

Key messages
Chapter 3.4 explores how pharmaceutical care is financed. Paying for medicines includes how the end-purchase of existing medicines is managed but also the way investment in R&D is handled. Key learning includes that:
Pharmaceutical innovation draws on substantial public and private resources.
The public sector primarily supports early-stage research, regulates the industry and incentivizes development.
The private sector is typically central to development, commercialization, manufacture and marketing. It seeks high profit margins and is not always transparent or responsive to public policy priorities.
Novel and specialized therapeutics as well as population ageing are likely to accelerate medicines expenditures. This requires careful management of pricing and reimbursement.
Policy-makers can leverage a mix of push and pull strategies to align industry efforts with societal need.
To optimize pharmaceutical policy policy-makers must consider:
○ clear communication of health system priorities;
○ transparent incentive and pricing systems and measures to enhance R&D efficiency;
○ payment mechanisms that foster equity and sustainability;
○ cross-country collaboration including on preparedness, procurement and pricing transparency.
Introduction
Pharmaceutical care is an important contributor to how health systems retain health gains and improve future outcomes. Pharmaceutical spending is one of the largest items in health expenditures for most health systems, while patient cost-sharing for pharmaceuticals is one of the main contributors to health care-related financial hardship. The rate of pharmaceutical spending growth seems to have slowed primarily due to cost containment measures implemented in response to the global financial crisis and the concurrent patent expiry of many popular outpatient medicines (OECD & EU, 2022; Belloni, Morgan & Paris, Reference Belloni, Morgan and Paris2016). However, the advent of specialized medicines and advanced therapy medicinal products (ATMPs) in conjunction with population ageing are likely accelerating spending again, a trend which is harder to track given that many of these newer therapies are used in inpatient settings, where data on pharmaceutical expenditure are harder to come by (Morgan & Xiang, Reference Morgan and Xiang2022). What is clear is that new medicines are entering the market at increasingly high price tags, a phenomenon which progressively hampers governments’ abilities to purchase the latest innovations for their citizens. Meanwhile, pharmaceutical companies are reaping significant financial benefits: 35 of the largest pharmaceutical companies generated a cumulative global revenue of US$ 11.5 trillion between 2000 and 2018, with greater profitability margins than other large Standard and Poor’s (S&P’s) 500 index companies (Cutler, Reference Cutler2020; Ledley et al., Reference Ledley2020). Thus, rethinking the balance between ensuring access and incentivizing innovation is becoming increasingly important.
In addition to paying for medicines which are already on the market, societies also spend on medicines in other ways. The high risk of failure in drug development, the controlled nature of clinical trials and the long interval between medicine discovery and market authorization mean that every stage in the medicine development pipeline comes at a substantial cost (Wong, Siah & Lo, Reference Wong, Siah and Lo2019). High prices for pharmaceuticals typically aim to offset pharmaceutical companies’ R&D investment and cover production and distribution costs. However, substantial amounts of public funding also flow into medicines’ discovery and development before consumer purchase (Galkina Cleary et al., Reference Galkina Cleary2018; Wizemann, Robinson & Giffin, Reference Wizemann, Robinson and Giffin2009). To fuel innovation and subsequently ensure equitable access to affordable medicines, governments adopt a range of policies to try and steer pharmaceutical R&D towards unmet health care needs and to influence the prices at which new medicines can be purchased after market authorization. As well as leveraging policy instruments to negotiate prices, governments must decide how best to allocate public funding and the level of co-payments, i.e. the extent to which they require their citizens to meet the cost of their own pharmaceutical care. These are decisions which may lead to considerable variation in pricing, coverage and access to medicines across different countries. Pricing and reimbursement decisions may be guided by evidence-based tools, including health technology assessment (HTA), to evaluate the clinical trial information submitted by manufacturers at the time of market authorization. Particularly for specialized medicines and biotechnology-based gene and cell therapies, repeated pricing and reimbursement assessments may be warranted over time, as further evidence on the cost–effectiveness of therapies becomes available.
This chapter explores how pharmaceutical care is financed, starting with investment in R&D for new medicines and then zooming in on the purchase of medicines by health systems, including the suitability of existing pricing and reimbursement models for novel types of therapeutics and the purchasing strategies adopted by different countries. Each of these two main sections follows a similar analytical structure and discusses the relative financial role of the public and private sectors (including patients), what the invested funds cover (particularly in terms of meeting patient needs), as well as the actual sums of money paid. Current pricing and reimbursement models and their suitability for novel types of therapeutics are also explored and we suggest potential ways the system could be improved.
Investing in R&D
R&D jointly make up the first stage in the pharmaceutical life-cycle and are concurrently the costliest and most failure prone. It can take several years for a new pharmaceutical target to become a real-world medicine and, on average, another eight years of clinical trials until marketing authorization and sales are underway (Wong, Siah & Lo, Reference Wong, Siah and Lo2019). It is estimated that only around 10–14% of novel medicine candidates are approved for launch, with considerably lower success rates in specific medical subfields such as oncology (Hay et al., Reference Hay2014; Thomas et al., Reference Thomas2016; Wong, Siah & Lo, Reference Wong, Siah and Lo2019).
Most medicine development starts with basic research (preclinical phase) into disease mechanisms with the objective of identifying potential therapeutic candidates for later development and commercialization. If a promising medicine candidate is selected for further advancement, its clinical efficacy and safety must be assessed under strictly controlled conditions in a sequence of clinical trials, which typically consist of three successive clinical trial phases (I–III) involving progressively larger participant groups (US FDA, 2018; Wong, Siah & Lo, Reference Wong, Siah and Lo2019). Phase III is the most expensive of the trial phases, but it is typically associated with a high rate of success (50–60%) (Hay et al., Reference Hay2014; Thomas et al., Reference Thomas2016; Wong, Siah & Lo, Reference Wong, Siah and Lo2019). Manufacturers apply for intellectual property protection during this process, which encompasses different types of incentives to drive pharmaceutical innovation (see Box 3.4.1). To be considered for marketing authorization, new medicines generally must yield positive results at the end of phase III trials. To obtain a marketing authorization, manufacturers are required to submit an application to the competent authorities in different countries or to the European Medicines Agency (EMA) for the centralized marketing authorization in the EU (the latter is currently the principal pathway for new medicines). Clinical trials also encompass a fourth phase termed pharmacovigilance, which unfolds after a medicine’s marketing authorization (phase IV) and serves to monitor its efficacy and safety under real-world conditions. Pharmacovigilance usually proceeds for several years after market entry and continues to inform policy processes on the pricing and reimbursement of medicines.
Intellectual property rights are one of the most widely established incentives used by governments to propel innovation in the pharmaceutical sector. They comprise a range of different advantages and rewards, which secure the financial viability of pharmaceutical companies in a system characterized by high rates of failure, while also protecting inventions from competition (for a set number of years) and so, potentially, steering research into areas of unmet clinical needs, which may ultimately benefit patients. The main types of intellectual property rights are as follows.
Patents
Patents are classified as a type of industrial property and are typically granted for 20 years. They provide a period of exclusivity for new inventions (including processes, technologies or active substances), during which time they may not be marketed and profited from by anyone other than the patent owner. Medicines can carry numerous different patents at any given time. Competition from other developers is only possible once all patents have expired.
Supplementary protection certificates
Manufacturers are required to file patents for new medicine candidates early in the R&D process. Since patent protection only starts when marketing authorization is obtained, in practice the time during which the medicine is protected from competition is shorter than the 20-year patent period, because of lengthy clinical trials required to prove the safety and efficacy of new medicines. The protection until market authorization is considered as “passive”. Supplementary protection certificates can restore part of the protection that has been lost, up to a maximum of five years.
Regulatory data protection
This form of intellectual property rights is separate from patent rights and protects data pertaining to a new medicine, including all data that are collected during the clinical trials leading up to market authorization, rather than the invention per se. After marketing authorization is granted, pharmaceutical companies generally have exclusive rights to this data for a certain number of years (eight years in the EU).
Market exclusivity/protection
This is another type of regulatory protection scheme which may apply in addition to patent rights. Market exclusivity/protection grants protection from other generics or biosimilars with the same therapeutic indication from being placed on the market for a defined amount of time after data exclusivity expires.
