage, such as oxidative stress, lipid peroxidation and protein/DNA degradation (Pirredda et al., Reference Pirredda, Fañanás-Pueyo, Oñate-Sánchez and Mira2024). However, it is important to note that not all molecular responses are equivalent between artificial aging and seed bank storage conditions (Gianella et al., Reference Gianella, Balestrazzi, Ravasio, Mondoni, Börner and Guzzon2022). Generally, accelerated aging experiments are used to understand plant-related processes during seed aging and have been shown to, e.g., impact the endophyte viability in grass–Epichloë species (Gundel et al., Reference Gundel, Martínez-Ghersa, Garibaldi and Ghersa2009, Reference Gundel, Martínez-Ghersa, Batista and Ghersa2010). However, their application to the broader associated seed microbiome remains largely unexplored.
In the present study, we have analysed the impact of accelerated aging stress on the seed bacterial community of oilseed rape (Brassica napus L.), one of the most important oilseed crops worldwide. Previous research on B. napus has shown that accelerated aging treatments can mimic the molecular changes observed in naturally aged seeds, particularly in terms of physio-chemical alterations (Yin et al., Reference Yin, He, Gupta and Yang2015). Here, we hypothesize that the stress imposed by accelerated aging will result in microbial diversity loss and taxonomic shifts towards stress-tolerant taxa. We exposed seeds of four different genotypes produced at two different field sites to the accelerated aging stress test and analysed the bacterial community after germination, on normal and abnormal seedlings, as well as on ungerminated seeds. Further, we aimed to identify taxa associated with the aging process and to find possible correlations with the reduced total germination.
Materials and methods
Experimental design and sample processing
Winter oilseed rape seeds of four different genotypes (named G1–G4) produced at two different field sites in Germany (Hohenlieth [HOH] and Hovedissen [HOV]), both sandy loam soil types, were used to investigate the effect of accelerated aging (Figure 1). Seeds were supplied by the NPZ Innovation GmbH (Germany). The seeds were harvested in July 2022 at both field sites (Supplementary Figure 1), and a subset of each accession received accelerated aging treatment in August 2022 at high temperature (45°C) and high humidity (>95%) for 48 h in a humidity-controlled incubator (Memmert, Germany). Seeds that were not exposed to accelerated aging treatment served as a control. Immediately after accelerated aging tests, total germination was evaluated under controlled conditions in two replicates per accession, where each replicate consisted of 100 seeds. Seeds were germinated for 6 days at 23°C and a 16/8-h photoperiod. Seeds were classified according to their germination phenotype: (i) normal or (ii) abnormal seedlings, following ISTA rules (ISTA, 2022), or (iii) ungerminated seeds.
Experimental design to investigate accelerated aging in B. napus seeds. Seeds from four genotypes (G1–G4) originating from two field sites in Germany (Hohenlieth and Hovedissen) were left untreated or exposed to accelerated aging (45°C, 95% relative humidity (RH) for 48 h). Following treatment, seeds were germinated. Germination phenotype was categorized as normal germination, abnormal germination, or ungerminated. Total DNA of seedlings from all germination types, field sites, and genotypes was extracted and subsequently processed for 16S rRNA gene amplicon sequencing. Additionally, bacteria were isolated from seedlings and tested for plant growth-promoting activities, including the production of AHLs, siderophore production, protease activity, and phosphate solubilization.

In addition, directly after accelerated aging, a subset of treated and untreated seeds was sent to Graz University of Technology where unsterilized seeds were germinated under sterile conditions on wet folded seed testing paper (surface weight 160 g/m2, width 110 mm, fold depth 20 mm, ROTILABO® Carl Roth GmbH, Germany) in plastic boxes (150 mm × 150 mm) with five seeds per fold. For the microbiome analysis, we aimed to obtain 7 replicates of 10 individual seedlings/ungerminated seeds per condition (genotype × field site × treatment × germination phenotype). However, most of the untreated control seeds germinated normally. Thus, we were not able to collect 7 replicates of 10 abnormal seedlings for all the control treatments. One control sample of ungerminated seeds was only available for G4, which exhibited a low total germination overall. Accelerated aging resulted in lower numbers of normal seedlings (HOH G2 and G4, 2 replicates; HOH G3, 4 replicates; HOV G2, 6 replicates; HOV G3, 3 replicates; and HOV G4, 4 replicates), as well as abnormal seedlings (HOH G3, 6 replicates; HOH G4, 2 replicates; HOV G1, 4 replicates; HOV G3, 6 replicates; and HOV G4, 2 replicates), thus, less replicates. Also here, all possible replicates for ungerminated seeds were included (HOH G1, 1 replicate; G2, 3 replicates; G3, 2 replicates; G4, 7 replicates; and HOV G4, 7 replicates). In total, we collected 197 samples (4 genotypes × 2 field sites × 2 treatments [accelerated aging and control] × 3 germination phenotypes × 1–7 replicates, with each replicate consisting of 10 individuals).
Here, it is important to mention that accelerated aging was chosen over the controlled deterioration method, which is recommended by ISTA. Preliminary trials on B. napus seeds confirmed a significant positive correlation between accelerated aging and controlled deterioration in terms of both final seed moisture content and post-treatment germination. Accelerated aging was therefore considered a robust and physiologically relevant stress treatment for assessing microbiome dynamics in response to seed aging.
