


The hypothesis that schizophrenia may represent a disorder of accelerated ageing was first proposed by Kirkpatrick et al in 2008. Reference Kirkpatrick, Messias, Harvey, Fernandez-Egea and Bowie1 Drawing on epidemiological data, we noted increased age-associated clinical features among individuals diagnosed with schizophrenia, including elevated mortality, early-onset cognitive decline and metabolic disturbances. This model reframed schizophrenia as a multisystem condition, not solely a brain disorder, Reference Kirkpatrick, Miller, García-Rizo and Fernandez-Egea2 and posited that affected individuals would exhibit abnormal levels of biological ageing markers. Over the past 15 years, numerous research groups have investigated this hypothesis using a range of biological ageing indicators. These investigations have included meta-analytic studies of telomere length, Reference Ayora, Fraguas, Abregú-Crespo, Recio, Blasco and Moises3 oxidative stress, Reference Jorgensen, Baago, Rygner, Jorgensen, Andersen and Kessing4 DNA-methylation-based measures of epigenetic age Reference Chrusciel, Orso, de Mattos, Fries, Kristensen and Grassi-Oliveira5 and brain ageing estimates derived from neuroimaging proxies. Reference Ballester, Romano, de Azevedo Cardoso, Hassel, Strother and Kennedy6 Despite this growing body of work, no synthesis has yet integrated findings across all biological systems.
Here, we present a systematic review of epidemiological and biological evidence linked to the accelerated ageing hypothesis in schizophrenia. We pay particular attention to meta-analytic data and studies involving minimally treated participants, including first-episode psychosis (FEP) cohorts. This focus enables us to address a common critique: that observed abnormalities may reflect confounding factors such as long-term antipsychotic use, sedentarism or substance use rather than accelerated ageing per se. Our aims were to evaluate whether schizophrenia was consistently associated with premature ageing across multiple biological domains and mechanisms; to appraise the strength, consistency and limitations of the current evidence base; and to set up future research directions.
Method
Protocol and registration
This study was PROSPERO registered (CRD42024574059) on 9 September 2024. The protocol was developed a priori and follows PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines. The PRISMA checklist is provided in the Supplementary Material available at https://doi.org/10.1192/bjp.2026.10606. The full protocol is available via PROSPERO.
Ethics statement
This was a systematic review of published and publicly available data. No new data were collected and no individual-level identifiable information was accessed. Ethical approval and participant consent were therefore not required.
Search strategy
The search strategy was structured according to the PEO (Population, Exposure, Outcome) method. Date restrictions were applied, so that only research published after 2009 was included. The search was further limited to studies that were published in English and appeared in peer-reviewed journals.
The PubMed search strategy included the following combinations of terms:
[accelerated] AND [ageing OR aging] AND [schizophrenia]
[accelerated] AND [ageing OR aging] AND [psychosis]
[early] AND [ageing OR aging] AND [schizophrenia]
[early] AND [ageing OR aging] AND [psychosis].
The following eligibility criteria were also applied:
-
(a) population: adults with schizophrenia or non-affective psychosis;
-
(b) design: observational (cross-sectional or longitudinal) or experimental studies;
-
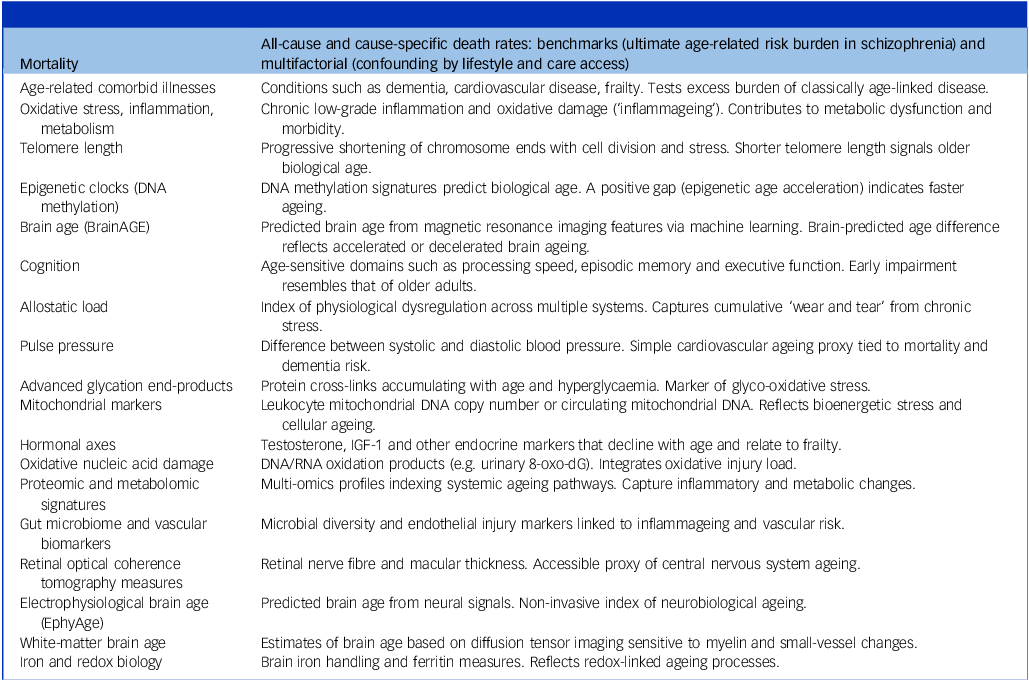
(c) exposure: studies assessing biological markers and mechanisms of ageing (e.g. telomere length, oxidative stress, inflammatory markers, mitochondrial dysfunction, epigenetic clocks or neuroimaging-based brain age) (see Table 1 for ageing terminology);
-
(d) comparators: where applicable, healthy controls matched by age and gender, free from major psychiatric or neurological illness or other conditions known to influence biological ageing;
-
(e) language: English only.
Epidemiological, clinical and biological indices of human ageing

Case reports, review articles, conference abstracts and studies lacking relevant ageing markers were excluded.
Participants were categorised by antipsychotic exposure: (a) drug-naïve or minimally treated (≤2 weeks exposure); or (b) prolonged treatment.
We searched PubMed and Google Scholar. In PubMed, predefined search strings were used. In Google Scholar, all articles citing the original 2008 article proposing schizophrenia as an accelerated ageing syndrome Reference Kirkpatrick, Messias, Harvey, Fernandez-Egea and Bowie1 were retrieved. The last search was conducted on 1 October 2024.
Records were screened using Rayyan (https://new.rayyan.ai), a web-based tool for systematic reviews. Two reviewers (E.F.-E. and C.G.-R.) independently screened titles, abstracts and full texts. Discrepancies were resolved through discussion, and unresolved cases were adjudicated by a third reviewer (B.K.).
Data extraction was performed independently by the same two reviewers using predesigned forms. Data fields included the following:
-
(a) study characteristics (authors, year, design);
-
(b) sample details (size, demographics, diagnostic criteria);
-
(c) medication status (drug-naïve, minimally treated or chronic);
-
(d) ageing biomarkers assessed;
-
(e) comparator group details, if applicable;
-
(f) outcome measures and reported results.
All decisions and extracted data were recorded in Rayyan.
Two authors (E.F.-E. and C.G.-R.) independently assessed the risk of bias in included studies, and a third author (B.K.) resolved disagreements. The Newcastle–Ottawa Scale Reference Stang, Jonas and Poole7 was used to grade the quality of observational studies, whereas meta-analyses were evaluated with AMSTAR 2. Reference Shea, Reeves, Wells, Thuku, Hamel and Moran8
Given the expected heterogeneity in study design and measurement approaches, a narrative synthesis was prioritised. Data were organised via subgroup analysis on the basis of ageing markers and illness stage.