Efficiency gains made during the R&D process can shorten the passive patent protection period and bring forward the moment when manufacturers start to benefit from market exclusivity and can maximize investment returns through sales of the new medicine. For example, in a marketing authorization process, termed rolling review, medicines may be considered for authorization by regulatory agencies while evidence is still being generated during clinical trials, thereby accelerating market entry (as was seen with vaccine development during the COVID-19 pandemic) (EMA, n.d. a; Marinus et al., Reference Marinus2022).
In the EU, new pharmaceuticals are subject to both data and market exclusivities (EU, 2001; 2004). The former protects preclinical and clinical trial data for a duration of eight years and hinders generic competitors from applying for marketing authorization. Data exclusivity clauses foresee public disclosure of dossiers after expiry; however, generic competitors may not be marketed for another two years as part of the market exclusivity rights granted to reference products under European legislation (European Commission, 2019; EMA, n.d. b). At the time of writing, the EU’s pharmaceutical legislation is currently being reviewed as part of a broader strategy to strengthen crisis preparedness and access to affordable and innovative medicines in the Union, with new proposals for a Regulation and Directive published in April 2023 (European Commission, 2023a; 2023b). The proposed legislation envisions changes that may alter the conditions under which additional regulatory protection schemes (data and market protection) are granted to applicant manufacturers in upcoming years, including possible extensions to the duration of data protection. Among several planned changes, the introduction of transferable data exclusivity vouchers aims to incentivize the development of priority antimicrobials by prolonging the duration of data protection and enabling developers to either use or sell vouchers to other marketing authorization holders (European Commission, 2023b).
Who pays for R&D?
The pharmaceutical industry funds the development, commercialization and manufacture of medicines. The public sector regulates the industry and incentivizes the development and production of medicines it typically neglects. It also pays for most of the initial research, which is what we deal with first.
Early-stage research: predominantly public funding
Most early-stage (preclinical) research is funded by public means (usually pooled from taxpayers’ money) and is typically conducted in academic institutions and research centres (Wizemann, Robinson & Giffin, Reference Wizemann, Robinson and Giffin2009). Gaining an understanding of biological processes and uncovering disease pathways lay the foundation for pharmaceutical innovation and the discovery of new medicine candidates. Investment in basic research comprises a large share of overall R&D spending. In the USA, the National Institutes of Health dedicates 50% of its budget to this cause and, between 2010 and 2016, funded research underlying (both directly and indirectly) all 210 new medicines approved by the United States Food and Drug Administration during this period (Galkina Cleary et al., Reference Galkina Cleary2018). The EU is another crucial funder of fundamental research, having dedicated nearly €100 billion to its current research programme, Horizon Europe (European Commission Directorate-General for Research and Innovation, 2021). Under the previous research programme, Horizon 2020, one quarter of health-related projects were in the area of basic research (Gallo et al., Reference Gallo2021). Research that is publicly funded has been reported to generate a distinctive share of products with important clinical effects (see Box 3.4.2), particularly in areas of unmet medical need such as rare diseases (Stevens et al., Reference Stevens2011).
The COVID-19 pandemic saw governments step out of their traditional roles as early-stage funders, contributing significantly to late-stage development and propelling the manufacture of COVID-19 countermeasures. Agarwal and Gaule (Reference Agarwal and Gaule2022) estimate that close to 70% of COVID-19 trials were started by public institutions, including universities and hospitals. Governments worldwide invested in the expansion of infrastructure and facilities to scale up production capacities, even before the completion of clinical testing of the vaccine candidates in question (Wouters et al., Reference Wouters2021). In 2020, the United States government alone invested between US$ 11 to US$ 19.5 billion in late-stage vaccine research and supported vaccine manufacturing capacity-building to ensure production could be expanded adequately within the country (known as Operation Warp Speed). Most high-income countries (HICs) stipulated prepurchasing agreements with multiple vaccine developers to secure vaccine doses for their populations. Beyond vaccine procurement, monoclonal antibodies now approved for treatment were developed using therapeutic research platforms and technologies which benefited from public sector support prior to the pandemic. The adaptation of these technologies to commercialize and develop an effective COVID-19 treatment yielded substantial additional investment from governments globally (Robinson, Reference Robinson2021).
Development: predominantly private funding with public sector incentives
In contrast to early research, the brunt of the cost for medicines’ development, commercialization and manufacture is borne by the private sector (Kalindjian et al., Reference Kalindjian2022). Additionally, the pharmaceutical industry spends substantial amounts on buying back shares and on marketing to ensure successful market penetration by newly developed medicines. These activities can surpass expenditure on R&D itself (Mazzucato et al., Reference Mazzucato2018) (Box 3.4.3).
The industry engages heavily in lobbying activities to propel its interests at policy level to ensure outputs surpass the R&D stage and successfully enter the market (EMA, 2022; Wouters, Reference Wouters2020). It also funds patient advocacy organizations working on indications in which they have marketed products and provides financial incentives to health care professionals, whose prescribing decisions have shown to be directly influenced by such industry payments (Mitchell et al., Reference Mitchell2021). The fact that the work of regulatory agencies is also funded through user fees paid for by the industry has led to discussions around the potential for regulatory capture (Wouters, Reference Wouters2020).
Over time, the industry’s structure has evolved to rely strongly on the commercialization of scientific discoveries and the entrepreneurial potential of scientific research, resulting in closer partnerships with academic institutions and strategic mergers with biotechnology firms through the financial support of private equity and venture capital (Malerba & Orsenigo, Reference Malerba and Orsenigo2015). However, the increased dependency on shareholder expectations and a stable performance in stock markets has largely favoured R&D investments with short-term returns and high profit margins. With a vision to sustain and increase its revenues, the industry has grown to become a skilled political and economic player in the health care arena.
Governments can try to regulate and incentivize the medicine development process to some extent to steer development to areas of unmet need. This includes leveraging so-called push and pull mechanisms (see chapters 3.9 and 3.10). These approaches incentivize private R&D investment at the entry and exit points of the pipeline, respectively. Box 3.4.4 showcases examples of how these tools work using the case of antibiotic development.
Push financing consists of R&D subsidies in the form of direct funding (e.g. project grants, fellowships and scholarships for researchers, funding for small and medium-sized enterprises (SMEs)), which can be conditional on research in specific domains or aimed at stimulating translational research along the entire medicine supply chain and so lower the risks of loss for private investors (Panteli & Edwards, Reference Panteli and Edwards2018). Push financing can also take the shape of indirect support such as tax benefits to encourage the participation of SMEs in the R&D process. While this lowers the risk of failure for the private sector, it can be a burden to public funders and does not guarantee delivery of the required output at the end of the pipeline. Similarly, it is frequently implemented on a national scale, limiting the potential global level benefits.
Conversely, pull mechanisms provide financial incentives once specific development milestones have been achieved. These must sufficiently offset the risk for SMEs to partake in the development process, while also requiring a longstanding political commitment given the protracted timeline of R&D for a new medicine. Such output-based financing tools can comprise lump sum or milestone prizes, advanced purchasing agreements, which entail committing to the acquisition of a new medicine at a specified price and/or volume prior to the completion of the R&D process (advanced market commitment). Alternatively, rewards-based tools can rely on performance indicators (pay for performance) such as medicine consumption levels or be awarded in exchange for intellectual property rights (patent buyouts) and unlimited patient access (payer licenses) (Panteli & Edwards, Reference Panteli and Edwards2018). These mechanisms reward the achievement of specific goals but are also susceptible to shifting political agendas. At the same time, the risk is borne entirely by the developer, which can deter smaller enterprises from participating in the R&D process. Pull mechanisms may require the pooling of resources across multiple countries to offer meaningful financial rewards for the delivery of a specific output without compromising cost–effectiveness (Panteli & Edwards, Reference Panteli and Edwards2018).
Beyond push and pull mechanisms, there are also important examples of initiatives blurring the traditional public and private sector roles and transcending the barriers to establish fruitful collaborations across sectors, including public–private partnerships such as the Innovative Medicine Initiative (now Innovative Health Initiative) and the Drugs for Neglected Diseases initiative (DNDi).