DNA extraction and 16S rRNA gene library preparation
Immediately after 6 days of germination, whole seedlings/seeds were homogenized by crushing them with a mortar in 0.85% NaCl under sterile conditions. Total DNA from seedling samples was extracted using the FastDNA SpinKit for Soil (MP Biomedicals, Illkirch, France) according to the manufacturer’s instructions, with the following modifications: three bead-beating steps were performed at the beginning. After the addition of protein precipitation solution, the samples were kept on ice for 5 min, and elution included an additional step at 55°C for 5 min after adding DES solution. 16S rRNA gene libraries were created in a one-step PCR targeting 16S rRNA gene region V4 (Caporaso et al., Reference Caporaso, Lauber, Walters, Berg-Lyons, Lozupone, Turnbaugh, Fierer and Knight2011) (515F [5′-GTGYCAGCMGCCGCGGTAA-3′] and 806R [5′-GGACTACHVGGGTWTCTAAT-3′]), including sample-specific Illumina barcodes with the Taq-&Go master mix for PCR (MP Biomedicals, Illkirch, France). To lower the amount of amplification of mitochondrial and plastidial PCR products, peptide nucleic acid (mPNA and pPNA) oligomers were added to the reaction (PNA Bio, Newbury Park, CA, USA). These PNA clamps bind to the targeted region of chloroplasts and mitochondria of the plant DNA and therefore reduce its amplification during the PCR (Kawasaki and Ryan, Reference Kawasaki, Ryan, Carvalhais and Dennis2021). For each sample, undiluted DNA was used in duplicates. The final PCR master mix contained 1× Taq-&GO master mix, 0.2 mM of each primer, 1.5 µM activated (55°C for 5 min) mPNA and pPNA, respectively, and 1 µL of template DNA. Amplification was performed with the following steps: 5 min at 96°C, 30 cycles at 96°C for 60 s, 78°C for 5 s, 54°C for 60 s, 74°C for 60 s, and 10 min at 72°C for final extension. The success of PCR amplification was verified through agarose gel electrophoresis.
After pooling the duplicates for each sample, the PCR products were purified with the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). DNA concentrations were estimated based on gel picture analysis (ImageJ, Fiji), and a random subset of samples was additionally measured via Qubit 4 Fluorometer (Invitrogen, Waltham, MA, USA). The combined results allowed for the analysis of the absolute quantity of DNA using equimolar amounts for all samples. 16S rRNA gene amplicon sequencing was conducted on an Illumina NovaSeq 6000 (PE250) at Novogene Company Limited (Cambridge, UK).
Bioinformatic analysis and statistics
Paired-end raw reads were demultiplexed, and primer sequences were removed using cutadapt (Martin, Reference Martin2011). The sequences were quality filtered, trimmed, denoised and merged, and chimeric sequences were removed using the DADA2 (Callahan et al., Reference Callahan, McMurdie, Rosen, Han, Johnson and Holmes2016) plugin for QIIME2 (Bolyen et al., Reference Bolyen, Rideout, Dillon, Bokulich, Abnet, Al-Ghalith, Alexander, Alm, Arumugam, Asnicar and Bai2019). The generated amplicon sequence variants (ASVs) were classified using the VSEARCH algorithm (Rognes et al., Reference Rognes, Flouri, Nichols, Quince and Mahé2016) with the SILVA database version 138.1 (Quast et al., Reference Quast, Pruesse, Yilmaz, Gerken, Schweer, Yarza, Peplies and Glöckner2013). The resulting ASV table was further cleaned using phyloseq (McMurdie et al., Reference McMurdie and Holmes2013) in RStudio version 4.2.3 (RStudio, Boston, MA, USA). Eukarya, unassigned ASVs, chloroplast, and mitochondria sequences were removed from the dataset (0.003%, 0.0007%, 2.27%, and 0.03%, respectively). DECONTAM (Davis et al., Reference Davis, Proctor, Holmes, Relman and Callahan2018) revealed no true contaminants based on the presence of ASVs in negative controls. ASVs were further filtered to a length between 230 and 270 bp.
Richness and Shannon diversity were estimated using phyloseq (random subsampling to the lowest amount of reads) and tested for significance with ANOVA and post hoc Tukey using the vegan package (Oksanen et al., Reference Oksanen, Blanchet, Friendly, Kindt, Legendre, McGlinn, Minchin, O’Hara, Simpson, Peter Solymos, Stevens, Szoecs, Wagner, Barbour, Bedward, Bolker, Borcard, Borman, Carvalho, Chirico, De Cáceres, Durand, Evangelista, FitzJohn, Furneaux, Hannigan, Hill, Lahti, Martino, Ouellette, Cunha, Smith, Adrian Stier, Braak and Weedon2019). Bacterial community composition was based on cumulative sum scaling normalization using metagMisc (Mikryukov, Reference Mikryukov2023) and Bray–Curtis dissimilarities. PERMANOVA was conducted to test for significant effects of the different sample groups with vegan (Wagner, 2019). Beta dispersion test of all samples based on Bray–Curtis dissimilarities and the specific permutation test (999 permutations) was performed using vegan. Relative abundances were obtained by normalization to compositional data using the microbiome package (Lahti and Shetty, Reference Lahti and Shetty2017), and the most abundant ASVs were filtered using MicEco (Russel, Reference Russel2024) and grouped on the genus level. Differential abundance analysis of taxa was conducted using DAtest with the accelerated aging as the predictor and field site and genotype as covariates (Russel et al., Reference Russel, Thorsen, Brejnrod, Bisgaard, Sørensen and Burmølle2018), and resulted in the implementation of DeSeq2 as the best representative method to determine differentially abundant taxa. Correlation analysis was performed using stats (R Core Team, 2023), effects (Fox and Weisberg, Reference Fox and Weisberg2019), and MicroViz (Barnett et al., Reference Barnett, Arts and Penders2021). Plots were visualized using ggplot2 (Wickham et al., Reference Wickham, Chang, Henry, Pedersen, Takahashi, Wilke, Woo, Yutani, Dunnington and van Den Brand2023).
Germination data were analysed with the dplyr, tidyr, ggplot2, ggpubr and FSA. Percentages of normal and abnormal germination and ungerminated seeds were compared between treatments using the Wilcoxon rank-sum test, and visualized as boxplots with jittered individual data points. Pairwise comparisons for genotype effects across all three phenotypes were conducted using Dunn’s test with Bonferroni correction for multiple testing.
Isolation and functional characterization of bacteria
To investigate the culturable bacterial fraction of the seedling microbiome, bacterial isolates were gained from genotype G4 (field site Hovedissen). Seeds of the control and accelerated aging treatments were germinated as described above. For each treatment, four replicates were taken from normal and abnormal seedlings. Due to poor germination after accelerated aging, only three replicates of abnormal seedlings were obtained, and no normal germinated seedlings. Each replicate consisted of 10 seedlings which were placed into Whirl-Paks (Carl Roth GmbH, Karlsruhe, Germany) to isolate epi- and endophytes. The samples were smashed carefully with 1.5 mL sterile NaCl solution (0.9%) using a mortar and pestle until homogenized. The suspension was transferred into 2 mL reaction tubes and a serial dilution until 10−5 was plated on Reasoner’s 2 Agar supplemented with 25 µg/mL Nystatin to inhibit fungal growth. Plates were incubated initially at 30°C for 24 h and an additional 48 h at 20°C. For specific isolation of Bacillus species, the homogenized seedling suspension was heated for 10 min at 80°C. A serial dilution of up to 10−3 was plated on Nutrient Agar II supplemented with 25 µg/mL Nystatin and incubated at 30°C for 24 h. Colony-forming units (CFUs) were determined per gram fresh mass (FM) of seedlings.
