In times past, an inquisitive physician-scientist must have pondered these questions: How do we unknowingly breathe? What brain structures control our breathing? Why is breathing so perfectly rhythmic? Is there a lung–brain communication, and if so, how? But an even more fundamental question must have been: How much brain injury can one sustain before breathing stops? The scientist most deserving of recognition for this line of inquiry must be Julien (Jean César) Legallois. His discovery, detailed in Expériences sur le Principe de la Vie, was a result of vivisection experiments in the early 1880s, actually undertaken to discover the structures needed to sustain breathing and thus life[Reference Legallois1]. It took two centuries (more or less) to answer the aforementioned questions and gradually add small pieces to a large (still incomplete) puzzle. We will notice that key pieces are still missing.
The respiratory center in the brainstem and its three classes of neural mechanisms—the central and peripheral chemosensitivity, central respiratory drive, and neuronal feedback from the muscles of respiration—were identified and characterized in the late 1800s and early 1900s. Similarly, the function of the respiratory muscles and its neural connection with cranial nerves (CN) became better known. For example, the muscles moving the alae nasi to decrease airway resistance (CN VII), the muscles opening the glottis (CN X), the major inspiration diaphragm muscle (cervical cord by the third to fifth cervical nerves merging into the phrenic nerve), and intercostal muscles from the dorsal spinal cord needed as backup during dyspnea were all identified.
At its most fundamental level, breathing acquires oxygen – our brains need it more than any other organ – and removes carbon dioxide. Many anatomists and physiologists worked on multiple aspects of the respiratory system, pulmonary function, and circulation under diverse circumstances.
This introductory chapter recounts the history of the neurology of breathing and, thus, the discovery of the respiratory center and the respiratory mechanics. Sections of the chapter review experimental and clinical discoveries of the central and the peripheral nervous system involved with breathing while acknowledging the interplay between the parts.
Understanding breathing requires knowledge of anatomy, physiology, and chemistry. Earlier experimenters needed sound knowledge of the work of others and expected to be checked by criticism of their results. Once these experiments were accepted, with results confirmed and known, investigators acquired increased understanding of how the nervous system controls our breathing and how disease can compromise, interrupt, and stop our breathing cycle. However, clinical observations of the neurology of breathing were not recognized concurrently with the early experimental studies on the course and regulation of breathing that emerged from nineteenth-century physiological laboratories. In fact, clinical acumen came much later, with the poliomyelitis epidemics and development of specialized ICUs to care for severe acute brain injury. Several perceptive neurologists and neurosurgeons deserve credit.
Central Segment of Breathing
The earliest experiments concentrated on the brain hemispheres and later more specifically on the anatomy of the brainstem after it was recognized as a major vital structure[Reference Wijdicks2]. Parallel to finding the anatomic centers of breathing were advances in understanding the role of oxygen and carbon dioxide in the seventeenth and eighteenth centuries, and several physiologists (such as Lavoisier and Haldane) stand out because they directed attention to the central nervous system and away from the peripheral nervous system and in particular the vagus nerve[Reference Proctor3].