Push mechanisms: Feeding the preclinical antibiotic pipeline
It has been estimated that over two thirds of the financial incentives to develop new antibiotics are provided in the form of push incentives (Simpkin et al., 2017). In recent years, many national and transnational programmes have been initiated across the USA and EU (Wasan et al., 2023). For example, the United States Biomedical Advanced Research and Development Authority (BARDA) launched a dedicated initiative in 2010 and established public–private partnerships aimed at developing new antibiotics and diagnostics. Between 2010–2019, nearly US$ 1 billion in grants, agreements and contracts were awarded by BARDA through these channels for the development of antibiotics (Wasan et al., 2023). At the EU level, the Innovative Medicines Initiative (predecessor of the Innovative Health Initiative), a public–private partnership co-funded by the EU and involving a range of stakeholders, including from academia and industry, established the New Drugs for Bad Bugs programme. With a budget of €650 million, the programme supported projects working towards solutions to overcome challenges impeding the development of new antibiotic treatments (Innovative Medicines Initiative, n.d.). For example, one of these projects (TRANSLOCATION) focused on finding new pathways for the delivery of antibiotics into bacteria (with a focus on Gram-negative pathogens such as Escherichia coli, which are subject to high rates of resistance and particularly challenging to penetrate for drugs) (Innovative Medicines Initiative, n.d.).
Pull mechanisms: The Longitude Prize
The Longitude Prize, a prize co-funded by Innovate United Kingdom (the United Kingdom’s national innovation agency) and a range of public and private entities including the BBC, Amazon and Glaxo Smith Kline, offers substantial financial rewards, up to several million pounds, to teams of innovators who develop novel solutions to address pressing societal problems. In the area of health, these so-called “challenge” prizes have addressed priority issues including dementia (i.e. how to help conserve the independence of affected individuals in their everyday lives) and antibiotics (i.e. how to halt the rise in resistance to antibiotics). For example, one prize, offered £8 million for the development of rapid diagnostic tests, which could be used by clinicians to identify the most suitable antibiotic therapies at the point-of-care and thereby promote better antibiotic stewardship in the clinical setting (Longitude Prize, https://amr.longitudeprize.org/).
For further information on the underlying issues of the antibiotics market and some of the initiatives operating to incentivize antibiotic R&D and tackle AMR, see Chapter 3.9.
Investment in R&D: does it produce the medicines we need?
As the primary proponent and cost carrier of medicines’ commercialization and manufacture, the pharmaceutical industry largely determines which medicine candidates enter the development pipeline. Private R&D investment choices have shown that expected profits and market growth are more important factors in pharmaceutical company decisions than unmet societal needs (Kalindjian et al., Reference Kalindjian2022). Risk of failure does not seem to be a prominent determinant of medicine development decisions, as particularly risky domains like oncology have experienced an important investment surge in recent years (Huang & Nambudiri, Reference Huang and Nambudiri2020).
As of late, the pharmaceutical sector has experienced a fall in numbers of new medicines brought to market, as medicine development has become increasingly specialized, complex and lengthy with the advent of biologicals and biotechnology-based gene and cell therapies (also known as ATMPs) (Hay et al., Reference Hay2014). R&D investment is increasingly concentrated in areas protected from competition, such as medicines for rare or neglected diseases and ATMPs. The area of oncology has seen a particular peak in authorizations over the last decade, with close to 50% of the global pipeline focused on oncology and immunology (Shimmings, Reference Shimmings2017). While cancer is a major public health issue, other chronic diseases such as cardiovascular conditions which cause millions of deaths each year have experienced a decline in investment and medicine development (Fordyce et al., Reference Fordyce2015). Moreover, evidence suggests at least one in five new cancer medicines do not bring any improvement to patients in terms of quality of life, overall survival, or safety benefits (Davis et al., Reference Davis2017; Salas-Vega, Iliopoulos & Mossialos, Reference Salas-Vega, Iliopoulos and Mossialos2017; Vokinger et al., Reference Vokinger2020; Reference Vokinger2022). Neglected and antimicrobial-resistant diseases are another area of public health concern, which is underrepresented in medicine development efforts, likely due to the limited scope for returns on investment (see chapters 3.9 and 3.10). Overall, R&D efforts do not appear to correlate with global disease burden (Cottingham, Kalbaugh & Fisher, Reference Cottingham, Kalbaugh and Fisher2014). Relatedly, incentives originally introduced to propel the development of medicines to cater for the unmet needs of rare disease patients have also had unexpected effects, oversteering investment in this area and contributing to the emergence of “niche” blockbuster products (Kumar Kakkar & Dahiya, Reference Kumar Kakkar and Dahiya2014). The Pharmaceutical Strategy for Europe and ongoing revisions of the EU’s general pharmaceutical legislation aim to address and correct some of these possible misincentives in the European context (European Commission, 2023a; 2023b).
Even in areas with substantial R&D investment like cancer, there is a disproportionate focus on projects of shorter duration, such as late-stage cancer, due to the immediacy of financial rewards and the limited profitability stemming from fixed patent duration (Budish, Roin & Williams, Reference Budish, Roin and Williams2015). The system’s reliance on patents as one of the primary incentives of innovation actively influences industry practices and hampers medicine innovation. In particular, it has created a market for new medicines which form part of the same therapeutic class as existing medicines (known as “me-too” medicines) and provide limited or no added clinical benefits due to their closely related molecular structures and modes of action. Me-too medicines build on existing knowledge and are hence faster to develop and commercialize. In addition, being subject to conventional intellectual property protection, they can represent profitable ventures for medicine developers once patents for other members of the same class expire (Gagne, Reference Gagne2011). These and similar approaches adopted by industry to extend the market protection and profitability of their products impede the introduction of generic alternatives which would improve the affordability of medicines for patients.
Research duplication and waste represent other important sources of costs, which can be challenging to fully ascertain. Evidence suggests that up to 80% of clinical trials produce redundant findings and approximately 50% never publish full reports of performed research and results (Glasziou & Chalmers, Reference Glasziou and Chalmers2016). The failure to disclose full records and clinical effectiveness information from clinical trials is accompanied by a reporting bias towards positive clinical outcomes (Mitra-Majumdar & Kesselheim, Reference Mitra-Majumdar and Kesselheim2022). As a result, a substantial proportion of medical research funding risks being misspent as research efforts are duplicated even when they have demonstrably yielded meaningless results. In addition, there are ethical implications of failing to report negative results of clinical trials. If these results were reported, it could prevent and protect future trial participants and patients from receiving therapies which are unlikely to generate clinical benefits (Nygaard, Reference Nygaard2017). Additionally, without full disclosure of all clinical trial results, public payers risk overpaying for medicines if they lack and cannot avail themselves of the full picture of clinical trial outcomes when entering pricing negotiations (Vogler, Reference Vogler2022a).
To enable fast access to medicines addressing unmet patient needs, the EMA provides manufacturers with the option of obtaining early scientific advice and accelerated assessments. This was systematized in its PRIME scheme, a voluntary scheme providing guidance on data and clinical trial requirements to participating medicines at early stages of development (EMA, n.d. c). Approaches such as this one are more permissive with regards to the standards of evidence required than traditional marketing authorization procedures and have shown to reduce average approval time by a few years (Panteli & Edwards, Reference Panteli and Edwards2018). This may result in efficiency gains in the R&D process leading up to authorization, although safety and efficacy should continuously be monitored as well as missing evidence substantiated to comply with post-marketing requirements. Concerns have been raised, however, that accelerated assessments may expose patients to safety risks and progressively lower evidentiary requirements if used inadequately, instead of effectively managing unmet needs (Panteli & Edwards, Reference Panteli and Edwards2018). Box 3.4.5 describes an additional way to accelerate the development of medicines to meet medical needs currently uncatered for, by repurposing existing medicines.
There are different ways in which a drug candidate or a marketed medicine can be repurposed to serve a new therapeutic use in addition to the one for which it was originally approved or developed. The development of repurposed medicines is associated with lower costs and shorter timelines for the developing entity than traditional clinical trials and can revive research on partially developed molecules which were never approved for use (Pushpakom et al., Reference Pushpakom2019); as a result, repurposing has been used as a commercial strategy for years (Botella, Reference Botella2022).
However, for conditions with unmet clinical needs and/or without viable treatment options, developing other clinical indications for administering and combining existing products can also present new effective treatment solutions and enhance patient access to them (Frail et al., Reference Frail2015). Numerous initiatives have been launched in this direction. For example, building on the work of the European Commission’s Expert Group on the Safe and Timely Access to Medicines for Patients, the EMA launched a repurposing pilot project to support academia and non-profit organizations in studying the use of existing medicines for new indications in 2021 (EMA, 2021a). Within the scope of the project, beneficiaries can obtain regulatory and scientific advice to support the development of an application for regulatory approval (EMA, 2021a).
How much money is spent on R&D?