Per replicate of the total culturable fraction, 10 randomly picked colonies were further cultured on Nutrient Agar II to create a culture collection. As the Bacillus plates revealed fewer colonies, five colonies per replicate were used for further cultivation. DNA from the isolated bacteria was extracted using the Monarch Genomic DNA Purification Kit (New England Biolabs GmbH, Frankfurt, Germany). BOX-PCR was performed to generate species-specific fingerprints (Martin et al., Reference Martin, Humbert, Camara, Guenzi, Walker, Mitchell, Andrew, Prudhomme, Alloing, Hakenbeck and Morrison1992). One representative of each fingerprint was used to amplify the partial 16S rRNA gene (Heuer et al., Reference Heuer, Kopmann, Binh, Top and Smalla2009) and sent for Sanger sequencing (LGC Genomics GmbH, Berlin, Germany). Sequences were quality-checked with MEGA 11 (Tamura et al., Reference Tamura, Stecher and Kumar2021), and if quality allowed, sequences were processed, and forward and reverse sequences were assembled using SeaView (Gouy et al., Reference Gouy, Guindon and Gascuel2010). The consensus sequences were used for taxonomic assignment using the basic local alignment search tool for nucleotides (NCBI, Bethesda, MD, USA). The first hit was used for the final taxonomic assignment. In case the second or third hit had the same percent identity as the first hit, it was noted as well.
Functional characterization of the isolates included assessment of protease activity (Weinert et al., Reference Weinert, Meincke, Gottwald, Heuer, Schloter, Berg and Smalla2010), siderophore production (Schwyn and Neilands, Reference Schwyn and Neilands1987) and phosphate solubilization (Nautiyal, Reference Nautiyal1999). A subset of isolates (from control seedlings; as these were assumed to have a higher proportion of Gram-negative strains) was used to determine the production of N-acyl homoserine lactones (AHLs) in a cross-streak assay using Chromobacterium violaceum cv026 for short-chain detection of C4–C8 AHLs (Durán et al., Reference Durán, Justo, Durán, Brocchi, Cordi, Tasic, Castro and Nakazato2016). All plate assays were incubated at 30°C for 24 to 48 h, and an additional time at room temperature if needed. Emerged clearing zones were categorized as follows: − = no activity, + = low activity, ++ = moderate activity, and +++ = high activity.
Results
Accelerated aging reduced the total germination
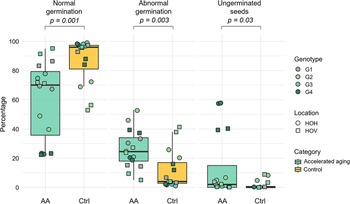
Accelerated aging significantly affected the proportions of different germination phenotypes compared to control seeds (Figure 2). Normal seedling development was reduced (median: 60.4% vs. 86.9%, p = 0.001), while both abnormal seedling rates (26.1% vs. 11.6%, p = 0.003) and the proportion of ungerminated seeds (13.5% vs. 1.6%, p = 0.03) increased significantly. Supporting data can be found in Supplementary Table 1. No significant effect of the field site was observed, while the impact of the genotype was significant for all germination phenotypes (normal germination: p = 0.01, abnormal germination: p = 0.007, and ungerminated seeds: p = 0.002), which is mainly due to differences between G1 and G2 and G2 and G3 (Supplementary Table 2). Genotype G4 was particularly sensitive to accelerated aging, showing a mean of 28.7% abnormal seedlings and 48.5% ungerminated seeds across both locations, while its performance under control conditions was comparable to other genotypes. However, the difference between G4 and the other genotypes was insignificant. In some cases, fungal infections were observed in seedlings following accelerated aging (Supplementary Table 1).
Boxplots illustrate the percentage of normal germination, abnormal germination and ungerminated seeds under accelerated aging (AA) and control conditions (Ctrl). Each point represents a biological replicate (n = 2 per genotype × location), with point colour corresponding to genotype (G1–G4) and point shape indicating field location (circle = Hohenlieth [HOH], square = Hovedissen [HOV]). The boxes represent the interquartile range (IQR) from the 25th to 75th percentile, the bold line within each box shows the median, and the whiskers extend to 1.5× the IQR. Statistical differences between treatments (AA vs. Ctrl) were assessed for each phenotype using the Wilcoxon rank-sum test, with exact p-values displayed above each comparison. All differences were statistically significant (p < 0.05). Y-axis values are germination percentages.

Structure of the B. napus seedling bacterial community
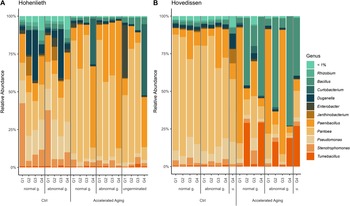
16S rRNA gene amplicon sequencing resulted in a final dataset containing 5,230,972 reads, assigned to 1484 ASVs. The main phyla inhabiting the oilseed rape seedlings were assigned to Proteobacteria, Firmicutes, and Actinobacteria, whereby accelerated aging increased the proportion of Firmicutes compared to the control. Generally, the most abundant genera (above 1% relative abundance; here averaged) belonged to Tumebacillus (HOV, 72%; HOH, 0.76%), Pseudomonas (HOV, 6.5%; HOH, 8.6%), Pantoea (HOV, 37.5%; HOH, 40.2%), Stenotrophomonas (HOV, 2.5%; HOH, 7.5%), Curtobacterium (HOV, 0.7%; HOH, 4.3%), Paenibacillus (HOV, 21.8%; HOH, 22.2%) and Bacillus (HOV, 17.4%; HOH, 2.7%) (Figure 3). While different genotypes and germination phenotypes resulted in similar bacterial communities for both field sites in control seedlings, the relative abundance of the genera changed after accelerated aging and was dependent on the genotype. For example, in Hohenlieth, genotype G4 exhibited a higher relative abundance of Paenibacillus, whereas in Hovedissen, G2 and G4 correlated with a higher relative abundance of Bacillus. G1 and G3 exhibited a higher relative abundance of Paenibacillus, whilst G2 and G4 showed a higher relative abundance of Tumebacillus.