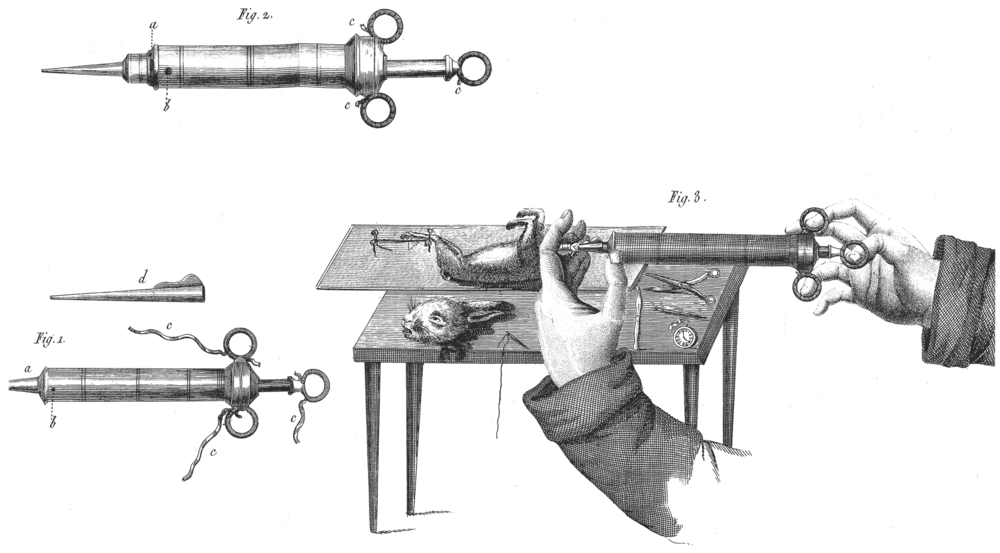
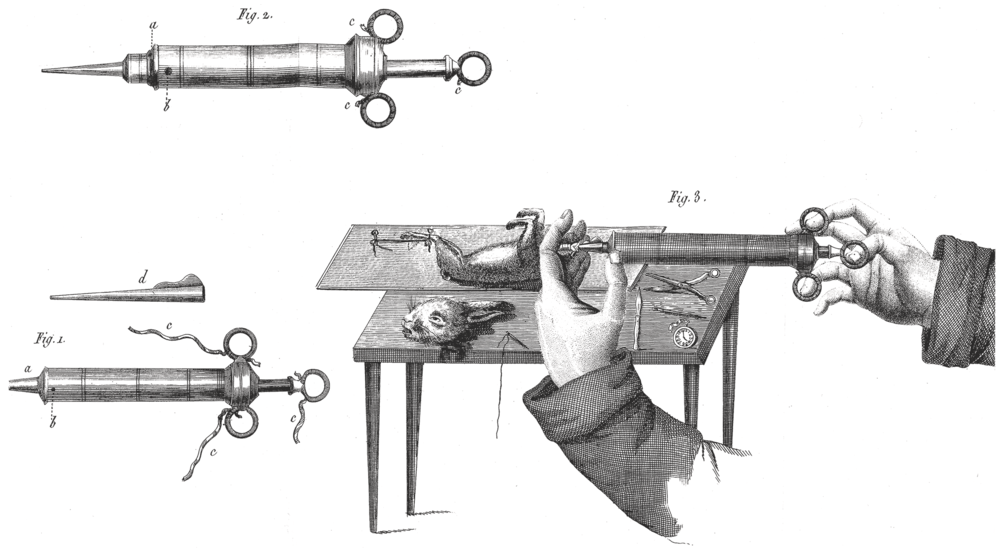
Legallois designed crude experiments to determine the minimum requirements to maintain life in a diminishing dissected portion of a rabbit. He sliced the brain caudally to the spinal cord and found that respiration stopped when he reached the medulla oblongata at the level of the eighth CN nucleus. He did not specifically seek to illustrate how certain parts of the brain interact to maintain breathing (Figure 1.1).
Legallois experiments.

Legallois experiments in decapitated rabbits.