Results
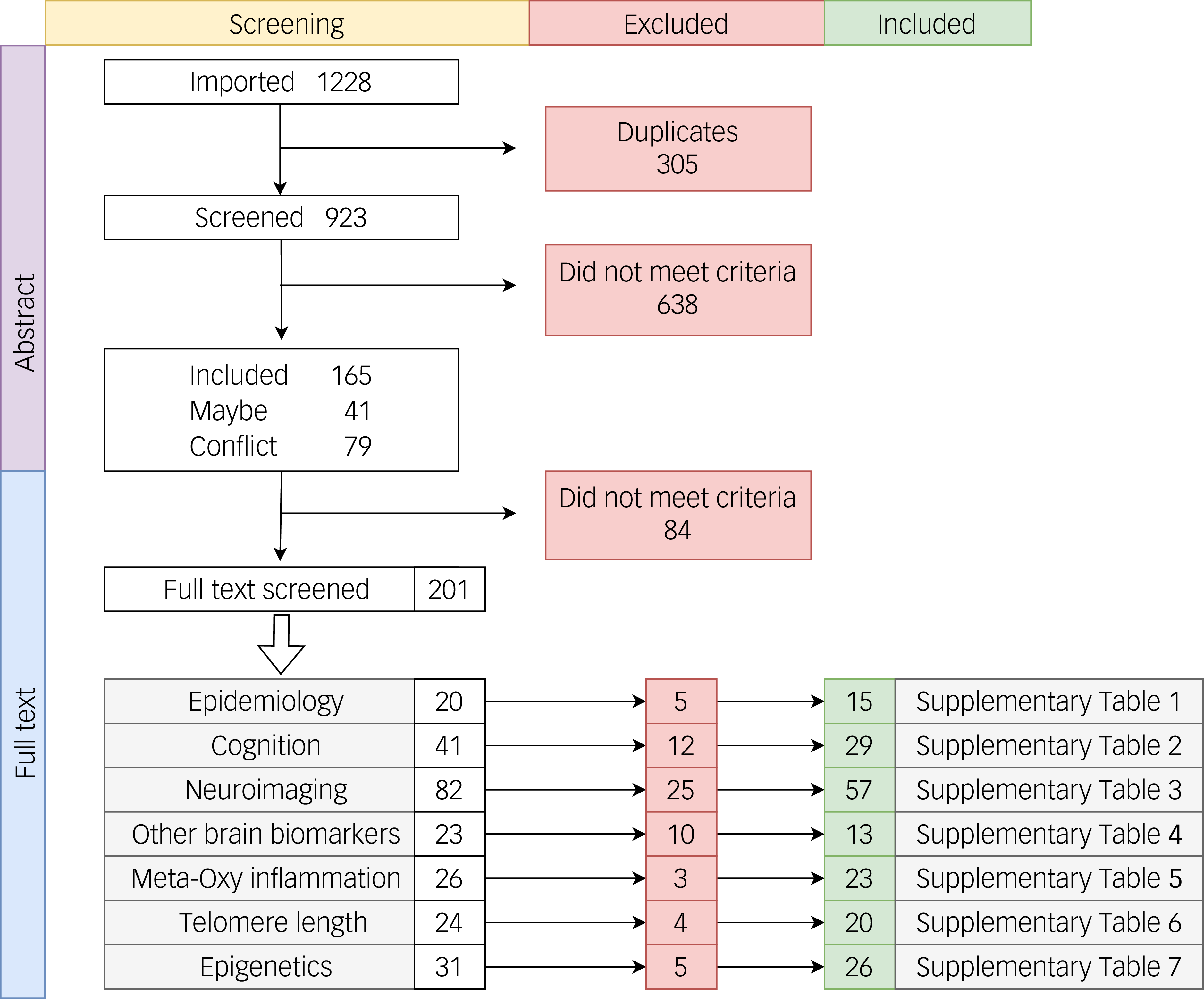
Figure 1 shows the study selection process. Of the 170 observational studies assessed with the Newcastle–Ottawa Scale, 93 of 170 (54.7%) were rated high quality (score 7–9), and 75 of 170 (44.1%) were rated moderate quality (score 5–6), with only 2 of 170 (1.2%) scoring <5. Across the meta-analyses appraised with AMSTAR 2 (n = 8), ratings were high (three of eight, 37.5%), moderate-to-high (one of eight, 12.5%) or moderate (four of eight, 50.0%), with none rated low or critically low (Supplementary Tables 1–7).
Flowchart of the 170 manuscripts included in the systematic review and classifications. Note that some manuscripts may have been included in more than one category (e.g. imaging and blood biomarkers).

Epidemiology
A meta-analysis of 11 cohorts found a relative risk of 2.52 for dementia in people with nonaffective psychosis. Reference Miniawi, Orgeta and Stafford9 Of these 11 samples, only one did not show a significant difference. Five studies were also found by our search strategy. Reference Kodesh, Goldberg, Rotstein, Weinstein, Reichenberg and Sandin10–Reference Stroup, Olfson, Huang, Wall, Goldberg and Devanand14 Two of the 11 studies provided data on the size of the differences between groups: dementia was diagnosed in 22% of people with schizophrenia before age 65 years versus 6% in the general population, Reference Ribe, Laursen, Charles, Katon, Fenger-Grøn and Davydow12 and the percentage of people with schizophrenia diagnosed with dementia by age 66 years was similar to the percentage diagnosed by age 88 years in the general population. Reference Stroup, Olfson, Huang, Wall, Goldberg and Devanand14 For eight of these cohorts, multivariate analyses were performed for potentially confounding factors, including antipsychotic medication history in some; with covariance, this group of studies yielded a relative risk of 2.2. An increased risk of dementia was found in two other large samples from the UK, Reference Stevenson-Hoare, Legge, Simmonds, Han, Owen and O’Donovan15 with hazard ratios of 2.87 and 4.46.
An increased mortality rate is consistent with accelerated ageing. The increased mortality found by Kodesh et al Reference Kodesh, Goldberg, Rotstein, Weinstein, Reichenberg and Sandin10 in people with very late onset of schizophrenia remained significant after covarying for other mortality risk factors.
In a large, population-based sample, there was no difference in the overall incidence of cancer. Reference Pettersson, Larsson, D’Onofrio, Almqvist and Lichtenstein16 A national cohort study found that cancer risk in people with schizophrenia was decreased for cancers that usually develop at an older age and increased for cancers that usually have a younger age at onset. Reference Lin, Lane, Chen, Wu, Wu and Wu17 One interpretation of this finding is that relatively few people with schizophrenia who are liable to develop cancer survive to an age at which they develop the cancers associated with greater age. Drevinskaite et al Reference Drevinskaite, Kaceniene, Patasius, Stukas, Germanavicius and Miseikyte18 found that total cancer incidence decreased in men but not in women with schizophrenia. Given the heterogeneity of cancer, considering all cancers together is probably not the best strategy for testing the hypothesis.
People with schizophrenia may have premature general physical disability, as measured by an early need for a basket of social services Reference Taube, Mentzel, Glue and Barak19 and a greater prevalence of physical impairments such as maintaining balance and rising from a chair. Reference Clouston, Jonas, Fochtmann, Bromet and Kotov20 These differences remained after covarying for other morbidities. A study of schizophrenia spectrum disorder and general population groups found no group difference in the prevalence of participants with two or more chronic physical illnesses, despite the greater average age of the general population group. Reference Šimunović Filipčić, Ivezić, Jakšić, Mayer, Grah and Rojnić Kuzman21 Renn et al Reference Renn, Yang, Chueh, Lin, Lan and Chou22 found that people with schizophrenia had decreased bone mass compared with control participants early in the course of schizophrenia, but bone loss did not progress more quickly in later years. In a national sample, people with schizophrenia aged 70 years did not differ from those without schizophrenia with respect to the number of chronic medical illnesses registered in health databases. Reference Brink, Green, Bojesen, Lamberti, Conwell and Andersen23
The most consistent epidemiological evidence supporting the accelerated ageing hypothesis was from studies of dementia and mortality. The evidence was quite mixed or negative for cancer and premature general physical disability. There was one study of allostatic load, which was consistent with accelerated ageing. The increased risk of advanced age was evident in several studies after covariance for a variety of confounding factors, sometimes including antipsychotic use.