Despite fewer new products being commercialized, the sector has experienced continued increases in investment with substantial incentives provided by public and private entities to spur innovation (Huang & Nambudiri, Reference Huang and Nambudiri2020). The pharmaceutical industry is characterized by elevated fixed R&D and marketing costs and a low marginal production cost. As explained above, it takes substantial investment to discover and launch a new medicine, while the cost of producing an extra unit is usually comparatively small (Cutler, Reference Cutler2020; Rennane, Baker & Mulcahy, Reference Rennane, Baker and Mulcahy2021). Several studies have deployed different methodologies and sources to attempt cost estimations of the R&D process, which range between US$ 314 million and US$ 2.8 billion for the development of new molecular entities (DiMasi, Grabowski & Hansen, Reference DiMasi, Grabowski and Hansen2016; Wouters, McKee & Lutyen, Reference Wouters, McKee and Luyten2020). Other publications report even broader ranges. Rennane and colleagues detected gaps of US$ 113 million to US$ 6 billion accounting for all new medicines, including those developed in specific cost-intensive therapeutic areas such as Alzheimer’s disease (Rennane, Baker & Mulcahy, Reference Rennane, Baker and Mulcahy2021). Similarly, a 2016 report by the United Nations Secretary-General’s High-Level Panel on Access to Medicines (2016) described estimates ranging between US$ 100 million to US$ 4.2 billion.
The costliest of the R&D stages are phase III clinical trials, with USA estimates suggesting that the cost of a pivotal trial alone lies somewhere between US$ 12 to US$ 53 million depending on therapeutic area studied. In the same study, cost estimates for phase II trials and phase I trials also varied importantly by therapeutic area being examined, with ranges of US$ 7 to US$ 20 million and US$ 1 to US$ 7 million, respectively (Sertkaya et al., Reference Sertkaya2016). Figures in a similar ballpark were reported by other USA-based investigations (Moore et al., Reference Moore2018; Reference Moore2020). The question remains whether pharmaceutical companies’ overall R&D investments are truly reflective of the high pharmaceutical prices charged at the end-users’ side. A few initiatives have been pressing for more transparency in this regard. For example, DNDi, the public–private partnership working towards treatment solutions for neglected diseases, has pledged to disclose the R&D costs of its products (DNDi, n.d. a; n.d. b; 2022), which as some evidence suggests, have been lower than those reported by pharmaceutical companies (Junod Moser et al., Reference Junod Moser2023; Moon, Reference Moon2017; Moon, Bermudez & ’t Hoen, Reference Moon, Bermudez and Hoen2012).
Pharmaceutical companies argue that the elevated failure rate and high costs incurred in the early stages of a medicine’s life-cycle necessitate the observed high prices of newly marketed products. However, the industry’s resistance to disclosing the full costs of medicine development undermines this argument and has prompted multiple investigations into the true costs of the R&D pipeline. Moreover, this argument fails to account for the substantial public investment in basic research as stated above. Going forward, enhancing the system’s transparency is key and could potentially support R&D priority-setting based primarily on societal needs rather than commercial viability (Panteli & Edwards, Reference Panteli and Edwards2018).
In this first half of the chapter, we have looked at the financing of medicines’ R&D. In the next half, we turn to look at how their purchase is funded and some of the processes involved.
Purchasing medicines
Once medicines have received marketing authorization and commercial production can begin, they are registered with relevant regulatory agencies who serve as intermediaries between pharmaceutical companies, prescribing health care professionals and patients, and ensure continued data collection and pharmacovigilance. Different payers are responsible for purchasing medicines depending on the organizational set-up of the health system. Against the backdrop of their budgetary constraints, payers face the challenge of providing comprehensive access to effective medicines while also providing appropriate incentives to continue fuelling the system with innovations. Health systems employ different mechanisms to determine reimbursement and pricing of medicines. At the early stage of marketing, evidence on the effectiveness and safety of medicines is usually limited, hampering payers’ abilities to robustly determine willingness to pay. Exclusivity rights and the resulting lack of competition (see Box 3.4.1) contribute to high pharmaceutical prices and expenditures (Kesselheim, Avorn & Sarpatwari, Reference Kesselheim, Avorn and Sarpatwari2016; Vincent Rajkumar, Reference Vincent Rajkumar2020). According to 2020 estimations, pharmaceutical spending was approaching 20% of net health expenditure in several European countries (OECD, 2020). Furthermore, official estimates of pharmaceutical expenditure commonly exclude the costs of medicines purchased and dispensed within hospitals and sometimes also those incurred by private households in the form of co-payments and out-of-pocket (OOP) payments. Since the majority of specialized and expensive medicines are used in hospital settings, current approximations of pharmaceutical expenditure are likely to gravely underestimate the true costs of medicines for health systems worldwide.
Who pays for the purchase of medicines?
Generally, pharmaceutical expenditure is covered by a mix of public and private funds. Solidarity-based health systems, which represent the most common model in Europe, leverage public funds to ensure access to essential health services, including essential medicines, for their populations. Primarily, this entails coverage via social health insurance models or a national health service, which rely on diverse revenue streams, including forms of taxation, employment and social security contributions (see Chapter 1.1). On average, 58% of pharmaceuticals are funded by government/compulsory schemes (39% are paid OOP and 3% through voluntary health insurance (VHI) schemes (see Chapter 1.3)) across Organisation for Economic Co-operation and Development countries, although there is considerable cross-country variation; at the two ends of the spectrum, over 80% of retail pharmaceuticals are funded publicly in France (83%), Ireland (82%), Germany (81%) and Croatia (80%) and fewer than 40% in Poland (35%), Bulgaria (24%) and Chile (20%) (OECD, 2023).
Although publicly funded and organized systems are designed to provide universal access, financial protection does not necessarily cover all pharmaceuticals, and patients are typically required to cover a part of the expenses for reimbursed (i.e. publicly funded) medicines in the community (i.e. outpatient sector). In countries with more limited progress towards universal health coverage, most medicines (in both outpatient and hospital sectors) are to be paid fully OOP by the patients. This may lead to a high financial burden for patients and their carers and can even result in catastrophic spending.
There are different types of co-payments (e.g. fixed prescription fee, percentage co-payments of the medicine price, and deductibles), with variable effects on the amount paid by the patients, and by the public payer (see Chapter 2.4). In solidarity-based health systems, vulnerable population groups may be fully or partially exempt from co-payments for outpatient reimbursable medicines (Panteli et al., Reference Panteli2016; Vogler, Zimmermann & Haasis, Reference Vogler, Zimmermann and Haasis2018; Vogler, Haasis & Zimmermann, Reference Vogler, Haasis and Zimmermann2021; WHO Regional Office for Europe, 2018). Evidence shows that patients in low- and middle-income countries (LMICs) are more frequently exposed to high OOP payments for pharmaceuticals with over 90% of the 150 million individuals incurring catastrophic health expenditure globally residing in these countries (WHO & IBRD, 2020). Even in the World Health Organization (WHO) European Region, estimates from 24 countriesFootnote 1comprising 18 HICs and six middle-income countries) suggest that up to 9% of households are impoverished and unable to pay for basic needs due to health spending, although large differences are observed among EU countries (estimates ranging between 0.3% to 5.9%) and non-EU countries (estimates ranging between 3.6% to 9.0%) (Thomson, Cylus & Evetovits, Reference Thomson, Cylus and Evetovits2019). Importantly, OOP payments for medicines are a key driver for this catastrophic and impoverishing health spending in Europe (Thomson, Cylus & Evetovits, Reference Thomson, Cylus and Evetovits2019; WHO & IBRD, 2020).
Purchasing medicines: what is covered?
Funding medicines implies compensating the actors in the supply chain for their services related to the product: the pharmaceutical manufacturer for the development, production and launch of the product; importers, wholesalers and other distributors for their logistics services; and pharmacies for services around dispensing the medicine. Usually, in price-regulated settings of solidarity-based systems, these services are remunerated through the different components of the medicine price: the ex-factory price to reward the manufacturer, the wholesale markup for the distributor, and the pharmacy markup for the pharmacy.Footnote 2 The costs attributable to medicines’ distribution can be considerable, but are beyond the scope of this chapter. The following paragraphs discuss which medicines are reimbursed by public payers, and how this is determined.
Which medicines are reimbursed by public payers?