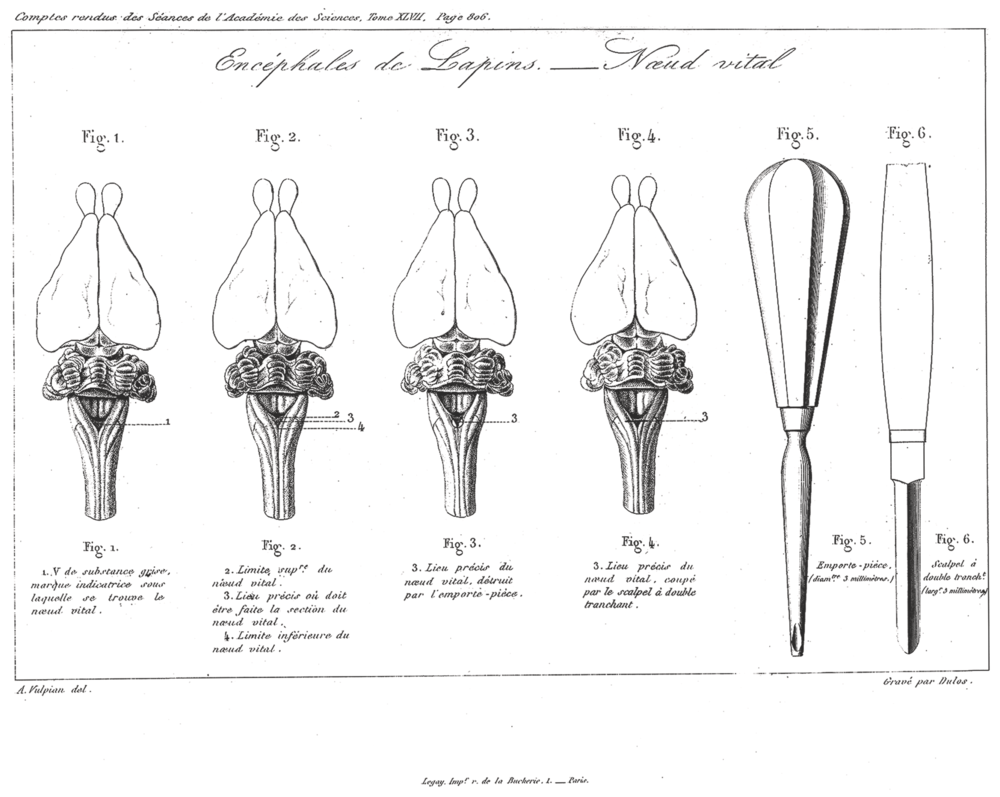
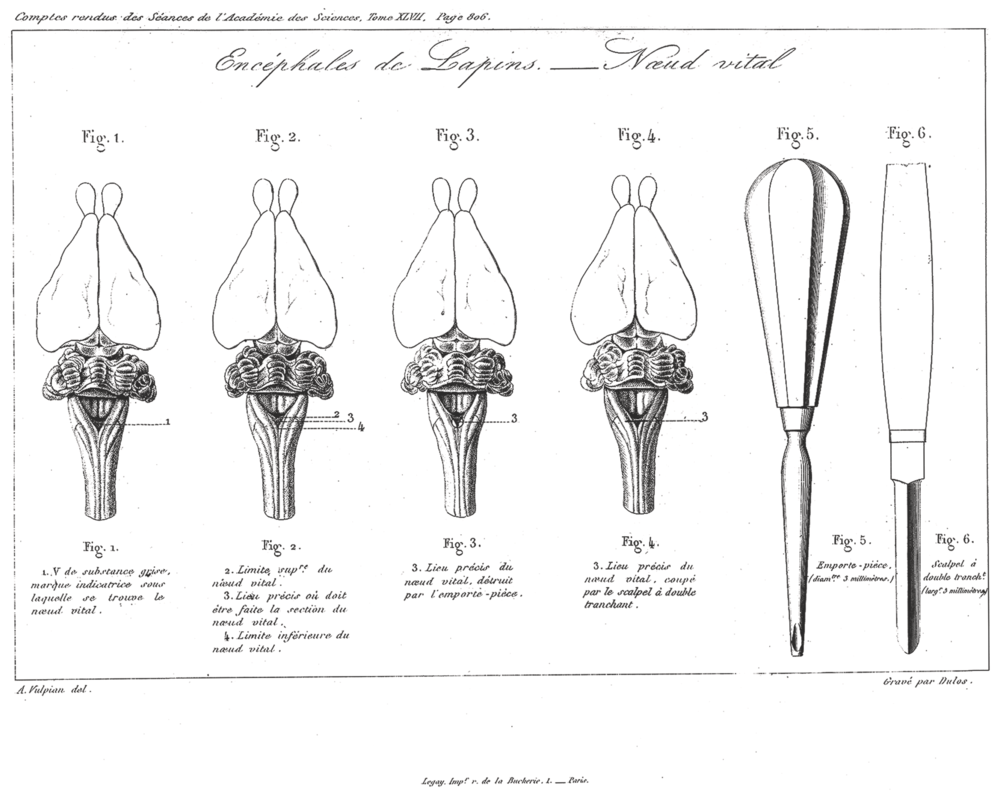
Pierre Flourens further localized possible “centers” and also performed surgical ablation experiments[Reference Flourens4–Reference Flourens6]. These experiments involved severing the spinal cord at several levels. Dividing the cord at C7 did not interrupt the respiratory drive but only diaphragmatic contraction with no motion of the chest wall. Higher up at C3, the respiratory motions stopped, but there were ineffective rescue muscles such as the sternocleidomastoid and trapezius muscles. He discovered that the spinal centers of the respiratory nerves were subsidiary to some center at the lower end of the medulla oblongata because a higher cut (above the acoustic striae of the cochlear nucleus) did not affect respiration. Flourens identified a pinhead area, which he called vital knot or node (noeud vital), which was reed-like tissue measuring 2.5 mm in diameter in the medulla oblongata occupying a third of the floor of the fourth ventricle at the caudal end, continuous with the medulla oblongata (Figure 1.2). In the mid-nineteenth century, he arrived at this discovery by making high cervical transections in rabbits and discovering the existence of respiratory movements in the rabbit’s muzzle without chest movements.