Cognition
No meta-analyses have tested the accelerated ageing hypothesis in relation to cognition in schizophrenia. Seven studies found that patients with early psychosis performed similarly to older healthy adults, particularly in terms of executive function and processing speed, using MATRICS (Measurement and Treatment Research to Improve Cognition in Schizophrenia), Reference Rajji, Voineskos, Butters, Miranda, Arenovich and Menon24–Reference Czepielewski, Massuda, Panizzutti, Grun, Barbé-Tuana and Teixeira26 the Brief Assessment of Cognition in Schizophrenia, Reference Mohan, Parekh, Lukose, Moirangthem, Saini and Schretlen27 the Wechsler Adult Intelligence Scale Reference Sponheim, Jung, Seidman, Mesholam-Gately, Manoach and O’Leary28 or a continuous performance task. Reference Mirzakhanian, Singh, Seeber, Shafer and Cadenhead29,Reference Smucny, Lesh, Iosif, Niendam, Tully and Carter30 Patients across these early-stage studies typically performed similarly to healthy individuals aged 5 to 10 years older, but there was no steeper age-related decline in those with schizophrenia compared with healthy controls, except in the study by Mohan et al. Reference Mohan, Parekh, Lukose, Moirangthem, Saini and Schretlen27
Longitudinal studies have described early but stable cognitive decline in schizophrenia. Zanelli et al Reference Zanelli, Reichenberg, Sandin, Morgan, Dazzan and Pilecka31,Reference Zanelli, Mollon, Sandin, Morgan, Dazzan and Pilecka32 followed patients from first-episode into midlife and reported declines in IQ and executive function, with patients in their 40s showing cognitive profiles comparable with those of healthy individuals more than 60 years old. Fett et al Reference Fett, Velthorst, Reichenberg, Ruggero, Callahan and Fochtmann33 conducted a 20-year prospective study and observed a cognitive performance gap between patients and controls that accelerated over time (>50 years old). Differences were most pronounced for verbal fluency and executive function, with performance gaps reaching up to 0.5 s.d., approximating a functional ageing shift of 10–15 years in some domains. Thompson et al Reference Thompson, Savla, Vahia, Depp, O’Hara and Jeste34 found that approximately 10% of older patients exhibited rapid cognitive decline, particularly those with negative symptoms and those in supported living. Cohen and Murante Reference Cohen and Murante35 reported that most older patients remained stable or improved across a 4.5-year follow-up, although a subgroup showed decline.
Studies in chronic schizophrenia described performance differences consistent with those observed in older adults. Wang et al, Reference Wang, Chan, Qing, Yang, Yu and Li36 Herold Reference Herold, Schmid, Lässer, Seidl and Schröder37 and Silver and Bilker Reference Silver and Bilker38,Reference Silver and Bilker39 found overlapping deficits on specific cognitive tasks in patients and older healthy adults. Dragovic et al Reference Dragovic, Fajgelj and John40 reported that patients in their 30s performed similarly to controls in their 70s on processing speed and fluency tasks. Hulstijn et al Reference Hulstijn, Cornelis, Morsel, Timmers, Morrens and Sabbe41 and Cornelis et al Reference Cornelis, De Picker, De Boer, Dumont, Coppens and Morsel42,Reference Cornelis, Picker, Hulstijn, Dumont, Timmers and Janssens43 described psychomotor slowing and implicit learning patterns comparable with those observed in elderly controls, whereas Mathias et al Reference Mathias, Knowles, Barrett, Leach, Buccheri and Beetham44 found that illness and age negatively influenced processing speed but in different ways. Malliaris et al Reference Malliaris, Angelopoulos, Dardiotis and Bonotis45 and Thuaire et al Reference Thuaire, Rondepierre, Bacon, Vallet, Jalenques and Izaute46 found greater impairments in executive and attentional tasks after midlife. Gawęda Reference Gawęda47 observed source monitoring deficits and age-related patterns in memory confidence.
Some studies included biological correlates of cognitive function. Sirivichayakul et al Reference Sirivichayakul, Kanchanatawan, Thika, Carvalho and Maes48 reported that elevated eotaxin levels were associated with memory and executive function deficits in individuals with schizophrenia; Huo et al Reference Huo, Zheng, Lu, Wu, Ning and Zhang49 found lower brain-derived neurotrophic factor levels in patients with attention and general cognitive deficits; Van Rheenen et al Reference Rheenen, Cropley, Fagerlund, Wannan, Bruggemann and Lenroot50 found that cognitive decline occurred in patients with low cognitive reserve and frontal cortical atrophy; and Wang et al Reference Wang, Kochunov, Sampath, Hatch, Ryan and Xue51 linked increased white matter brain age with cognitive performance. Feng et al Reference Feng, Palaniyappan, Robbins, Cao, Fang and Luo52 identified anterior cingulate inefficiency during working memory tasks in antipsychotic-naïve and first-episode schizophrenia patients; these patterns were similar to those seen in older adults and were linked to dopaminergic gene expression.
Overall, cognitive data support an early deficit consistent with a 5–10-year ageing shift, with a modest additional decline in a subset of patients in mid-to-late life, rather than universal accelerated deterioration.
Neuroimaging
Two meta-analyses examined BrainAGE in schizophrenia. The ENIGMA Schizophrenia Working Group Reference Constantinides, Han, Alloza, Antonucci, Arango and Ayesa-Arriola53 pooled data from 26 cohorts (2803 patients and 2598 controls) and found a mean brain-predicted age difference (brain-PAD) of +3.55 years in schizophrenia (95% CI: 2.91–4.19), with a moderate effect size (Cohen’s d = 0.48). This elevation was consistent across sites and unrelated to illness duration, symptom severity or antipsychotic exposure. A smaller meta-analysis of 18 studies compared patients with three psychiatric disorders with healthy volunteers Reference Ballester, Romano, de Azevedo Cardoso, Hassel, Strother and Kennedy6 and found that patients with schizophrenia showed the largest brain age gap (BAG; 3.08 [95% CI: 2.32–3.85], compared with 1.93 [95% CI: 0.53–3.34] for patients with bipolar disorder and 1.12 [95% 0.41–1.83] for those with major depressive disorder).
Twenty studies evaluated BrainAGE in individuals with early psychosis or those in clinical high-risk states, with 14 providing clear support for advanced brain age. Cross-sectional studies typically reported significant BAGs from +1.4 to +6.9 years, primarily driven by grey matter changes in frontal and temporal regions. Reference Ballester, Romano, de Azevedo Cardoso, Hassel, Strother and Kennedy6,Reference Hajek, Franke, Kolenic, Capkova, Matejka and Propper54–Reference Shahab, Mulsant, Levesque, Calarco, Nazeri and Wheeler62 Longitudinal studies showed initial BAG elevations (+0.7 to +1.7 years), although some evidence suggested reversibility after treatment initiation. Reference Xi, Wu, Cui, Bai, Gan and Jia63,Reference Hua, Abram, Loewy, Stuart, Fryer and Vinogradov64 Obesity was identified as a moderating factor, exacerbating accelerated ageing effects in early psychosis cohorts. Reference Kolenic, Franke, Hlinka, Matejka, Capkova and Pausova65,Reference Mcwhinney, Kolenic, Franke, Fialova, Knytl and Matejka66 Additional studies highlighted associations of elevated BAG with early illness onset, diagnostic uncertainty and subtle clinical symptoms. Reference Kim, Heo, Maeng, Shen, Tsogt and Odkhuu59,Reference Koutsouleris, Davatzikos, Borgwardt, Gaser, Bottlender and Frodl67–Reference Demro, Shen, Hendrickson, Arend, Disner and Sponheim70
Nine studies assessed BrainAGE in chronic schizophrenia, with eight supporting advanced brain age and one offering partial support moderated by cognitive reserve. Nenadić et al Reference Dietzek, Langbein, Sauer and Gaser60 reported a BrainAGE score of +2.56 years in schizophrenia, versus −0.22 in controls and −1.25 in bipolar disorder. Klaus et al Reference Klaus, Nguyen, Thomas, Liou, Soontornniyomkij and Mitchell71 found a +6.5-year brain-PAD, with higher TNF-α levels linked to greater ageing. Ryan et al Reference Ryan, Hong, Hatch, Gao, Chen and Haerian72 found large ageing deviations (d = 1.42), independent of cardiometabolic factors. Teeuw et al Reference Teeuw, Ori, Brouwer, de Zwarte, Schnack and Hulshoff Pol73 identified a +4.0-year BAG that was related to polygenic risk. Wang et al Reference Wang, Kochunov, Sampath, Hatch, Ryan and Xue51 reported a white matter BAG of +5.9 years associated with cognitive deficits. Cetin-Karayumak et al Reference Cetin-Karayumak, Biase, Chunga, Reid, Somes and Lyall74 observed accelerated white matter decline after early maturation peaks, especially in callosal fibres. Schnack et al Reference Schnack, Haren, Nieuwenhuis, Pol, Cahn and Kahn75 found a baseline BAG of +3.4 years, increasing by +1.24 years over follow-up, with the greatest acceleration during early illness. Ballester et al Reference Ballester, Suh, Ho, Liang, Hassel and Strother76 found a greater contribution to BAGs of grey matter volume compared with white matter. Van Rheenen et al Reference Rheenen, Cropley, Fagerlund, Wannan, Bruggemann and Lenroot50 showed exaggerated brain ageing in schizophrenia but found cognitive decline only in patients with low cognitive reserve, suggesting resilience in some groups.