Relative abundance of the most abundant bacterial genera (>1%) in accelerated aging (AA) and control (Ctrl) seedlings of the two field sites, Hohenlieth (A) and Hovedissen (B). Three different germination (g.) phenotypes were analysed (normal, abnormal, and ungerminated [u.]).

Seed aging affected bacterial diversity and community composition
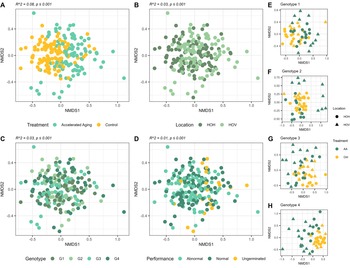
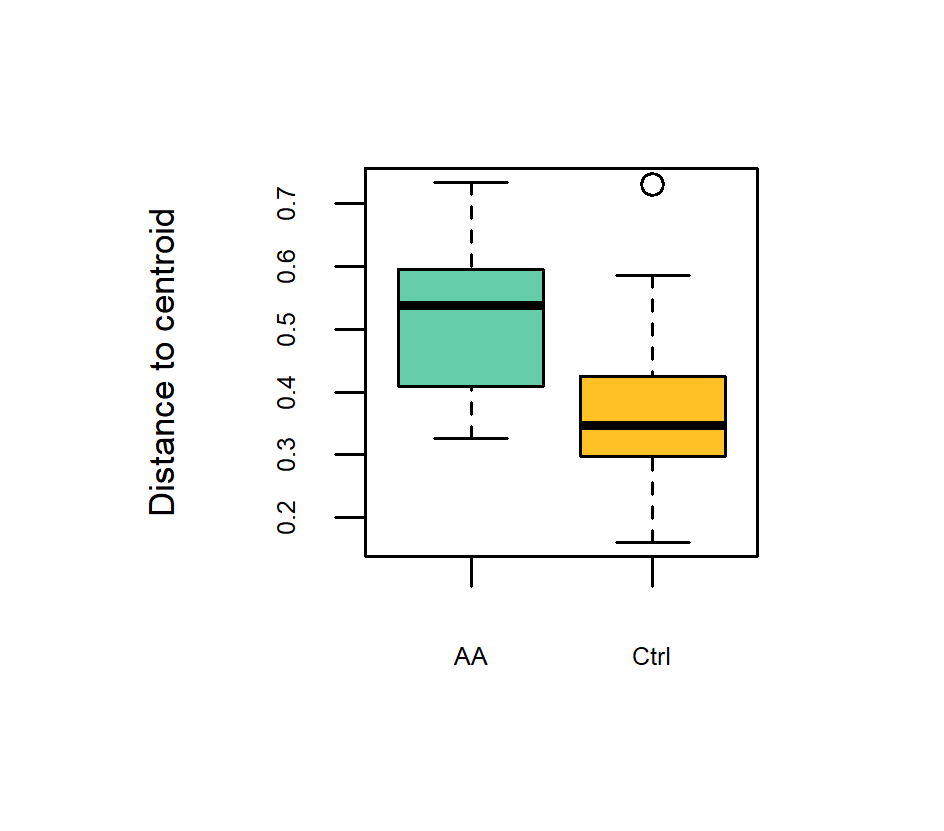
Accelerated aging stress exhibited the highest impact on the bacterial community structure (R 2 = 0.08, p ≤ 0.001), followed by field site (R 2 = 0.03, p ≤ 0.001), genotype (R 2 = 0.03, p ≤ 0.001), and germination phenotype (R 2 = 0.01, p ≤ 0.001; Figure 4A–D). However, the impact of accelerated aging on the seedling bacterial community was higher for seedlings from the field site Hohenlieth (R 2 = 0.11, p ≤ 0.001; Supplementary Figure 2) compared to Hovedissen (R 2 = 0.10, p ≤ 0.001; Supplementary Figure 2). Moreover, the influence of the oilseed rape genotype on the bacterial community structure was also higher for seedlings from Hohenlieth compared to Hovedissen (Supplementary Figure 2). In contrast, the impact of the germination phenotype (normal/abnormal/ungerminated) was similar at both sites (Supplementary Figure 2). Considering accelerated aging and control seedlings separately (Supplementary Figure 3), the bacterial community revealed a significant impact for both the field site and the genotype. In detail, for seedlings from the control, a clear separation of the seedling bacterial community between the two field sites was visible (R 2 = 0.06, p ≤ 0.001; Supplementary Figure 3) and less pronounced in the accelerated aging seedlings (R 2 = 0.04, p ≤ 0.001; Supplementary Figure 3). The genotype effect was higher for the control seedlings compared to the accelerated-aged seedlings (Supplementary Figure 3). Overall, a higher variation in beta diversity was visible for the bacterial community composition of the accelerated-aged seedlings for each of the four genotypes (Figure 4E–H). This trend was validated by a beta dispersion analysis (Figure 5) showing a significant difference between the two treatments independent of the genotype, field site, or germination phenotype (p ≤ 0.01).
Non-metric multidimensional scaling (NMDS) of the bacterial community of oilseed rape seedlings based on Bray–Curtis dissimilarities. Panels (A), (B), (C), and (D) visualize the samples grouped by treatment, field site (Hohenlieth, HOH; Hovedissen, HOV), genotype (G1–G4), and germination phenotype, respectively. Statistical results based on PERMANOVA are reported at the top of each panel. NMDS stress: 0.2246. Panels (E–H) visualize the samples of each genotype separately, distinguishing between accelerated aging (AA) and control (Ctrl). NMDS stress (E) 0.2142, (F) 0.2132, (G) 0.2134, and (H) 0.2248.