Flourens experiments “finding” the noeud vital using scalpel and pastry cutter.

A number of physiologists, including Charles-Édouard Brown-Séquard, did not accept Flourens’ conclusions[Reference Aminoff7]. Brown–Sequard claimed to have seen rhythmic respiratory movements in decapitated animals, postulating a respiratory center localized in the spinal cord, but other contemporary physiologists felt those movements were simply agonal and asphyxial. However, Brown-Séquard was supported by a number of other experimenters such as B. W. Richardson, who claimed to find a similar node in decapitated crocodiles, and most notably, by the experiments of Oskar Langendorff, who also strongly believed respiratory centers were present in the spinal cord[Reference Langendorff8, Reference Langendorff9]. The controversies on the role of the spine in regulating respiration and how it led to errors of interpretation in a number of experiments by many physiologists were summarized in detail by Porter[Reference Porter10].
Max Marckwald’s work in The Movements of Respiration introduced new theories about the action of respiratory centers, the rhythm of respiration caused by the vagi, and the action of the glossopharyngeal nerves and others on the respiratory center[Reference Marckwald11]. Separation of the medullary center slowed respirations to two to four per minute, with inspiration much longer than expiration, and thus nothing else remained other than respiratory muscle spasms. Mid-pontine decerebration resulted in slow – all or nothing – rhythm and was suppressed by cortical, upper-brainstem, or vagus input. Decerebration at the pontomedullary junction produced another rhythm with ataxic, irregular breathing in the animal medulla oblongata.
Much interest focused on how increased intracranial pressure-controlled ventilation (and, thus, patterns of breathing and gas exchange). As early as Cushing’s experiments in 1902, both increased and decreased respiratory drive were noted with increased intracranial pressure (ICP). Persistent hyperventilation at high ICP was reported in some – but not all – animal experiments. Ventilation was often depressed with diminishing perfusion pressure, although this could have been a secondary effect of displacement, rather than ischemia, of the brainstem[Reference Cushing12].
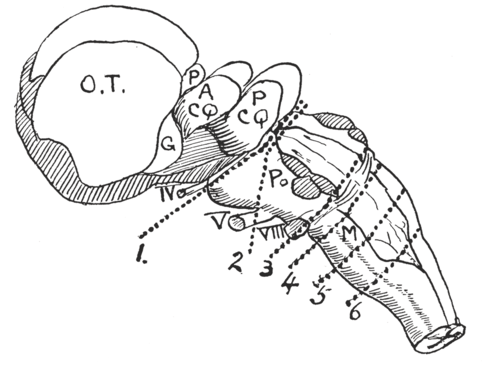
Further characterization of the respiratory centers had to wait until the 1920s, when Lumsden carried out transection experiments (Figure 1.3). Lumsden concluded there were pneumotaxic (ventral pons), apneustic (dorsal pons), and expiratory and gasping (medulla oblongata) areas. Lumsden’s main technique was more selective damage to the brainstem, and he added a stimulant such as hypoxemia or hypercapnia. Eupneic breathing required intact pons and medulla oblongata. Transecting caudally to the rostral pons produced apnea with a sustained inspiratory hold (apneusis). Transection at the pontine medullary junction caused gasping rather than apnea. Transections to the obex stopped all chest and abdomen movements. Multiple transection and stimulation experiments located the rough outlines of a respiratory center underlying the caudal third of the fourth ventricle floor. Further morphologic experiments identified inspiratory and expiratory centers and later the pneumotaxic center. Lumsden’s sectional experiments demonstrated a prolonged, inspiratory type of breathing produced by sectioning dorsal to the posterior quadrigeminal bodies. The “convulsive and incoordinate” apnea in the area he called “the apneustic center” was distinct from the so-called “gasping center” in the lower part of the spinal bulb. Lumsden observed that in asphyxia the nervous centers become afunctional caudally – first pneumotaxic center (apnea) followed by gasping as the last movement[Reference Lumsden13, Reference Lumsden14].