Studies directly testing the accelerated ageing hypothesis using different methodology
Nine studies using non-BrainAGE neuroimaging in early psychosis largely supported disrupted age-related brain trajectories. In a 5-year study, van Haren et al Reference Pol, Schnack, Cahn, Brans and Carati77 found accelerated grey matter loss and ventricular expansion before the age of 45 years, suggestive of early neurodegeneration. Pham et al Reference Pham, Sasabayashi, Takahashi, Takayanagi, Kubota and Furuichi78 reported progressive frontotemporal gyrification loss in individuals with schizophrenia but not in those with schizotypal disorder. Dukart et al Reference Dukart, Smieskova, Harrisberger, Lenz, Schmidt and Walter79 found grey matter decline in both first-episode patients and those in at-risk states. Lin et al Reference Lin, Li, Zhou, Deng, Ma and Wang80 observed steeper cortical thinning with age in drug-naïve patients. Sheffield et al Reference Sheffield, Rogers, Blackford, Heckers and Woodward81 and Kuo et al Reference Kuo, Lee, Hung, Liu, Lee and Chung82 demonstrated functional declines in key cognitive networks (Sheffield et al observed an accelerated decline in the functional integration and efficiency of the frontoparietal and cingulo-operatic networks, whereas Kuo et al used structural covariance networks to demonstrate associations of schizophrenia with advanced ageing across multiple systems, notably within the default mode network and salience network). Shivakumar et al Reference Shivakumar, Kalmady, Rajasekaran, Chhabra, Anekal and Narayanaswamy83 reported telomere shortening associated with hippocampal volume loss in drug-free patients. Chiapponi et al Reference Chiapponi, Piras, Fagioli, Girardi, Caltagirone and Spalletta84 identified accelerated microstructural degeneration in the hippocampus, whereas Thormodsen et al Reference Thormodsen, Rimol, Tamnes, Juuhl-Langseth, Holmén and Emblem85 noted absent normative cortical thinning in early-onset schizophrenia. An exception was the study of Bose et al, Reference Bose, Mackinnon, Mehta, Turkheimer, Howes and Selvaraj86 which found increased white but reduced grey matter decline.
Fifteen 15 studies provided mixed support for accelerated ageing in patients with chronic schizophrenia. Man et al Reference Man, Ding, Chai, An, Liu and Qin87 found a small but significant BAG, and Rowland et al Reference Rowland, Krause, Wijtenburg, McMahon, Chiappelli and Nugent88 reported steeper declines in medial frontal γ-aminobutyric acid. Longitudinal cortical thinning with downregulation of specific genes was observed by González-Peñas et al, Reference González-Peñas, Alloza, Brouwer, Díaz-Caneja, Costas and González-Lois89 whereas De Picker et al Reference Picker, Ottoy, Verhaeghe, Deleye, wyffels and Fransen90 reported age-related microglial activation. Kochunov et al Reference Kochunov, Chiappelli, Wright, Rowland, Patel and Wijtenburg91,Reference Kochunov, Ganjgahi, Winkler, Kelly, Shukla and Du92 described accelerated white matter decline independent of perfusion, and Sheffield et al Reference Sheffield, Repovs, Harms, Carter, Gold and Macdonald93 found worsening network inefficiency. Additional studies showed selective age-related changes in grey or white matter: Wright et al Reference Wright, Kochunov, Chiappelli, McMahon, Muellerklein and Wijtenburg94 confirmed white matter ageing, and Kochunov et al Reference Kochunov, Glahn, Rowland, Olvera, Winkler and Yang95 linked this to genetic risk. Only Voineskos et al Reference Voineskos, Lobaugh, Bouix, Rajji, Miranda and Kennedy96 reported no evidence of accelerated frontotemporal tract decline.
Indirect imaging evidence relating to accelerated ageing
Although not specifically designed to test the accelerated ageing hypothesis, 12 imaging studies identified through PROSPERO offered relevant indirect evidence. A meta-analysis by Hua and Mathalon Reference Hua and Mathalon97 reported widespread morphometric abnormalities consistent with advanced brain age in patients with early psychosis. Longitudinal work by Pruessner et al Reference Pruessner, Lepage, Collins, Pruessner, Joober and Malla98,Reference Pruessner, Bechard-Evans, Pira, Joober, Collins and Pruessner99 showed hippocampal volume loss linked to hypothalamic–pituitary–adrenal axis dysfunction, whereas Hua et al Reference Hua, Cummings, Roach, Fryer, Loewy and Stuart100 found progressive disruption in structural connectome integrity. Other studies of early psychosis, such as that of Skudlarski et al, Reference Skudlarski, Schretlen, Thaker, Stevens, Keshavan and Sweeney101 identified white matter abnormalities, but findings from the studies of Schuster et al, Reference Schuster, Schuller, Paulos, Namer, Pull and Danion102 Mamah et al, Reference Mamah, Chen, Shimony and Harms103 Feng et al Reference Feng, Palaniyappan, Robbins, Cao, Fang and Luo52 and Seitz et al Reference Seitz, Rathi, Lyall, Pasternak, del Re and Niznikiewicz104 were less conclusive. Koutsouleris et al Reference Koutsouleris, Davatzikos, Borgwardt, Gaser, Bottlender and Frodl67 observed atrophy compatible with accelerated ageing, although this was confounded by illness duration and medication.
Miscellaneous neurobiomarkers
Neurophysiological markers were examined using electroencephalogram- and magnetoencephalogram-derived brain age models. Sarıisik et al Reference Sarisik, Popovic, Keeser, Khuntia, Schiltz and Falkai105 reported an increased electrophysiological age gap (EphysAGE). Abram et al Reference Abram, Roach, Holroyd, Paulus, Ford and Mathalon106 found no significant group differences in event-related potential-derived brain age, although greater depressive symptoms were associated with higher estimated brain age in the schizophrenia group. Chen et al Reference Chen, Lee, Yang, Landau, Chang and Chen107 observed no early P300 abnormalities in drug-naïve individuals but reported a steeper age-related increase in P300 latency.
Retinal imaging studies used optical coherence tomography to assess structural markers. Blose et al Reference Blose, Lai, Crosta, Thompson and Silverstein108 and Domagata Reference Domagała, Domagała, Kopiś-Posiej, Harciarek and Krukow109 found age-related thinning of the retinal nerve fibre layer, macular volume and ganglion cell–inner plexiform layer in individuals with schizophrenia spectrum disorders, with these changes persisting after adjustment for diabetes and hypertension. Wagner et al, Reference Wagner, Cortina-Borja, Silverstein, Zhou, Romero-Bascones and Struyven110 using a large sample of 465 patients and 100 931 controls, reported reduced retinal thickness in schizophrenia, with associations between retinal measures, cognitive performance and brain integrity.