In principle, all medicines that have been granted marketing authorization can be paid for by public payers. In practice, however, to ensure responsible use of public money and eventual sustainability for the health systems, not all medicines are included in public spending. Reimbursed medicines are mainly prescription medicines, but several countries also include at least some nonprescription medicines in their benefit baskets (see Chapter 2.2). In the EU, out of every €4 spent on outpatient medicines, €3 are used to pay for prescription medicines and the rest mainly cover nonprescription medicines (OECD & EU, 2022). In the next few paragraphs, we look at the differences in pharmaceutical benefits baskets across countries and outline the underlying decision-making processes, including those for high-cost novel therapeutics.
In the outpatient sector, selected medicines are included in reimbursement, following an evaluation process as described below. Most European countries apply the concept of a positive list, which compiles medicines that are considered eligible for public funding, whereas negative lists which explicitly specify medicines excluded from reimbursement are rare (WHO Regional Office for Europe, 2018; Vogler, Haasis & Zimmermann, Reference Vogler, Haasis and Zimmermann2019). HICs tend to have large benefit baskets since they usually have more resources they can spend on medicines. In general, medicines that are considered essential are included in public funding. While WHO’s model essential medicines list, which has grown from 208 medicines in 1977 to 460 medicines in 2021 (WHO, n.d.), aims to guide decision-makers in defining which medicines are considered essential, the essential medicines concept has deliberately been designed to be flexible so as to allow consideration of national particularities and priorities. Adjustments to specific situations (e.g. different needs resulting from the country’s geographical area, such as tropical versus Nordic zones) are encouraged. Policy-makers are urged to ensure that essential medicines are “available within the context of functioning health systems at all times, in adequate amounts, in the appropriate dosage forms, with assured quality, and at a price the individual and the community can afford” (Quick et al., Reference Quick2002).
The range of medicines included in reimbursement lists thus varies significantly across countries and regions. For instance, within the EU context, Spain included 21 703 medicines in its reimbursement list in 2020 (including different pharmaceutical forms and dosages) compared to 11 845 medicines in Italy (in 2020), 7399 medicines in Austria (in 2019) and 1733 medicines in Cyprus (in 2020) (Vogler, Reference Vogler2021b; Vogler, Haasis & Zimmermann, Reference Vogler, Haasis and Zimmermann2019; Reference Vogler2020; Zimmermann & Haasis, Reference Zimmermann and Haasis2021). Differences in reimbursement are even more striking in and compared to other central and eastern European countries outside the EU where the number of medicines included in reimbursement lists in 2018 (information provided for the outpatient sector and per active substance) ranged from 460 International Nonproprietary Names (INNs) in Belarus to 399 INNs in Kazakhstan, 148 INNs in Moldova, 50 INNs in Kyrgyzstan and 23 INNs in Ukraine (Vogler et al., Reference Vogler, Haasis and Zimmermann2020).
In addition, public funding may also be provided through additional programmes or funds, outside the regular reimbursement systems (e.g. alternative funding sources, alternative inclusion criteria and processes) to cover specific groups of medicines. In some countries, including some in central Asia, so-called vertical programmes fund medications for diseases such as cancer, diabetes, hepatitis C, HIV/AIDS, tuberculosis (TB) and multiple sclerosis (Vogler et al., Reference Vogler, Haasis and Zimmermann2020). In HICs, dedicated funds offer patients access to medicines with high price tags, such as ATMPs, other cancer medicines and/or medicines for rare/neglected diseases. A well-known example is the English Cancer Drug Fund which finances, for a maximum of two years, cancer medicines which have not yet been deemed as cost-effective. In 2021, the creation of another similar fund called Innovative Medicines Fund for potentially life-saving new treatments (for example, in the areas of spinal muscular atrophy and cystic fibrosis) was announced in the English National Health Service. Since 2017, Italy has been operating two innovation funds (one for innovative cancer medicines and one for innovative non-cancer medicines, each worth €500 million) which were merged into a single fund in 2022 (Vogler, Reference Vogler2022b).
How do public payers decide which medicines to pay for?
Policy-makers leverage a range of policies to ensure that medicines covered by statutory money will be carefully selected (reimbursement policies) and that publicly funded medicines are purchased at affordable prices (pricing and procurement policies) to reduce waste, curb pharmaceutical spending and enable equitable patient access. In practice, there is, to a large extent, interlinkage between reimbursement and pricing policies, since whether or not a medicine is included in public funding frequently also depends on its price.
Criteria that countries in the WHO European Region apply when they decide on the inclusion of medicines in reimbursement comprise the (added) therapeutic benefit of a medicine, medical necessity/priority, safety, cost–effectiveness and budget impact (WHO Regional Office for Europe, 2018). The main tool which supports policy-makers in taking more evidence-based decisions about which medicines to cover is HTA, which is defined as “a multidisciplinary process that uses explicit methods to determine the value of a health technology at different points in its life cycle” (O’Rourke et al., Reference O’Rourke2020) (see Chapter 2.2 for more information on health benefits packages and HTA).
The way in which assessment of pharmaceuticals is built into coverage decision-making varies across countries (Fontrier, Visintin & Kanavos, Reference Fontrier, Visintin and Kanavos2022; Panteli et al., Reference Panteli2016). While most European countries use HTA, or components of it, for reimbursement (and pricing) decisions (37 of 53 countries of the WHO European Region), only some countries (including Denmark, Germany, Norway, Sweden, United Kingdom) apply HTA systematically (Vogler, Reference Vogler2022b). In addition, different assessment methodologies can be applied (Panteli et al., Reference Panteli2016; Robinson, Panteli & Ex, Reference Robinson, Panteli and Ex2019; Emanual et al., Reference Emanuel2020; The Kings Fund, 2020). An outcome of HTA may be that a medicine shows only limited or no added (therapeutic) value. This does not automatically equate to exclusion from reimbursement, but the medicine is usually covered at a lower price. For instance, statutory health insurers in France reimburse new medicines which are deemed to offer no additional clinical benefits at 5–10% lower prices than comparator medicines on the market (Emanuel et al., Reference Emanuel2020). In Australia, both clinical effectiveness and cost–effectiveness considerations play a role. If therapeutic potential is deemed equivalent to standard treatment options, a new medicine is priced to match the least expensive medicine in the medicine comparison group (Emanuel et al., Reference Emanuel2020). When there are high degrees of uncertainty, HTA can result in a recommendation for a conditional reimbursement, or a managed-entry agreement can be put in place (see pricing mechanisms in the How are purchase prices determined? section).
Several European countries also apply such a policy to cluster therapeutically equivalent or similar medicines into a reference group, within which the same amount is reimbursed for all medicines. This can reflect the price of the least expensive medicine within the group or may be calculated based on more complicated processes. If patients wish to access a higher-priced medicine from the same reference group, they must usually cover the difference themselves; thus, this policy, called a reference price system, incentivizes the use of lower-priced alternatives (usually generics) whenever they are available. With such systems, often manufacturers price medicines below or just in line with the chosen reference price limit to ensure their product is covered by the insurance funds and is consequently chosen by health care providers and patients; the majority of these medicines are generics (Robinson, Panteli & Ex, Reference Robinson, Panteli and Ex2019). The underlying idea of including medicines of same or similar therapeutic value within the scope of reimbursement is to enhance competition and thus to offer medicines at affordable prices (for payers and patients). However, it is important that these lower-priced medicines are actually prescribed, dispensed and used; this can be supported by demand-side measures targeted at prescribers (e.g. prescribing by the INN), pharmacists (e.g. generic substitution), and consumers (e.g. financial incentives for requesting lower-priced equivalent in pharmacies, information campaigns to inform and build trust in generic and biosimilar medicines) (Vogler et al., Reference Vogler, Haasis and Zimmermann2021b).
Below, as with R&D, we set out the actual sums of money involved in the purchase of medicines, as well as the share of it paid by patients. We then go on to examine how the purchase prices for medicines are determined, and in particular how public payers decide what they are willing and able to pay, and the resultant national variations in purchase price.
How much money is spent on the purchase of medicines?