Beta dispersion test of accelerated aging (AA) and control (Ctrl) B. napus seed samples showing the sample variance based on Bray–Curtis dissimilarities (p ≤ 0.01). The average distance to the median is 0.5142 for the accelerated aging group and 0.3653 for the control group. Samples represent the whole dataset.

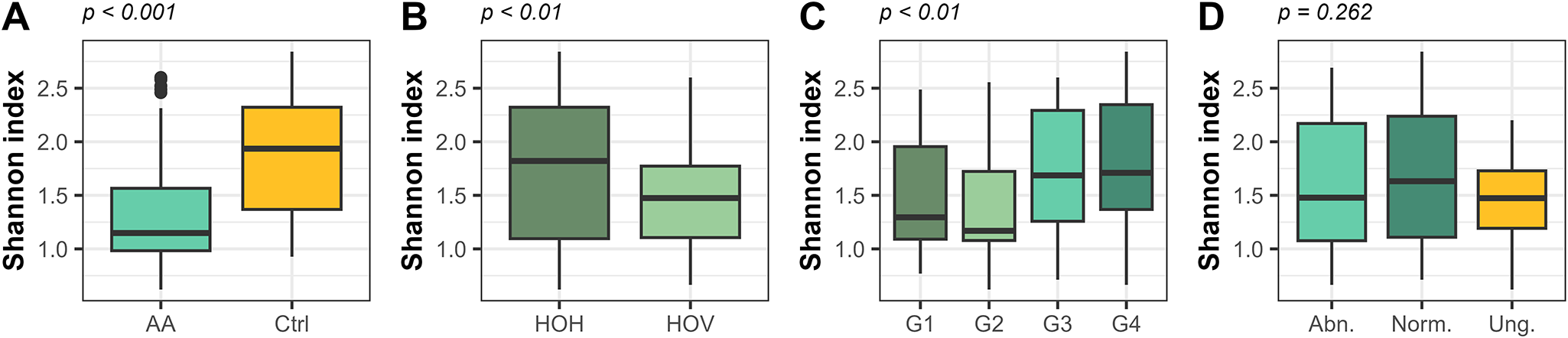
Accelerated aging significantly reduced bacterial alpha diversity (p ≤ 0.001). Field site (p ≤ 0.01) and genotype (p ≤ 0.01) showed a significant but lower impact, while the effect of germination phenotype was insignificant (p = 0.262; Figure 6). The bacterial diversity was analysed separately for the two field sites as well. For field site Hohenlieth, accelerated-aged seedlings showed significantly reduced bacterial diversity compared to the control seedlings, whereas for field site Hovedissen, the impact of accelerated aging had no significant effect due to high variation within replicates (Supplementary Figure 4). Regarding the impact of the genotype, G3 and G4 showed a higher bacterial diversity than G1 and G2 in both field sites for both normal and abnormal germinated control seedlings (Supplementary Figure 4).
Bacterial alpha diversity in oilseed rape seedlings, presented as the Shannon diversity index. Panels (A), (B), (C) and (D) represent the samples grouped by treatment (AA: accelerated aging; Ctrl: control), field site (Hohenlieth, HOH; Hovedissen, HOV), genotype (G1–G4) and germination phenotype (abnormal/normal/ungerminated), respectively. Results of statistical significance tests, based on one-way ANOVA, are presented on top of each panel.

Indeed, seedlings and seeds are different physiological states and comparing them directly can bias the results. Therefore, we repeated the alpha and beta diversity analysis without the samples of ungerminated seeds (Supplementary Figure 5). Those data revealed the same statistical significances as reported above, where ungerminated seeds are included.
Distribution and occurrence of highly abundant taxa were influenced by accelerated aging and field site
DeSeq2 was used to identify which bacterial genera showed differential abundance between the control and the accelerated aging-treated seedlings (p-value ≤ 0.05 and log2-fold change ≥ 2). In general, Tumebacillus, Paenibacillus and Bacillus were highly enriched in accelerated-aged seedlings, as well as one ASV each affiliated to Ralstonia, Burkholderia and Variovorax. Sphingomonas, Pedobacter, Rhizobium, Methylobacterium, Duganella and Burkholderiales were more abundant in control seedlings (Figure 7). For the field site Hohenlieth, mainly Pantoea, Paenibacillus, Bacillus and Curtobacterium were enriched in accelerated-aged seedlings, whereas in control seedlings, mainly Rhizobium, Pedobacter and Chryseobacterium were enriched compared to accelerated aging (Supplementary Figure 6A). For the field site Hovedissen, Pantoea, Pseudomonas, Rhizobium and Sphinogmonas were significantly more abundant in control seedlings, whereas mainly Bacillus and Tumebacillus were significantly more abundant after accelerated aging (Supplementary Figure 6B). Whereas different genotypes and germination phenotypes resulted in similar bacterial communities for both field sites in control seedlings, the relative abundance of the genera changed after the accelerated aging and was dependent on the genotype.
Significantly enriched or depleted amplicon sequence variants (ASVs) in accelerated aging samples versus control according to DeSeq2 for the complete dataset (field site and genotype as covariates). The dataset was filtered based on a p-value ≤ 0.05 and a log2-fold change ≥ 2.

Correlation of highly abundant taxa with seed germination potential
We detected an increased abnormal germination after accelerated aging, as well as a reduced total germination (Supplementary Table 1). Correlations between the germination potential (in percent) and ASV richness, Shannon diversity, and highly abundant genera (Pantoea, Paenibacillus, Bacillus, and Tumebacillus) were calculated (Figure 8). Both Shannon diversity and ASV richness showed a positive correlation (p ≤ 0.001) with the oilseed rape germination potential. For the tested highly abundant genera, only Tumebacillus and Bacillus showed a significant negative correlation with the germination potential (p ≤ 0.001).
Correlation of total germination of B. napus seeds with (A) Shannon diversity (estimate +27.54, p ≤ 0.001), (B) ASV richness (estimate +1.73, p ≤ 0.001), and relative abundance of (C) Tumebacillus (estimate −134.303, p ≤ 0.01) and (D) Bacillus (estimate −58.611, p ≤ 0.05).