Other biological measures were also explored. A meta-analysis found no significant increase in levels of amyloid-β plaques or neurofibrillary tangles in cognitively impaired patients with schizophrenia compared with controls, Reference Wilson, Liu, Jones, Mahmood, Arya and Howard111 and these levels were lower than those in people with Alzheimer’s disease. Several studies examined specific Brodmann areas: Uranova et al Reference Uranova, Vikhreva and Rakhmanova112 identified microglial degeneration in the prefrontal cortex (BA10), characterised by lipofuscin accumulation and mitochondrial disruption, with associations with age and illness duration. Mizutani et al, Reference Mizutani, Saiga, Yamamoto, Uesugi, Takeuchi and Uesugi113 examining Brodmann area 24, found increased neurite curvature in individuals with schizophrenia, highlighting structural changes opposite to those observed in normal ageing. Tang et al Reference Tang, Chang, Lanigan, Dean, Sutcliffe and Thomas114 identified transcriptomic overlap between early-stage schizophrenia and normative ageing in Brodmann area 46. Other relevant findings include those of Herold et al, Reference Herold, Lässer, Seidl, Hirjak, Thomann and Schröder115 who found a steeper increase in neurological soft signs with age in patients compared with controls; Tang et al, who reported transcriptomic overlap between early-stage schizophrenia and normative ageing in BA46; Arakelyan et al, Reference Arakelyan, Avagyan, Kurnosov, Mkrtchyan, Mkrtchyan and Zakharyan116 who observed age-shifted gene expression patterns, with profiles resembling those of older healthy individuals; and Torkamani et al, Reference Torkamani, Dean, Schork and Thomas117 who described age-related dysregulation in developmental gene networks but in an opposite direction to that seen in ageing.
Blood biomarkers
Oxidative stress, inflammation and metabolism
Evidence from meta-analytic and early-course data was robust, suggesting systemic physiological dysregulation. Jorgensen Reference Jorgensen, Baago, Rygner, Jorgensen, Andersen and Kessing4 meta-analysed oxidative-stress-induced nucleic acid damage (82 studies, comprising 10 151 patients and 10 532 controls), describing elevated levels in patients with psychosis (medium to large effect) and suggesting involvement in the pathophysiology of age-related medical conditions. Notably, these markers appeared to be intrinsic rather than solely treatment driven as in FEP treatment-naïve samples. Garcia-Rizo described abnormal glycaemic homeostasis Reference Garcia-Rizo, Fernandez-Egea, Bernardo and Kirkpatrick118 and higher blood cell counts, Reference Garcia-Rizo, Casanovas, Fernandez-Egea, Oliveira, Meseguer and Cabrera119 whereas Fernandez-Egea described reduced biologically active testosterone. Reference Fernandez-Egea, García-Rizo, Miller, Parellada, Justicia and Bernardo120 These findings in treatment-naïve participants indicate that metabolic and hormonal dysregulation occurs before the onset of psychosis, representing a pre-existing risk that may contribute to earlier morbidity and subsequent mortality.
In addition to these core findings, the literature converged on the following four distinct mechanistic domains:
-
(a) Mitochondrial function: mitochondrial signatures suggest widespread impairment in cellular energy homeostasis, with Shivakumar et al describing reduced DNA copy number variations, Reference Shivakumar, Rajasekaran, Subbanna, Kalmady, Venugopal and Agrawal121 whereas Garcia de la Cruz observed significantly elevated levels of circulating cell-free mitochondrial DNA. Reference Juarez-Rojop, Tovilla-Zarate, Nicolini and Genis-Mendoza122
-
(b) Oxidative stress: alongside nucleic acid damage, specific markers of ageing and oxidation are altered. Lotan et al described a reduction in physiological age-dependent iron accumulation and decreased ferritin, implying an imbalance that theoretically implies oxidative damage; Reference Lotan, Luza, Opazo, Ayton, Lane and Mancuso123 Jørgensen et al described higher urinary excretion of systemic DNA and RNA oxidation markers independent of treatment; Reference Jorgensen, Broedbaek, Fink-Jensen, Knorr, Greisen Soendergaard and Henriksen124 and Zhang described decreased levels of vitamin B12 and folate-dependent enzyme methionine synthase forms. Reference Zhang, Hodgson, Trivedi, Abdolmaleky, Fournier and Cuenod125 In addition, Hagen described a higher marginal accumulation rate of advanced glycation end products over a 2-year follow-up in a treated FEP sample. Reference Hagen, Sutterland, Edrisy, Tan and de Haan126 However, discrepancies arose; Rebouças found no changes in glutathione peroxidase or protein carbonyl content, Reference Sartori, Librenza-Garcia, Rabelo-da-Ponte, Massuda and Czepielewski127 and Busch found no difference in hair cortisol concentrations in forensic autopsies. Reference Busch, Wang, Lynnerup, Jacobsen, Jørgensen and Linnet128
-
(c) Multi-omics, inflammation and metabolism: broader profiling revealed disruption in inflammatory and metabolic pathways. Campeau et al performed a multi-omics analysis, finding altered proteomic and metabolomic signatures that overlapped with frailty and metabolic ageing. Reference Campeau, Mills, Stevens, Rossitto, Meehan and Dorrestein129 Postmortem studies aligned, with Yu et al describing an ‘older’ lipidome state in the prefrontal cortex Reference Yu, He, Zubkov, Huang, Kurochkin and Yang130 and Weissleder et al describing decreased IGF1 mRNA levels involved in metabolic regulation. Reference Weissleder, Webster, Barry and Shannon Weickert131 Other studies described higher levels of inflammatory markers (CrP, chemokines, TNF-α, IL-6, insulin–glucose homeostasis parameters) associated with psychopathology, chronicity and ageing endophenotypes, including increased eotaxin levels Reference Sirivichayakul, Kanchanatawan, Thika, Carvalho and Maes48,Reference Hong, Lee, Martin, Soontornniyomkij, Soontornniyomkij and Achim132–Reference Squassina, Manchia, Pisanu, Ardau, Arzedi and Bocchetta137 accompanied by increased allostatic load. Reference Nugent, Chiappelli, Rowland and Hong138 Allostatic load (Table 1), which is defined by several physiological measures that reflect the cumulative physiological effect of stress, increases from roughly ages 20 to 60 years, after which it is usually relatively constant. Reference Crimmins, Johnston, Hayward and Seeman139
-
(d) Vascular endothelium and gut microbiota: Nguyen et al, in separate studies, analysed the gut microbiota, describing differences in beta-diversity associated with microbial metabolic pathways linked to atherosclerosis and immunity Reference Nguyen, Kosciolek, Daly, Vázquez-Baeza, Swafford and Knight140 and in levels of vascular endothelial biomarkers, which are key players in the intersection of inflammatory response and vascular risk. Reference Nguyen, Dev, Chen, Liou, Martin and Irwin141
The confluence of increased inflammatory levels, oxidative stress biomarkers and metabolic load suggests a complex interplay underlying the ageing process in schizophrenia; however, interpretation requires some caution. The evidence gathered is cross-sectional and vulnerable to confounders. Markers could have been influenced by lifestyle habits or environmental factors, such as childhood trauma, that promote worse insulin–glucose homeostasis.