Pharmaceutical expenditure is the product of the volume of medicines consumed and their price (Fig. 3.4.1). For comparative purposes, pharmaceutical expenditure is usually expressed in one of three ways: per capita, as a share of national gross domestic product (GDP), or as a component of total health spending. Per capita annual spending for outpatient pharmaceuticals in the EU was estimated at €462 in 2020, with considerable variation detected across Member States (OECD & EU, 2022). At €660 per capita, Germany was the highest spender, while Denmark lay well below the EU average at €258 per capita (see Panteli et al. (Reference Panteli2016) on the relevant context for these figures). In the wider European Region, disparities in spending are even more extreme. Adjusted for purchasing power parity, current expenditure on medical goods ranged from €1281 per capita in Germany to €221 per capita in Moldova in 2020 (WHO, 2020). Lastly, if measured as a share of GDP, pharmaceutical expenditure usually corresponds to current health expenditure (as a share of GDP) times the share of health expenditure spent on outpatient pharmaceuticals, and in the EU ranged from 0.6% (of GDP) in Luxembourg to 2.87% (of GDP) in Greece in 2020 (OECD, n.d.; Panteli et al., Reference Panteli2016). As explained above, there is a lack of data on the costs incurred from hospital pharmaceuticals in health systems and sometimes also from private households, making it challenging to paint a comprehensive picture of total pharmaceutical expenditure. Generally, the share of pharmaceutical expenditure out of total health expenditure tends to correlate negatively with national income level. This is observed in many LMICs, where spending on pharmaceuticals can reach up to 60% of health care spending (Milne & Kaitin, Reference Milne and Kaitin2019). One reason for this is that HICs have a stronger price negotiating power.
At the EU level, 29% of outpatient medicines were financed by private households in 2020 (OECD, 2022), while public expenditure accounted for 70% and VHI for 1% of total expenditure on outpatient pharmaceuticals. The shares of public and private expenditure vary significantly across different Member States, with particularly high public shares observed in Cyprus (85%), Germany (82%) and France (82%) and low shares in Bulgaria (24%), Poland (35%), and Latvia (41%). About half of what is spent OOP by patients in the EU went towards medical goods (including outpatient pharmaceuticals and therapeutic appliances) in 2020 (OECD, n.d.). The comprehensiveness of the benefit basket, the level of prices as well as the type of co-payments impact actual patient expenditure.
Pharmaceutical expenditures are composed of the volume of medicines consumed times their prices
OTC: over the counter; VAT: value-added tax; VHI: voluntary health insurance.

Figure 3.4.1 Long description
The first graphic explains the costs when prescriptions are covered. The individual pharmaceuticals include Ex-factory, Wholesale, Retail, and VAT, which together form the Cost-sharing and Reimbursement price. Only the reimbursement price is multiplied by the Consumption volume, which results in the (Measured) public expenditure. This equals the Rebates (average X percent). The second graphic explains the costs when medications are O T C and not covered. The individual pharmaceuticals include Ex-factory, Wholesale, Retail, and VAT, which together form the Direct payment and V H I. This is multiplied by the Consumption volume to get the (Measured) private expenditure.
How are purchase prices determined?
Pharmaceutical manufacturers frequently argue that it is high R&D costs that render high medicine prices necessary to sustain future investment in innovation. However, evidence suggests that even larger proportion of pharmaceutical company budgets is spent on advertising and stakeholder awareness-raising activities to increase market share in the later stages of the pharmaceutical lifecycle (as mentioned above) and that manufacturer prices are determined based on what the market is willing or likely to bear.
In the US market, total annual spending on advertising and marketing was estimated at $29.9 billion in 2016. Marketing to medical professionals makes up the largest proportion, yet direct-to-consumer advertising has rapidly increased over the past 20 years, making up one third of the total marketing costs in 2016 (Schawartz & Woloshin, Reference Schwartz and Woloshin2019). While direct-to-consumer advertising of prescription medicines is not permitted in Europe, payments to health professionals and organizations advocating for patient rights are common practice and still poorly regulated across many European countries (Fabbri et al., Reference Fabbri2018). Several countries, including the UK, have opted for a self-regulatory mechanism in which pharmaceutical companies disclose financial support provided to health professionals in public databases (e.g. Disclosure UK). This is one of few means to estimate the extent of payments, which amounted to an annual £50 million in 2015 and 2016 in the UK alone (Mulinari & Ozieranski, Reference Mulinari and Ozieranski2018).
Payers employ a range of mechanisms to determine the prices they are willing and able to pay for pharmaceuticals, against the backdrop of balancing necessary considerations of cost containment with the need to retain a healthy R&D pipeline (Box 3.4.6). Approaches include value-based pricing, managed-entry agreements, external reference pricing, internal price referencing and centralized procurement. We explain each of these briefly below.
While most countries only regulate prices for reimbursed medicines, some (e.g. Lithuania, Türkiye) statutorily determine (or negotiate) the prices of all medicines (Vogler, Zimmermann & Haasis, Reference Vogler, Zimmermann and Haasis2018). The rationale for this scope of price regulation is to protect patients from excessive prices even if they pay for their medicines OOP.
Value-based pricing
Increasingly used to determine what health systems are prepared to pay, this approach entails setting medicine prices on the basis of evaluations of clinical and economic benefits. For instance, new medicines with additional benefit compared to available alternatives may be awarded with premium prices (Panteli et al., Reference Panteli2016). Pricing approaches that consider the comparative effectiveness of new medicines struggle with new therapeutics that offer curative options where only symptomatic treatment was available before. A well-known example is that of hepatitis C therapies based on direct-acting antivirals such as sofosbuvir (Sovaldi®), which started entering pharmaceutical markets in 2014. These medicines have evident clinical benefits and improved cost–effectiveness compared to interferon-based treatment, but given their high prices and sizeable target populations, providing access had important budgetary implications for health systems. Indeed, the total cost of treating every patient affected by hepatitis C was estimated to represent a potential threat to the sustainability of national pharmaceutical budgets in many countries (Iyengar et al., Reference Iyengar2016). In order to mitigate the effect on public expenditure, some countries initially restricted access to certain patient groups, but this obviously poses an ethical dilemma in the long run. For ATMPs with the potential to cure complex medical conditions that have so far been treated symptomatically, target populations are usually smaller, but prices for these too have the potential to be so high that the budget impact will be considerable (Gonçalves, Reference Gonçalves2022) (Box 3.4.7).
At a price of around €2 million per dose, Zolgensma® was the most expensive medicine on the market at the time of its launch (Nuijten, Reference Nuijten2022). It is a single-dose gene therapy medication used to treat spinal muscular atrophy in children, a disease which causes life-threatening muscle wasting and weakness leading to a very short life expectancy (EMA, 2021b). After being shown to improve survival and reduce the need for artificial ventilation, Zolgensma® received a conditional marketing authorization from the EMA in 2020 (EMA, 2021b). Due to the rarity of the condition and lack of comparator treatments, most of the studies conducted prior to authorization have been single-arm clinical trials comprising small patient groups, as is the case for many gene therapies. Additional evidence on the therapy’s benefits and risk profile is continuously collected and shared with marketing authorization agencies in an effort to fill remaining knowledge gaps, including on the long-term outcomes and curative potential of this treatment.
Governments worldwide are struggling to balance the potential benefit for these patients with the staggering costs for health systems and concerns for equity. Within Europe, countries have limited access to specific patient age groups and types of spinal muscular atrophy based on the quality of available clinical evidence, while also adopting differing price negotiation and reimbursement approaches to secure access to Zolgensma®. Italy has approved its reimbursement using a “payment at results” contract with payments due at checkpoints of 12, 24, 36 and 48 months of treatment and confidential discounts on the ex-factory price applied for public health facilities and facilities accredited by the Italian national health system (Agenzia Italiana del Farmaco, 2021). Belgium, Ireland and the Netherlands participated in a joint HTA process through the Beneluxa Initiative and subsequently joined forces to negotiate a mutual agreement on pricing in late 2021 (Beneluxa, n.d.; Besley et al., Reference Besley2022).
Managed-entry agreements
Related to value-based pricing are managed-entry agreements. These allow payers to mitigate the risks of purchasing new medicines whose benefits to patients remain highly uncertain by stipulating contractual agreements with manufacturers which set out specific conditions for their reimbursement. Commonly, managed-entry agreements constitute two types of agreement: performance-based (also called outcome-based), whose public funding is conditional on achieving specific health outcomes, or finance-based, which are not subject to these conditions. The scope of managed-entry agreements varies, but they always have confidential features, for instance negotiated discounted prices are always kept secret (Webb et al., Reference Webb2022). This type of agreement is commonly employed in specialties where new pharmaceuticals with limited evidence but high expected value are marketed at high prices, such as oncology, immunology or rheumatology. Performance-based pricing models have been successfully negotiated between payers from several countries and the manufacturers of the first two approved CAR-T cell therapies for treatment of cancer. Although the specific clinical indicators considered are not always disclosed, the reimbursement of CAR-T therapies is conditional on clinical outcomes in several European countries, including France, Germany, Italy, Spain and the United Kingdom; some are also subject to future reassessments based on longer-term follow-up data as part of managed-entry agreements (Jørgensen, Hanna & Kefalas, Reference Jørgensen, Hanna and Kefalas2020).