Brainstem centers.
Once the brainstem was isolated, more animal experiments followed. The most celebrated of these were by Gesell[Reference Gesell, Bricker and Magee15, Reference Gesell16] among others, who were able to make microelectric recordings and found a large number of active neurons in the dorsal medulla but very few “respiratory cells” in the suprapontine structures. He was unable to separate inspiratory or expiratory centers. “In many experiments, we were unable to find any respiratory potentials whatever, even with the most searching efforts. Such scarcity of potentials seemed to indicate that relatively few cells are needed in the medulla to activate enough ventral horn cells to carry on eupneic breathing”[Reference Gesell, Bricker and Magee15].
Later studies further differentiated the neuronal buildup of these centers – how individual cell groups could generate a “pacemaker” type action and how neurons could inhibit expiration and expiration. Moreover, Heymans found chemoreceptors in carotid and aortic bodies in 1927 and 1930 with afferent fibers in vagus and glossopharyngeal nerves followed by the identification of ventrolateral chemoreceptors of the medulla oblongata[Reference Heymans and Bouckaert17, Reference Heymans and Heymans18].
Most notable was the discovery that arterial PCO2 could stimulate the carotid chemoreceptor through acidosis. However, a major advance came with Leusen’s experiments[Reference Leusen19, Reference Leusen20]; he found that PCO2 and pH of the cerebrospinal fluid (CS)F stimulate drive by denervating the peripheral chemoreceptors. While the pH of CSF does not exactly correspond with arterial pH as a result of the blood–brain barrier, increasing PCO2 results in CO2 diffusion, quickly altering CSF pH and making ventilatory drive responsive to ion change. A threshold for peripheral ventilatory effect of PCO2 is between 30 and 40 mm Hg and linear in its effect. (The central ventilator effect for arterial PCO2 was similar in range.) The retro trapezius nucleus consists of a cluster of chemoreceptor neurons that detect changes in CSF pH. These neurons adjust breathing rate, inspiratory and expiratory muscle activity, and resistance of the upper airway.
Research in the mid-twentieth century (and onwards to the next millennium) discovered a number of new respiratory neuronal groups in the pons and medulla oblongata that were characterized in the regulation of breathing. These included the Kölliker–Fuse nucleus, the parabrachial nuclei, the retrotrapezoid nucleus, the Bötzinger and pre-Bötzinger nuclei, the rostral and caudal ventral ventilatory group, and the nucleus ambiguus with the nucleus retroambiguus and nucleus tractus solitarius[Reference Guyenet and Bayliss21–Reference Chen and Guyenet23]. Neuroscientists actively continue trying to understand how these neuronal groups with their connections precisely interact., However, it remains unclear how further elucidation of their function could actually assist clinicians treating patient with acute brain injury.
This book has separate chapters on the pathophysiology and clinical management of primary breathing disorders in which the neural control of breathing is impaired including congenital central hypoventilation syndrome and central and obstructive sleep apnea. Neurologic disorders that commonly affect respiration include epilepsy, demyelinating disorders, stroke, and a number of well-defined neurodegenerative diseases, and readers will find comprehensive discussion on the mechanisms in each of these disorders and how to manage them clinically.