Telomere length
Four meta-analyses described a significant decrease in telomere length. Reference Ayora, Fraguas, Abregú-Crespo, Recio, Blasco and Moises3,Reference Rao, Kota, Li, Yao, Tang and Mao142–Reference Russo, Prinzi, Proietti, Lamonaca, Frustaci and Boccia144 Ayora et al (22 studies, 4145 patients and 4184 controls) described a small-to-medium effect (standardised mean difference (SMD) = −0.388)], Reference Ayora, Fraguas, Abregú-Crespo, Recio, Blasco and Moises3 and Russo et al Reference Russo, Prinzi, Proietti, Lamonaca, Frustaci and Boccia144 (18 studies, 1827 patients and 1741 controls) reported a medium effect size (SMD = −0.52), whereas Rao et al Reference Rao, Kota, Li, Yao, Tang and Mao142 and Polho et al Reference Polho, De-Paula, Cardillo, dos Santos and Kerr143 performed similar main analyses (11 studies, 1243 patients and 1274 controls) and found a medium effect size (SMD = −0.48). Another meta-analysis involving a wide array of mental diagnoses including schizophrenia (six studies, 772 patients and 763 controls) described a smaller effect size for leukocyte telomere length shortening (Hedge’s g = −0.23). Reference Darrow, Verhoeven, Révész, Lindqvist, Penninx and Delucchi133
Several individual studies supported a reduction in telomere length: Fernandez-Egea et al reported this finding in an antipsychotic-naïve FEP sample, Reference Fernandez-Egea, Bernardo, Heaphy, Griffith, Parellada and Esmatjes145 Zakharova et al in individuals with schizophrenia specifically with suicidal risk, Reference Zakharova, Bravve, Mamedova, Kaydan, Ershova and Martynov146 Squassina et al in patients with treatment-resistant schizophrenia Reference Squassina, Manchia, Pisanu, Ardau, Arzedi and Bocchetta137 and Russo et al in patients below 50 years of age. Reference Russo, Prinzi, Proietti, Lamonaca, Frustaci and Boccia144 Van Mierlo et al, in a study of post-mortem brain, described a significant decrease in telomere length in the superior temporal gyrus, Reference van Mierlo, Wichers, He, Sneeboer, Radstake and Kahn147 suggesting that cell senescence was affected in white but not grey matter. Czepielewski et al, Reference Czepielewski, Massuda, Panizzutti, Grun, Barbé-Tuana and Teixeira26,Reference Czepielewski, Massuda, Panizzutti, da Rosa, de Lucena and Macêdo148 Rao et al Reference Rao, Kota, Li, Yao, Tang and Mao142 and Galletly et al. Reference Galletly, Dhillon, Liu, Balzan, Hahn and Fenech149 described shorter telomere lengths in a male sample.
Other authors reported no differences or an increase in telomere length. In individuals with antipsychotic-naïve FEP, Shivakumar et al (studying a mixed sample of antipsychotic-naïve and antipsychotic-free individuals) described no differences; however, males displayed a non-significant reduction in telomere length compared with female and control male participants, with worse psychopathology associated with reduced telomere length. Reference Shivakumar, Kalmady, Rajasekaran, Chhabra, Anekal and Narayanaswamy83 Li et al described no differences in telomere length; Reference Li, Hu, Zong, He, Wang and Dai150 however after 8 weeks, treatment responders showed a trend towards longer telomere length compared with non-responders, and Talarico et al described longer telomere lengths. Reference Talarico, Xavier, Ota, Spindola, Maurya and Tempaku151
Çevik et al described a non-significant reduction in telomere length, whereas childhood adversities were associated with reduced telomere length. Reference Çevik, Mançe-Çalışır, Atbaşoğlu, Saka, Alptekin and Üçok152 Riley et al described no differences in telomere length, but significant gender differences arose in the relationship of early trauma with telomere length (in male patients). Reference Riley, Perrin, Vaez-Azizi, Ruby, Goetz and Dracxler153 Monroy-Jaramillo et al described no differences in telomere length; however, when participants were stratified by metabolic risk due to antipsychotics, patients receiving high-metabolic-risk treatment presented reduced telomere length. Reference Monroy-Jaramillo, Rodríguez-Agudelo, Aviña-Cervantes, Roberts, Velligan and Walss-Bass154 Wolkowitz et al reported no diagnostic differences; however, they analysed the effects of biological gender and described prematurely shortened telomere length in women with schizophrenia, with a differential gender effect in the pattern of decline pattern. Reference Wolkowitz, Jeste, Martin, Lin, Daly and Reuter155 Pisanu et al reported mixed results but found that specific pleiotropic variants may contribute to the cellular ageing processes. Reference Pisanu, Congiu, Meloni, Paribello, Patrinos and Severino156
Overall, the current literature offers moderate support for telomere shortening as a marker of accelerated ageing in schizophrenia. All four meta-analyses consistently showed significant small-to-medium size effects (ranging from −0.39 to −0.52); however, individual studies showed some inconsistencies and confounders, particularly those due to differential aspects of biological gender, influence of antipsychotic treatment, stressful environmental events and psychopathology (suicidal behaviour), which seemed to promote telomere shortening.
Epigenetics
The epigenetic landscape in schizophrenia is complex, with results heavily dependent on the methodology and tissue type used. A recent systematic review and meta-analysis of eight studies (N = 6490) by Chrusciel Reference Chrusciel, Orso, de Mattos, Fries, Kristensen and Grassi-Oliveira5 found no significant difference in biological age between patients and controls when all epigenetic clocks globally were analysed globally. However, this global null result masked significant heterogeneity. When participants were stratified by specific clock type, a small but significant ‘accelerated ageing’ effect emerged for the PhenoAge and Hannum clocks (SMD = 0.29). Unpacking this heterogeneity requires distinguishing between illness stages and the specific biological dimensions captured by different clocks.
With respect to markers related to the trajectory, evidence suggests a dynamic shift in epigenetic status over the course of the illness. In contrast to the accelerated ageing hypothesis, early-stage and at-risk cohorts often displayed an unexpected ‘epigenetic deceleration’ or youthfulness. Naïve FEP samples, Reference Talarico, Xavier, Ota, Spindola, Maurya and Tempaku151,Reference Li, Zong, Li, He, Tang and Hu157 youth at familial risk and small-sample cohorts Reference Jeremian, Malinowski, Chaudhary, Srivastava, Qian and Zai158 have shown decelerated ageing according to Horvath’s clock. Epigenetic deceleration (or ‘youthfulness’) refers to a biological clock that seems younger than chronological age owing to a relative maturational delay in epigenetic changes, rather than a reversal of DNA methylation errors.
However, this pattern seemed to reverse with illness chronicity and treatment. Longitudinal data indicated that although patients may appear ‘decelerated’ at baseline, an accelerated pattern emerged after 8–10 weeks of antipsychotic treatment. Reference Talarico, Xavier, Ota, Spindola, Maurya and Tempaku151,Reference Li, Zong, Li, He, Tang and Hu157 This shift was consistent with findings in chronic populations, in which older adults – particularly females at high genetic risk – exhibited accelerated PhenoAge and pace-of-ageing (DunedinPACE). Reference Caspi, Shireby, Mill, Moffitt, Sugden and Hannon159 Higgins-Chen et al Reference Higgins-Chen, Boks, Vinkers, Kahn and Levine160 suggested that this reversal may be driven by slowing of ‘mitotic clocks’ (linked to cell division and immunity), while ‘mortality clocks’ accelerate owing to accumulated damage. An exception to this chronic acceleration pattern was found by Okazaki et al, Reference Okazaki, Numata, Otsuka, Horai, Kinoshita and Sora161 who reported decreased epigenetic age (Hannum) in patients undergoing long-term hospital stays, although replication results were mixed.
Regarding the role of lifestyle confounders, the ‘acceleration’ observed in chronic stages seemed to be strongly driven by secondary lifestyle factors rather than intrinsic disease mechanisms. The ‘GrimAge’ clock, a strong predictor of mortality, showed accelerations of up to 5 years in schizophrenia, but this effect was largely attenuated after controlling for smoking and age-associated proteins. Reference Higgins-Chen, Boks, Vinkers, Kahn and Levine160 Teeuw et al Reference Teeuw, Ori, Brouwer, de Zwarte, Schnack and Hulshoff Pol73 found that accelerations in PhenoAge often did not survive correction for smoking, whereas Rosoff et al Reference Rosoff, Hamandi, Bell, Mavromatis, Park and Jung162 suggested that smoking, rather than a direct genetic link to longevity, explained the apparent ageing effects. Despite these confounders, sensitivity analyses still linked Hannum’s clock to mortality: Reference Kowalec, Hannon, Mansell, Burrage, Ori and Ophoff163 Muntané et al Reference Bosch, Martorell, Navarro and Vilella164 described a polygenic overlap between schizophrenia and shorter lifespan, whereas Segura et al Reference Segura, Prohens, Mezquida, Amoretti, Bioque and Ribeiro165 found accelerated patterns using DNAmTL in patients experiencing clinical relapse. Previous findings have suggested that distinct clocks capture different signals: some measure intrinsic biological ageing (often normal or decelerated in patients with schizophrenia), whereas others capture cumulative environmental stress (accelerated).