Taxonomic and functional profile of bacterial isolates
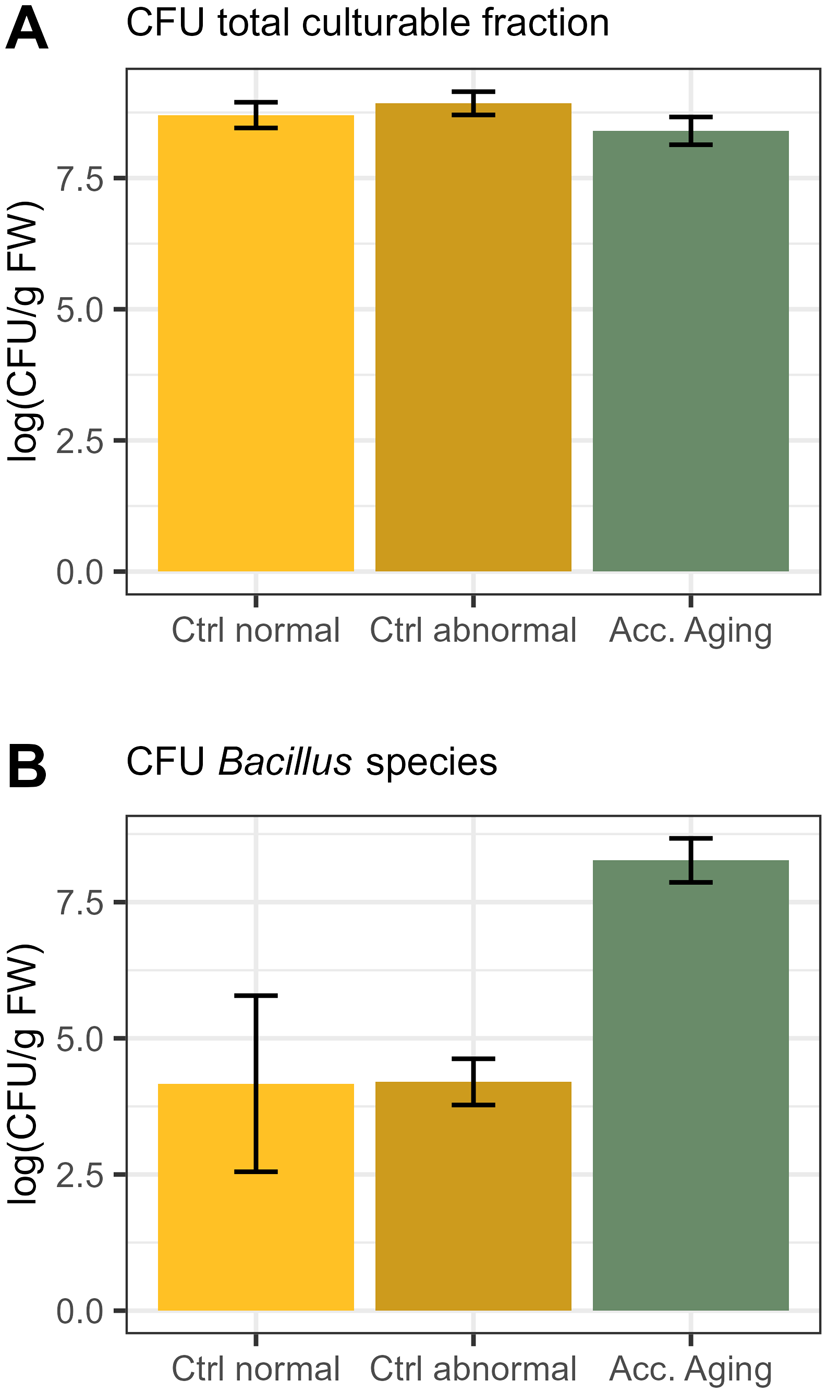
As seeds of the genotype G4 showed the lowest total germination after accelerated aging (Supplementary Table 1), the bacterial communities of seeds that still germinated after accelerated aging were also characterized with culture-dependent approaches. Therefore, bacteria were isolated from emerged seedlings from the control and accelerated aging. In addition to the total culturable fraction, Bacillus species were specifically isolated, as the high abundance of Bacillus was common for both field sites (Figure 3). The CFUs per gram FM of the total culturable fraction from oilseed rape seedlings revealed similar values for each treatment (8 × 108 logCFU/g FM for control and 4 × 108 logCFU/g FM for accelerated aging; Figure 9). The number of Bacillus isolates was much lower for the control (2 × 104 logCFU/g FM) compared to accelerated aging (2 × 108 logCFU/g FM; Figure 9). Among the 109 randomly isolated bacteria from the total culturable fraction, the majority were assigned to the genera Bacillus, Curtobacterium, Frigoribacterium, Pantoea, Pseudomonas, and Pseudarthrobacter (Table 1). The functional profile revealed that isolates from the control showed fewer phosphate solubilization traits and siderophore production than isolates from the accelerated aging samples (Table 1). For the specific Bacillus isolates (51), 10 different BOX fingerprints (Supplementary Figure 7) were selected and identified as members of Bacillaceae with different functional abilities (Table 1). BOX fingerprints served as a DNA fingerprinting technique and enabled, e.g., distinguishing isolates of Bacillus based on highly conserved repetitive sequences in their genome. A partial 16S rRNA gene sequence of one representative of each BOX fingerprint was sequenced and used to identify the members of the culture collection.
Log-transformed colony-forming units (CFUs) per gram of fresh weight (FW) for seedlings of B. napus genotype G4 (field site Hohenlieth), of (A) total culturable fraction and (B) Bacillus species of different germination phenotypes (normal, abnormal germination), for both control (Ctrl) and accelerated aging (Acc. Aging). Error bars represent standard deviations.