Although many countries use these agreements, the extent to which they deliver on their promise in different health care systems is still unclear. Evaluations are difficult to perform, given the confidential data that are required (Gerkens et al., Reference Gerkens2017). It is accepted that their administrational burden is considerable. Italy is one of the earliest adopters of performance-based agreements, investing millions of euros into an infrastructure to follow up on real-world outcomes in individual patients treated with expensive medicines, such as acetylcholinesterase inhibitors for Alzheimer’s disease. Despite substantial investment, the Italian system of managed-entry agreements has been shown to be expensive and slow at generating the evidence required to track meaningful clinical outcomes. It is burdensome in practical terms, as health care professionals are exposed to additional administrative tasks, such as filing the required forms on clinical outcomes. Evidence suggests that finance-based agreements have generated some savings in the Italian setting (Dabbous et al., Reference 441Dabbous2020).
External reference pricing
External reference pricing consists of setting the price of a new medicine according to a benchmark determined based on prices charged for the same medicine in other countries (WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies, n.d.). This approach is commonly applied to new medicines, either as the sole method of determining the maximum price statutory systems will pay or serving as a starting point for negotiating pricing and reimbursement conditions with pharmaceutical companies. For instance, in Norway the maximum price for a new medicine is set by averaging the lowest three prices applied among nine other European countries (Austria, Belgium, Denmark, Finland, Germany, Ireland, Netherlands, Sweden, United Kingdom) (Emanuel et al., Reference Emanuel2020; Weise, Festoy & Hagen, Reference Weise, Festøy and Hagen2018). Several European countries use external reference pricing as the main mechanism for medicine pricing. Others, including Belgium, Finland, Germany, Italy, Poland and Spain, use it as a supportive mechanism (European Commission Directorate-General for Health and Food Safety, et al., 2015; Schneider & Vogler, Reference Schneider, Vogler and Vogler2019; Toumi et al., Reference Toumi2014). The widespread use of this method of pricing incentivizes pharmaceutical manufacturers to keep high the official list prices used for it and then offer confidential discounts on the list price to payers. This creates an overall lack of transparency and concerns that the actual prices paid for pharmaceuticals are not in line with what different health systems can afford (see Webb et al., Reference Webb2022).
Internal price referencing
For pharmaceuticals with one or more therapeutic alternatives already on the market, internal price referencing is applied by several countries (Panteli et al., Reference Panteli2016; WHO Regional Office for Europe, 2018), intended as both a cost containment and competition-stimulating measure. Internal price referencing has two variants: the first features the reference price system in which medicines are classified into a reference group, within which all medicines are reimbursed at the same price by public payers. This can be understood as a reimbursement policy (more than a pricing policy), as described previously. A second type of internal price referencing is exercised in the form of a so-called price link (generic price link or biosimilar price link), by which the price of a generic or a biosimilar medicine is set in relation to the price of the originator or biological originator medicine (WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies, n.d.). In some countries, price link approaches also affect the price of the originator medicine. Generic and biosimilar price links are used in several European countries (Vogler et al., Reference Vogler, Haasis and Zimmermann2021b).
(Centralized) tendering
The procurement of medicines in the inpatient sector usually involves tendering for competitive offers. In European countries, hospital procurement is conducted by individual hospitals or through group purchasing. However, an increasing number of countries have started to introduce centralized procurement which can take place at national and regional levels (European Health and Digital Executive Agency et al., 2022). Different variants of centralized procurement through tendering can be found in countries such as Denmark, Norway, Portugal or Serbia (Bartels, Reference Bartels2016; Vogler, Bauer & Habimana, Reference Vogler, Bauer and Habimana2022). In 1990, Denmark established the public sector organization Amgros owned by the country’s five regional authorities. Amgros is tasked with procuring all medicines for public hospitals in Denmark. It conducts cost-effective purchasing using different types of tendering contracts to attract bids from diverse suppliers and identifies which expensive medicines to acquire with the support of treatment recommendations issued by another competent body, the Danish Council for the Use of Expensive Hospital Medicines (Bartels, Reference Bartels2016).
A study on public procurement of medicines has shown that countries with strategic approaches in medicines procurement were able to generate lower purchase prices. Their approaches involve applying a mix of policies and procedures such as pooling volumes and capacity (e.g. through centralized procurement and targeted procurement techniques (e.g. framework agreements)) which are aligned to their medicine procurement objectives; awarding the most economically advantageous tender and not the lowest-priced one; multiple-winner bids; and making use of supplementary tools such as HTA or horizon scanning. In addition, procurement can be used strategically to achieve other policy objectives such as security of supply, environmental protection, or crisis preparedness (European Health and Digital Executive Agency et al., 2022). There are also tendering-like procedures in the outpatient sector in a few European countries (e.g. Denmark, Germany, Netherlands) in which reimbursement is granted to the medicine out of a group of comparable medicines (e.g. the same active substances or therapeutic equivalents) for which the lowest price was offered; this is often supported by demand-side measures such as generic substitution. It has also been shown to effectively contain costs (Vogler, Gombocz & Zimmermann, Reference Vogler, Gombocz and Zimmermann2017).
National variations in purchase price
The mix of different mechanisms employed by countries along with factors such as purchasing power and market size contribute to a considerable price variation across health systems globally. For example, studies examining the costs of new hepatitis C medicines based on direct-acting antivirals sofosbuvir (Sovaldi®) and ledipasvir/sofosbuvir (see the Value-based pricing section) show considerable variation in originator prices across countries (e.g. the median list price for a 12-week treatment course was of US$ 40 502 in 2020 with a range from US$ 10 730 in Argentina to US$ 91 461 in Italy (Barber et al., Reference Barber2020).
Evidence suggests that national income levels correlate negatively with prices – at least with official list ones. Although evidence on medicine prices in LMICs remains limited, over the past few years higher prices in LMICs compared to HICs have been documented for the same treatments (Moye-Holz & Vogler, Reference Vogler2022). This pattern is not limited to territories outside Europe, as similar discrepancies have been demonstrated between EU Member States of different income levels (Moye-Holz & Vogler, Reference Vogler2022; Vogler, Vitry & Babar. Reference Vogler, Vitry and Babar2016). This appears to be linked to lower health care budgets and less power to negotiate discounts with manufacturers, who are attracted to markets in wealthy countries amenable to purchasing expensive specialty medicines, such as oncology medicines. Evidence from Latin American countries suggests ineffective or lack of pricing regulation in these countries may result in higher prices in comparison to prices paid in HICs for these products (Moye-Holz & Vogler, Reference Vogler2022).
International price comparisons face a number of methodological challenges (Panteli et al., Reference Panteli2016), not least in terms of meaningfulness of results due to the widespread use of confidential discounts. One of the few studies that has published confidential price data (for cancer medicines) highlighted differences of up to 58% in discounted prices across 15 European countries (van Harten et al., Reference van Harten2016). Countries with large markets such as Italy and Spain are more likely to be granted higher discounts for medicines than other countries, for instance in central and eastern Europe (Vogler, Reference Vogler2021a). Furthermore, there is no explicit and generally accepted definition of what constitutes an “affordable” or a “fair” price for a medicine; this means that while comparisons can highlight differences, they cannot automatically guide policy decisions.
Policy relevance and conclusions
The previous sections have described the financing of pharmaceutical care, focusing on the R&D investment required to create new products and the purchasing process that makes them available to patients. We have also highlighted the challenges budget-constrained health systems face when trying to provide sufficient financial means to stimulate innovation and make the resultant new products available to patients. An ideal pharmaceutical system building on the current model would simultaneously: allow industry to maintain a profit margin to cover the expenses and risks incurred when bringing a new medicine to the market; meet the expectations of shareholders and investors providing the necessary capital for R&D; continuously spur the development of novel medicine candidates attuned to unmet needs; and enable the marketing of effective products that are affordable to public purchasers and accessible to patients. This is easier said than done.