With respect to divergence in brain tissue, peripheral blood findings did not necessarily mirror brain ageing. Direct examination of post-mortem brain tissue generally failed to support the accelerated ageing hypothesis. Large analyses of blood and brain tissue by McKinney et al Reference McKinney, Lin, Ding, Lewis and Sweet166 and Wu et al Reference Wu, Ye, Wang and Zhao167 reported no differences or delayed acceleration. In direct brain analyses, Voisey et al Reference Voisey, Lawford, Morris, Wockner, Noble and Young168 and McKinney et al Reference McKinney, Lin, Ding, Lewis and Sweet169 described no significant methylation differences. Liu et al Reference Liu, Qi, Cheng, Meng, Yang and Pan170 described decelerated epigenetic ageing in the cerebellum of aged patients However, although methylation clocks remained neutral, transcriptomic studies revealed functional shifts. Arakelyan et al Reference Arakelyan, Avagyan, Kurnosov, Mkrtchyan, Mkrtchyan and Zakharyan116 found that the gene expression profiles of patients aged 50–65 and 65–80 years closely resembled those of healthy controls more than 85 years old. Other studies have identified disrupted age-related gene expression networks Reference Kim, Jo, Webster and Lee171,Reference Sabunciyan172 and histone modifications Reference Tang, Dean and Thomas173 that imply an ‘older’ functional state. Conversely, Torkamani et al Reference Torkamani, Dean, Schork and Thomas117 described age-related dysregulation in developmental gene networks in an opposite direction to that associated with ageing.
Overall, current epigenetic evidence does not support a uniform ‘accelerated ageing’ phenotype. Instead, the data point to a decoupled process, with intrinsic deceleration or delay in early development and brain tissue, overlaid by a secondary lifestyle-driven acceleration in peripheral markers that accumulates with chronicity and treatment exposure.
Discussion
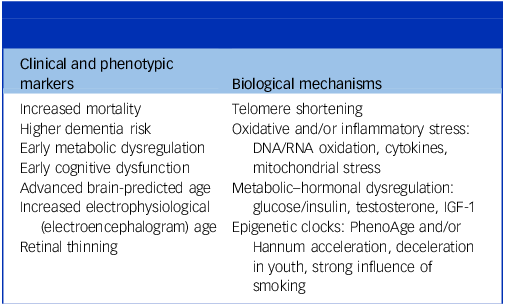
This review supports the view that schizophrenia is associated with a modest but consistent shift towards advanced biological ageing across several systems (see Table 2 for a summary of findings). The strongest evidence comes from clinical end-points or whole-organism phenotypes markers such as increased mortality, dementia risk, brain age elevation and cognitive decline. Biological mechanisms of ageing, which are commonly used as biological measures of ageing, were also disrupted, with stronger evidence for shortened telomere length and primary metabolic dysfunction compared with epigenetic changes. Although heterogenous, findings from meta-analyses and large cohorts support an early ageing shift, detectable from first episode and reinforced by immune–metabolic and neurobiological abnormalities.
Schizophrenia: multisystem advanced biological ageing disordera

IGF-1, insulin-like growth factor-1.
a. Most domains showed an advanced (level-shift) pattern; true acceleration was confined to selected brain and clinical subgroups (please note there is no intended correlation between left and right columns).
This review extends the hypothesis originally proposed by Kirkpatrick and colleagues, Reference Kirkpatrick, Messias, Harvey, Fernandez-Egea and Bowie1 which anticipated multisystem ageing signals in schizophrenia, including cardiovascular stiffness, metabolic dysregulation and cognitive profiles resembling those of older adults. Our synthesis confirms those early predictions and incorporates data from other biomarkers, including imaging-derived brain age, telomere length and epigenetic clocks that were unavailable at the time of the initial formulation. The strength of some of this evidence is undermined by the potential for confounding, especially by antipsychotic treatment with accompanying metabolic risks, and bias, including survival and protopathic bias. The evidence from studies of participants with very early psychosis suggests these issues do not fully account for the changes outlined above.
Our findings align well with evidence from other studies. Meta-analytic and large cohort data have indicated increased dementia risk Reference Stafford, Chung, Sommerlad, Kirkbride and Howard13 and excess age-related morbidity and mortality in schizophrenia; Reference Kodesh, Goldberg, Rotstein, Weinstein, Reichenberg and Sandin10 these effects persisted after covariate adjustment, consistent with an ageing shift. Importantly, the impact of anticholinergic burden remains a potential confounder in evaluations of the increased risk of dementia and cognitive impairment in schizophrenia. Recent evidence suggests that cumulative anticholinergic exposure may be responsible for a proportion of cases of early-onset cognitive decline and subsequent dementia diagnoses.
Early cardiovascular markers predicted by the original hypothesis, such as increased pulse pressure or prediabetes at illness onset, have empirical support in antipsychotic-naïve samples. Reference Fernandez-Egea, Bernardo, Heaphy, Griffith, Parellada and Esmatjes145,Reference Fernandez-Egea, Bernardo, Donner, Conget, Parellada and Justicia174 However, we found no studies of other major phenotypic or clinical makers of ageing, such as increased hair loss, hearing loss, early presbyopia, arthritis, increased susceptibility to infection or early menopause.
Neuroimaging-derived brain age was not suggested in the initial formulation of the hypothesis but was found here to be robustly and consistently elevated. The ENIGMA meta-analysis reported an approximately +3.5-year difference in brain predicted age (Cohen’s d ≈ 0.5). First-episode cohorts have already shown BAGs, often localised to the frontal and temporal association cortex, and longitudinal studies have suggested early divergence with later heterogeneity in trajectories, with obesity consistently exacerbating the brain age signal. White matter studies have shown steeper age-related declines in fractional anisotropy in patients with schizophrenia than in controls. Together, these findings support an early and distributed brain ageing phenotype, with little subsequent acceleration. Cognition also maps on to this pattern. Early psychosis cohorts often performed similarly to healthy individuals 5–10 years older, particularly with on processing speed and executive tasks; long-term cohort studies have suggested widening gaps into midlife for subsets of patients, with floor effects limiting sensitivity in severe illness. Overall, this profile is consistent with an ageing shift rather than an acceleration of decline. Other age-sensitive markers align with this, including retinal optical coherence tomography studies showing early age-linked retinal thinning and electroencephalogram-based ‘EphysAGE’.
Mechanisms of ageing, defined as the molecular and cellular regulators of ageing and often also used as biological markers of ageing, were examined in only three domains: telomere attrition; oxidative stress, inflammation and/or metabolic dysregulation (including SIRT1-linked pathways); and epigenetic methylation. Not all mechanisms of ageing seemed to be uniformly affected. Telomere meta-analyses consistently showed small-to-moderate leukocyte shortening effects with age, gender, trauma and adversity, and treatment-related heterogeneity. Epigenetic clocks showed clock-specific effects, with small PhenoAge/Hannum effect sizes, whereas other clocks showed deceleration in youth and acceleration in adult women with high genetic risk. Mortality-linked clocks were accelerated and largely driven by smoking and age-related proteomic signatures; measures of pace of ageing (DunedinPACE) were elevated. Antipsychotic-naïve first-episode cohorts already showed impaired glucose regulation, higher inflammatory cell counts and lower free androgen index values, whereas multi-omics and postmortem work indicated oxidative stress, mitochondrial dysfunction, ‘older’ lipidomes, reduced IGF signalling, and microbiome and/or vascular signatures tied to cardiovascular risk. Other canonical mechanisms (e.g. autophagy dysregulation) were not represented in the studies included. Reference Kirkpatrick and Kennedy175
Across modalities, the evidence converges towards a biological age excess of approximately 3–5 years in the brain and 5–10 years in terms of cognitive performance. These differences represent only a fraction of the observed reduction in life expectancy of 15–20 years, suggesting that accelerated ageing may constitute a specific vulnerability that interacts with lifestyle and treatment-related factors such as smoking, physical inactivity, poor diet and antipsychotic exposure. Conceptually, this framework reinforces our understanding of schizophrenia as a systemic disorder rather than purely a brain disorder, and hence a developmental rather a neurodevelopmental disorder. The evidence also provides a rationale for the incorporation of preventive and restorative interventions targeting somatic health – such as cardiometabolic risk modification, smoking cessation, weight control and enhancement of cognitive reserve – as strategies to mitigate ageing-related decline. Mechanistically, it encourages research into targeted modulation of oxidative stress and telomere maintenance pathways as potential therapeutic avenues.