Isolated bacteria and their functional profile (genotype G4, Hovedissen) from (A) the total culturable fraction of control (Ctrl) and accelerated aging (AA) B. napus seedlings and (B) isolates from the targeted Bacillus isolation. The hit with the highest percent identity revealed by the nblast algorithm of NCBI was used for taxonomic identification; in case several genera showed the same percent identity, they are indicated by /. Functional profile is categorized as follows: − indicates no activity, +, ++, and +++ indicate slight, moderate, and strong activity, respectively. / indicates differences in the isolates tested from the same genus/box fingerprint. Short-chain AHL production was only tested for selected isolates

Discussion
Our study provides new insights into the accelerated aging test, a technique that has been relied upon for more than 50 years to estimate the storability of seed lots (Delouche and Baskin, Reference Delouche and Baskin2021), by introducing the microbiome as a mostly overlooked component. Although molecular studies on plants have shown distinct differences between artificial aging and long-term seed bank storage conditions (Gianella et al., Reference Gianella, Balestrazzi, Ravasio, Mondoni, Börner and Guzzon2022), B. napus has been reported to exhibit certain similarities in physio-chemical properties under both accelerated and natural aging (Yin et al., Reference Yin, He, Gupta and Yang2015). The present study indicates a shift in the composition of the bacterial community towards reduced bacterial diversity and specificity, and an increase in potentially heat-resistant strains, such as Bacillus species. Our study did not consider the fungal component of the seed microbiome, although fungi can also be vertically transmitted, influence seed and seedling performance, and interact with bacterial communities (Hodgson et al., Reference Hodgson, de Cates, Hodgson, Morley, Sutton and Gange2014; Bastías et al., Reference Bastías, Bustos, Jáuregui, Barrera, Acuña-Rodríguez, Molina-Montenegro and Gundel2022).
In the present study, we proved that accelerated aging affected bacterial community structure, composition and diversity in B. napus seeds (Figures 3, 4 and 6). For all parameters characterizing the bacterial community, accelerated aging was identified as the main driver. We assume that the observed taxonomic shifts (Figures 3 and 7) are largely due to the hot and moist conditions of accelerated aging. In addition to those external stressors, the plant response also plays a role, e.g., reduction of enzyme activities, such as superoxide dismutase, catalase, ascorbate peroxidase and glutathione peroxidase (Yin et al., Reference Yin, Xin, Song, Chen, Zhang, Wu, Li, Liu and Lu2014; Ebone et al., Reference Ebone, Caverzan and Chavarria2019). Amongst others, they can lead to the enrichment of reactive oxygen species (Bailly, Reference Bailly2004; Gill and Tuteja, Reference Gill and Tuteja2010), which might impact the seed microbiome. Reactive oxygen species can have a dual role in the seed, releasing from dormancy, as well as causing oxidative damage. However, in the study by Yin et al. (Reference Yin, He, Gupta and Yang2015), reactive oxygen species were not increased in accelerated-aged seeds of B. napus; instead, increased abscisic acid levels were observed.
Interestingly, the observed differences in the bacterial communities caused by accelerated aging were highly dependent on the field site where the seeds were produced, and slightly, on the genotype (Supplementary Figure 3). The impact of the field site reflects the location-specific bacterial fingerprint of the soil, meaning that since seeds were not surface sterilized, our dataset also includes seed epiphytes derived from the environment, known to influence the seed microbiome composition of B. napus (Morales Moreira et al., Reference Morales Moreira, Helgason and Germida2021). The plant genotype, in contrast, significantly determines the seed endophyte communities (Rybakova et al., Reference Rybakova, Mancinelli, Wikström, Birch-Jensen, Postma, Ehlers, Goertz and Berg2017; Chen et al., Reference Chen, Krug, Yang, Li, Yang, Berg and Cernava2020; Bziuk et al., Reference Bziuk, Maccario, Straube, Wehner, Sørensen, Schikora and Smalla2021; Wassermann et al., Reference Wassermann, Abdelfattah, Wicaksono, Kusstatscher, Müller, Cernava, Goertz, Rietz, Abbadi and Berg2022), where the host genotype most likely contributes to the vertically transmitted microbiome inside the seeds (Newcombe et al., Reference Newcombe, Harding, Ridout and Busby2018; Abdelfattah et al., Reference Abdelfattah, Tack, Lobato, Wassermann and Berg2023). Gram-positive bacteria, particularly Bacillus species, were enriched after accelerated aging (Figures 3 and 9), as expected due to heat-induced spore formation (Gould, Reference Gould2006). The effect of accelerated aging on the bacterial community was found to be field site dependent, but it also led to an overall higher variability in the bacterial community structure (Figure 5). Microbial survival in seeds can be governed by diverse strategies. Certain bacteria may persist through dormancy, spore formation, or protective associations with seed tissues, while others may be more vulnerable to oxidative stress, desiccation, or nutrient depletion during storage (Khan et al., Reference Khan, Shaw, Kabiraj, Paul and Bandopadhyay2025). These differential survival mechanisms likely contribute to the observed shifts in community structure in accelerated-aged seedlings. In addition, this observation supports the ‘Anna Karenina Principle’ for microbiomes, which suggests that microbiome responses to perturbations are often stochastic, leading to transitions from stable to unstable community states. As a result, dysbiotic individuals tend to exhibit greater variability in microbial composition than healthy ones, mirroring Tolstoy’s dictum that ‘all happy families are alike; each unhappy family is unhappy in its own way (Zanefeld et al., Reference Zaneveld, McMinds and Vega2017). Moreover, under stress conditions, the microbiome can contribute to the holobiont’s resilience by deterministic selection of beneficial microorganisms (Arnault et al., Reference Arnault, Mony and Vandenkoornhuyse2023). This was shown in our study as well by enrichment of potentially beneficial members of Firmicutes, e.g., Bacillus and Tumebacillus.
However, Bacillus and Tumebacillus were furthermore found to negatively correlate with oilseed rape germination phenotype (Figure 8). Bacillus is a versatile genus containing strains that produce a broad range of antimicrobial compounds and are highly competitive in the plant microbiome (Boro et al., Reference Boro, Sannyasi, Chettri and Verma2022). Usually, members of Bacillus are known for their plant-beneficial characteristics, increasing plant health (Hauschild et al., Reference Hauschild, Orth, Liu, Giongo, Gschwendtner, Beerhues, Schloter, Vetterlein, Winkelmann and Smalla2024), including enhanced seed germination under abiotic stress (Li et al., Reference Li, Yue, Li, Liu, Zhang, Wang and Jiang2021). Also in seeds, Bacillus species are frequently detected (Figueiredo Dos Santos et al., Reference Figueiredo Dos Santos, Fernandes Souta, de Paula Soares, Da Oliveira Rocha, Luiza Carvalho Santos, Grativol, Fernando Wurdig Roesch and Lopes Olivares2021). The presence of Bacillus in our dataset is, thus, expected. Not much is known about Tumebacillus as a member of the plant microbiome; it was mainly isolated from water systems or soils (Her et al., Reference Her, Srinivasan and Lee2015; Zhang et al., Reference Zhang, Gao, Du and Wang2023). The loss of seed microbial diversity and evenness and the dominance of Tumebacillus might lead to reduced growth of seedlings after accelerated aging. However, both Tumebacillus and Bacillus were found to be enriched in plants receiving salt stress (Mukhtar et al., Reference Mukhtar, Mehnaz, Mirza, Mirza and Malik2018; Han et al., Reference Han, Zhang, Han, Ren, Wang, Zhu, Sun, Wang, Qu, Lu and Yuan2023). Seed germination phenotype of accelerated-aged seeds was also found to correlate with microbial diversity (Figure 8), underlining the importance of microbial diversity for plant health (Berg et al., Reference Berg, Köberl, Rybakova, Müller, Grosch and Smalla2017).