On the side of R&D funding, clear communication of priorities on behalf of health systems and transparent and aligned incentive systems combining push and pull elements as described earlier in the chapter are crucial for addressing the commercial unattractiveness of certain medicines and steering efforts in their direction (see Anderson, Panteli & Mossialos, Reference Anderson, Panteli and Mossialos2023). At the same time, measures to ensure efficiency in R&D practices should be strengthened (Panteli & Edwards, Reference Panteli and Edwards2018). For instance, a large proportion of research on new pharmaceutical candidates is discarded before it is ever published or shared with the scientific community (Glasziou & Chalmers, Reference Glasziou and Chalmers2016). This often causes inconclusive research or negative research findings to be excluded from public databases and official communication channels, and results in substantial duplication and consequent waste of research in the system. According to a Transparimed report from 2017, medical research that is wasted amounts to US$ 170 billion each year (Bruckner, Reference Bruckner2017). Research registration systems and the development of reporting guidelines have started to spur transparent research practices; however, this needs to be further sustained to ensure knowledge generated from research is openly disclosed and fewer resources are wasted in the early stages of the pharmaceutical pipeline. The EU Clinical Trials Regulation, which will be fully implemented after a transition period lasting until 2025, aims to increase transparency of trial information through a new portal and database (Vogler, Reference Vogler2022a). Nondisclosure of R&D costs complicates public payers’ understanding of how to best invest in supporting innovation and the extent to which high pharmaceutical prices are justified. There are many who argue for more explicit ways to account for public “return on investment” for medicines that have been developed with substantial support from public funds (Mazzucato et al., Reference Mazzucato2018; McKee et al., Reference McKee2021; Torreele, Mazzucato & Li, Reference Torreele, Mazzucato and Li2021).
On the spending side, we have shown the high variability across countries and the related impact on equity. Overall, countries with less advanced regulatory systems, lack of HTA capacities, and lower price negotiating power are susceptible to high pharmaceutical expenditure which frequently translates into higher OOP contributions and an elevated financial burden for the general population. However, even where there are well-established pricing and reimbursement systems, payers face acute sustainability concerns, particularly in the light of the increasing number of new medicines with extremely high price tags. Reviewing current payment mechanisms is essential. For example, managed-entry agreements where financial compensation is contingent on the provision of additional evidence on clinical outcomes over time can have direct influence on how the medicine’s price evolves. This can, in theory, relieve the initial costs of medicines, while also ensuring that public money is allocated to medicines with a genuine added value for the patient and the health system involved. However, the evidence on the impacts of managed-entry agreements in terms of actually boosting affordability and access to innovative medicines is still limited (Vogler, Reference Vogler2022b), and their administrative burden is high. External reference pricing, while prevalent, is limited by both the lack of transparency due to confidential discounts and the lack of consideration of the “appropriateness” of referenced prices.
To mitigate these limitations, newer approaches need to be considered and piloted further. A range of different approaches including those which aim to reduce the upfront costs of treatment have already been discussed or tested. For instance, payment models that delink manufacturer revenue from the price and volume of medicines used entail agreements between payers and manufacturers that foresee a regular pre-set payment for (a range of) medicines. This procurement approach is known as the subscription or Netflix model. Such models can be used for costly therapies which should be given to all patients who need them, as in the case of hepatitis C medicines (Dore, 2021; Vogler, Reference Vogler2022b). They can also be suitable for medicines with larger target populations, or where the goal is to retain alternatives but limit unnecessary use (such as in the case of antibiotics, Vogler, Reference Vogler2022b). In this sense, subscription models can also function as pull mechanisms to incentivize manufacturers to engage in R&D efforts for medicines that are needed but would otherwise be financially unattractive (see Anderson, Panteli & Mossialos, Reference Anderson, Panteli and Mossialos2023). Subscription models can be combined with other provisions (e.g. with managed-entry agreements in the case of the hepatitis C in Australia, or updated HTA methodology to assess novel antibiotics in England) to strengthen their effectiveness. However, due to their novelty, evidence on the effectiveness of these models is still limited. Other suggested options entail paying in instalments and reclassifying costs for the procurement of ATMPs as investments rather than current expenditure (Active Citizenship Network, 2022); while these options would potentially improve access, they would not necessarily contribute to the sustainability of health budgets.
Even though their health systems have both important budgetary constraints and less elaborate price regulation options, LMICs have more limited access to new medicines and are exposed to higher prices than HICs. Equity-based tiered pricing is a mechanism that has the potential to mitigate these disparities. Such approaches entail setting differential prices based on countries’ economic abilities to pay for medicines working off a benchmark determined by involved governments. While equity-based tiered pricing could substantially improve access to innovative medicines in LMICs, its effective adoption requires careful design and, most importantly, solidarity and political will from leaders in HICs (Nemeth et al., Reference Nemeth2022). A further prerequisite is the reconsideration of the current practice of confidential discounts: an equity-based official price for a HIC that gets an ex post discount to a level below the equity-based price for a lower-income country would defeat the purpose of the initiative.
Strengthening cross-country collaboration in general is another potential avenue for improving access to innovative medicines. Collaborative approaches and pooling scientific expertise and negotiation capacities can reduce the number of resources that individual countries must invest. At the same time, this can create economies of scale and strengthen smaller and/or lower-income countries’ abilities to access pharmaceutical products at affordable prices. In recent years, several voluntary collaborations between governments in different geographical constellations have emerged, such as the Beneluxa Initiative, the Nordic Pharmaceutical Forum or the Valletta Declaration. Within these initiatives, countries aim to collaborate on a range of activities, including joint horizon scanning activities, HTA, joint pricing negotiations with industry and, in some cases, joint procurement of medicines (WHO Regional Office for Europe, 2020). Due to differences in national legal and organizational policy frameworks and the need for new working structures and governance processes, such collaborations can be resource-intensive and challenging, particularly at the start. As such, the success of joint negotiations and procurements is not guaranteed (WHO Regional Office for Europe, 2020). Nevertheless, there are some first positive outcomes of joint tenders and negotiations (Vogler, Haasis & Zimmermann, Reference Vogler, Haasis and Zimmermann2021). The potential of joint purchasing initiatives at the EU level was demonstrated by the procurement of over 4 billion COVID-19 vaccines on behalf of all Member States (European Commission, 2022b) as well as COVID-19 therapeutics (European Commission, 2022a). This EU experience also demonstrated the importance of the design of such mechanisms, as the advance purchase agreements for vaccines were considered less constrained by unilateral priorities compared to tenders under the Joint Procurement Agreement. Furthermore, EU-level collaboration on HTA has been ongoing for many years and was cemented in the 2021 Health Technology Agreement Regulation which foresees joint assessments for new medicines starting in 2025. Next to the described benefits for payers, streamlining HTA, pricing negotiations and procurement can also benefit industry by simplifying the processes involved and accelerating the entry of novel therapies to markets.
Finally, strengthening the preparedness and improving the transparency of the pharmaceutical system can have important benefits for equitable and affordable access to medicines. Horizon scanning activities entail active collection of information on medicines (and other health technologies) in development to enable and facilitate priority-setting and long-term planning and shape pricing and reimbursement decisions. Few countries in the WHO European Region have introduced horizon scanning systems at the national level (Vogler, Reference Vogler2022c), though collaborative approaches like the International Horizon Scanning Initiative are starting to operate (Vogler, Reference Vogler2022b). The lack of transparency around pharmaceutical prices discussed in this chapter contributes to inequities and reduces accountability and trust; however, evidence on the potential effects of introducing measures to improve price transparency is very limited. Efforts to establish platforms for exchanging price information, such as the European Integrated Price Information Database (EURIPID), and clearing-house approaches demonstrate potential for further expansion and consideration, respectively. Similarly, clearing houses managed by trusted third-party actors may be best suited to collect pricing information anonymously from payers, including details on confidential discounts (Webb et al., Reference Webb2022).
The selection of policy actions described in this section is by no means exhaustive. No single one of the approaches is a magic remedy to the complex issues emerging in the pharmaceutical and innovation sector if implemented on its own. Payers at different levels need to consider these options according to their context, and cross-country collaboration can strengthen action to address these global issues with local specificities.Footnote 3
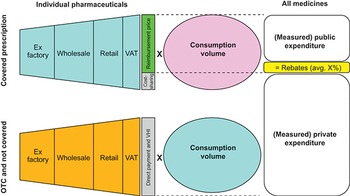

 Open access
Open access