It is also important to highlight the consistency of our findings with the concept of schizophrenia as a neurodevelopmental disorder, Reference Howes and Murray176 and with other lines of research. Widely replicated risk factors for schizophrenia, including stressful events during the intrauterine Reference Entringer, Epel, Kumsta, Lin, Hellhammer and Blackburn177 or childhood period, Reference Bourassa, Caspi, Brennan, Hall, Harrington and Houts178 are also risk factors for metabolic outcomes such as type 2 diabetes when they are associated with accelerated ageing and occur in the presence of telomere shortening. Studies in animal models Reference Garcia-Rizo and Bitanihirwe179 have demonstrated that perinatal stressful events prime offspring for a broad range of systemic ‘epiphenomena’ that mirror ageing features, including neurostructural abnormalities, cognitive deficits and glucose–insulin abnormalities, leading to visceral obesity and type 2 diabetes. Reference Garcia-Rizo and Bitanihirwe180 Evidence from these other lines of research suggests that in affected cases, the observed ageing phenotype in schizophrenia may represent a consequence of early life stress, acting as a common antecedent for both the psychiatric diagnosis and biological decline and conferring vulnerability to physical complications after onset of the disorder. In this context it makes sense that abnormal ageing would be apparent early in the course of schizophrenia. The multisystem ‘ageing’ observed in schizophrenia may essentially reflect the long-term trajectory of early developmental ‘scars’, Reference Çevik, Mançe-Çalışır, Atbaşoğlu, Saka, Alptekin and Üçok152,Reference Entringer, Epel, Kumsta, Lin, Hellhammer and Blackburn177–Reference Garcia-Rizo and Bitanihirwe179 which might also confer increased vulnerability leading to progressive degeneration, both physical and mental.
A further reflection regarding terminology
As several domains show early level differences with parallel subsequent slopes of decline rather than a consistently steeper decline, ‘advanced ageing’ or ‘premature ageing’ may be a more accurate and falsifiable label than ‘accelerated ageing’ in many contexts. In this sense, we distinguish between advanced ageing, characterised by an early biological ‘level-shift’ followed by a normal rate of decline – a pattern we found to be most consistent with global neuroimaging and cognitive data – and accelerated ageing, which involves a continuously steeper slope of decline and a widening gap over time. True acceleration seems to be less common and may be confined to specific systems, such as oxidative stress accumulation or the rapid brain volume loss observed in the first 5 years of illness.
Strengths and limitations
This review has both strengths and limitations. Its strengths lie in the use of a preregistered protocol and a priori definitions, as well as the distinction drawn between markers and mechanisms of ageing. Particular emphasis was placed on minimally treated and early-psychosis samples, and longitudinal data were prioritised over cross-sectional findings. Limitations are also evident. By restricting inclusion to studies that explicitly cited the accelerated ageing hypothesis, we inevitably excluded high-quality meta-analyses and cohorts that could have been reframed within this paradigm, such as studies of cardiometabolic comorbidity Reference Pillinger, D’Ambrosio, McCutcheon and Howes181 or decline in dendritic density, Reference Howes, Cummings, Chapman and Shatalina182 meta-analysis of early metabolic disturbances, Reference Greenhalgh, Gonzalez-Blanco, Garcia-Rizo, Fernandez-Egea, Miller and Arroyo183 studies of inflammatory markers in early psychosis Reference Miller, Buckley, Seabolt, Mellor and Kirkpatrick184 or our work that led to development of the accelerated ageing hypothesis. Reference Fernandez-Egea, Bernardo, Donner, Conget, Parellada and Justicia174 In addition, the validity of biological ageing proxies (e.g. BrainAGE and epigenetic clocks) in severe mental illness is uncertain: algorithms trained on healthy cohorts may capture illness-, treatment- and lifestyle-related effects, such that these measures may reflect cumulative biological burden rather than ageing per se. It is also relevant that small mechanistic studies are especially vulnerable to reporting bias. Interpretation was further constrained by methodological heterogeneity across epigenetic clocks, telomere assays and brain-age algorithms, which complicated inference about rates versus levels of ageing. Although the exclusion of grey literature (such as conference abstracts, preprints or unpublished reports) may be seen as a limitation, this was a deliberate choice to ensure the methodological rigour of the included data.
Future research directions
Across biological, cognitive and epidemiological domains, schizophrenia is associated with a measurable shift toward older biological status, detectable from illness onset and only partly explained by treatment or lifestyle factors. The most reproducible signals – elevated brain-predicted age, shorter telomeres and immune–metabolic dysregulation – suggest a convergent pathophysiology linking oxidative stress, inflammation and neurodevelopmental vulnerability. While the degree of acceleration is modest compared with the 15–20-year reduction in life expectancy, these findings provide a biological framework for early physical health interventions and mechanistic studies. Conceptualising schizophrenia as a systemic disorder of advanced ageing integrates brain and body pathology within a unified model and raises the possibility of a shift from purely neuropsychiatric to whole-body treatment and prevention. Reference Kirkpatrick and Kennedy175
Supplementary material
The supplementary material is available online at https://doi.org/10.1192/bjp.2026.10606
Author contributions
E.F.-E, C.G.-R. and B.K. designed the study. E.F.-E. completed the search. E.F.-E. and C.G.-R. screened the documents and reached the initial decisions. All documents included were reviewed by the three authors. Each author was responsible for writing a section of the results. The initial draft of the introduction and discussion was written by E.F.-E. All authors contributed to refining, amending and enhancing the text.
Funding
E.F.-E. is supported by the 2022 Medical Research Council/National Institute for Health and Care Research (NIHR) Clinical Academic Research Parternships award (MR/W029987/1), and all research in the Department of Psychiatry at the University of Cambridge is supported by the NIHR Cambridge Biomedical Research Centre (NIHR203312) and the NIHR Applied Research Collaboration East of England. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. C.G.-R. is supported by the Maria i Núria Cunillera legacy (FCRB_CU1_2024) and projects ‘PI20/00661; PI24/00196’, funded by the Instituto de Salud Carlos III and co-funded by the European Union.
Declaration of interest
E.F.-E. has received consultancy honoraria from Boehringer-Ingelheim (2022), Atheneum (2022) and Rovi (2022–2025); speaker fees from Adamed (2022–2025), Otsuka (2023) and Viatris (2024); and training and editorial honoraria from the Spanish Society of Psychiatry and Mental Health (2023–2025). E.F.-E. is a member of the British Journal of Psychiatry editorial board but did not participate in the review process for this manuscript for the journal. C.G.-R. has served as a consultant, advisor or speaker for the following entities: Adamed, Angelini, Casen-Recordati, Janssen-Cilag, Lunbeck and Newron. B.K. has received (a) licensing royalties from WCG Clinical for use of the Brief Negative Symptom Scale by for-profit groups (these fees are donated to the Brain and Behavior Research Foundation) and an honorarium for presenting on negative symptoms to an online audience; (b) an honorarium and travel expenses from Rovi Pharmaceutical Laboratories for participating in an educational programme; and (c) consulting fees and travel expenses from Minerva Neurosciences. He is also Chief Executive Officer of and has stock in Quantic Innovations, which provides consulting on digital phenotyping to the pharmaceutical industry.



 Open access
Open access
eLetters
No eLetters have been published for this article.