In contrast to accelerated aging samples, in total 26 bacterial ASVs were significantly more abundant in the control samples, with seven ASVs up to two times more abundant (Figure 7). Simonin et al. (Reference Simonin, Briand, Chesneau, Rochefort, Marais, Sarniguet and Barret2022) conducted a large-scale meta-study on the seed microbiome, including 50 plant species from agricultural and natural environments. In this study, the seed core microbiome consisted of 13 ASVs that were assigned to Pantoea, Pseudomonas, Rhizobium, Sphingomonas, Methylobacterium, and Paenibacillus, in descending abundance. Except for Pantoea and Paenibacillus, all of them were also identified as core taxa for control samples in our dataset. Particularly, strains from the genera Pseudomonas, Rhizobium, and Sphingomonas have been described to possess beneficial traits for plant health (Zamioudis and Pieterse, Reference Zamioudis and Pieterse2012; Matsumoto et al., Reference Matsumoto, Fan, Wang, Kusstatscher, Duan, Wu, Chen, Qiao, Wang, Ma and Zhu2021; Tanveer et al., Reference Tanveer, Akhtar, Ilyas, Sayyed, Fitriatin, Perveen and Bukhari2023; Hauschild et al., Reference Hauschild, Orth, Liu, Giongo, Gschwendtner, Beerhues, Schloter, Vetterlein, Winkelmann and Smalla2024), which were detected in lower abundance after accelerated aging (Figure 7; Supplementary Figure 6). On that basis, accelerated aging caused a reduction of specific, and presumably plant-beneficial, microbiomes that are natively associated with seeds. Further, there is a complex relationship between microbial diversity and its ability to increase resilience (Shade et al., Reference Shade, Peter, Allison, Baho, Berga, Bürgmann, Huber, Langenheder, Lennon, Martiny, Matulich, Schmidt and Handelsman2012), suggesting that the accelerated aging stress and the connected loss of diversity are reducing the plant’s resilience to its environment.
In the cultivation-dependent approach, we confirmed a significantly higher abundance of Bacillus species in accelerated-aged seedlings (Figure 9). The reduced microbial diversity of accelerated-aged seedlings in the amplicon dataset (Figure 6) was reflected among the isolated bacteria (Table 1), affecting both the total culturable bacteria and specifically Bacillus species. Isolated bacteria from the accelerated-aged seedlings, presenting the same genus reflected by BOX fingerprints, possessed a higher plant beneficial potential with a more diverse functional profile compared to the control (Table 1), suggesting a different functional profile influenced by the accelerated aging. As the sampled oilseed rape seedlings were still able to germinate, although germinating abnormally, bacteria associated with them should be explored as potential plant biostimulants. Both groups of microorganisms are important to ensure either health or resilience. In general, the accelerated aging stress shifted the microbiome lifestyle, as more copiotrophic bacteria were isolated from seedlings that received the accelerated aging stress compared to the control (e.g., Bacillus, Pseudomonas; Table 1). Interestingly, a higher functional variation was found for bacteria isolated from accelerated-aging seedlings, again confirming the Anna Karenina Principle for the plant microbiome, recently reviewed by Arnault et al. (Reference Arnault, Mony and Vandenkoornhuyse2023). It seems that the disruption caused by accelerated aging leads to more variable and less predictable microbiomes.
While we observed significant and clear responses of the seed bacterial community to accelerated aging, our study lacks a comparison to naturally aged seeds. Such experiments are of great importance to get deeper insights into the effect of seed aging on the microbiome. Overall, more mechanistic studies are needed to better understand the communication and interaction between the plant and its microbiome and the impacts of stress conditions, including long-term storage. In addition, modelling approaches that combine plant origins, genetics, the microbiome, and storage conditions could help to predict the storability of seed lots.
Conclusion
This study demonstrates that accelerated aging affects the seed-associated bacterial community in B. napus. In particular, we observed an increase in Gram-positive bacteria, which correlated with reduced germination rates. These microbial shifts are likely driven by the elevated temperature and humidity conditions characteristic of the accelerated aging protocol. While this test induces physiological symptoms similar to those observed in naturally aged seeds, it may not fully replicate the microbiome changes associated with natural seed aging. Future research should directly compare the microbial dynamics of naturally aged seeds with those subjected to various artificial aging methods. Since the seed microbiome is crucial for germination and plant health, integrating microbial assessments into seed storage tests is increasingly important. Moreover, identifying storage practices that preserve both seed viability and microbial integrity will help maintain optimal plant performance even after long-term storage.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0960258526100099.
Acknowledgements
We thank the reviewers for their constructive comments and suggestions, which helped to improve the clarity and quality of this manuscript. We also thank Isabella Kögl (acib) for assistance during laboratory work. Supported by TU Graz Open Access Publishing Fund.
Funding statement
The COMET centre: acib: Next Generation Bioproduction is funded by BMK, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria and Vienna Business Agency in the framework of COMET – Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency, FFG.
Competing interests
The authors declare no conflicts of interest.
Data availability
The 16S rRNA gene amplicon sequences are deposited in the EMBL Nucleotide Archive (ENA; ebi.ac.uk/ena) under the accession number PRJEB89949.
Ethical standards
The authors confirm that they have adhered to the ethical policies of the journal.







 Open access
Open access