Peripheral Segment of Breathing
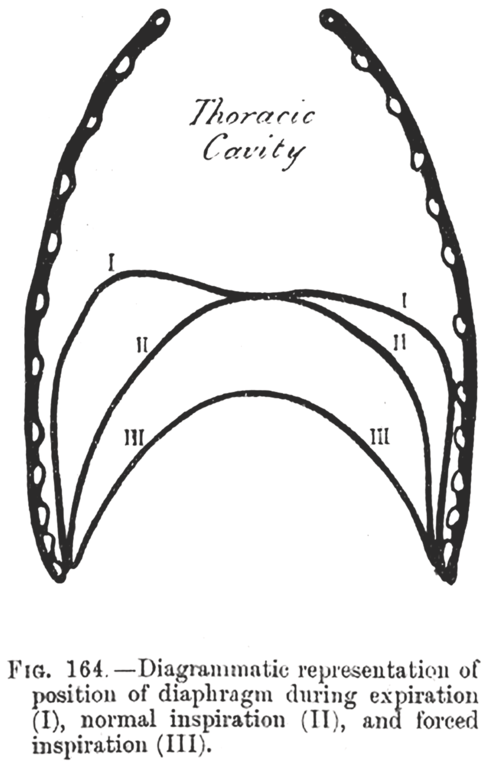
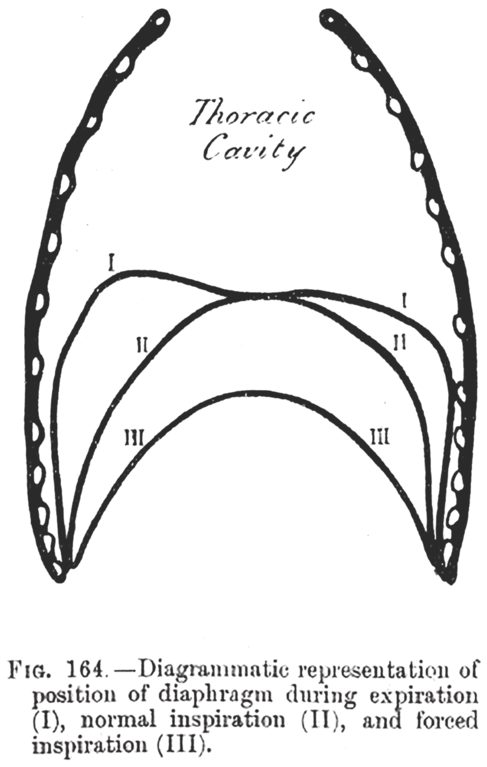
Traditionally, “peripheral” implies the trajectory of the phrenic nucleus to the muscle fibers of the diaphragm. The history of lung mechanics started before there was even a basic idea of central nervous system regulation. For example, Galen (Claudius Galenus ad 131–201) in On the Natural Faculties[Reference Galen and Brock24] was unclear and conflicted on the purpose of breathing (considered by some to cool inborn heat), but he identified the diaphragm as a major muscle not only separating the chest from the abdomen but also as a muscle of respiration. Since the early 1800s, biologists recognized that the cavity of the thorax enlarges in all directions during inspiration. The lungs expand to take up the created space. When the muscles relax, the gravitational force and elastic reaction of the thoracic wall and pulmonary tissue brings the thorax back to its original size. All mammals breathe with this mechanism; inspiration requires muscle contraction, and expiration is largely passive.
The great anatomist, physiologist, and illustrator Leonardo da Vinci was greatly interested in lung mechanics and lung inflation, and his drawings showed some preliminary concept of nervous control and the biomechanics of muscles[Reference Zammattio, Marinoni and Brizion25]. These early observations were not followed by any new research for centuries. In the late 1800s, the physiologist Henry Newell Martin refocused on the muscles of respiration; he attempted to elucidate the additional role of the internal intercostal muscles and their function as accessories when the diaphragm fails and during extreme dyspnea. “The diaphragm, when the apnea passed off, made a few contractions without any activity of the intercostal muscle: but this latter soon began to contract in regular alternation with the diaphragm and before the occurrence of expiratory convulsions; in fact with the commencement of dyspnea”[Reference Martin and Hartwell26]. These muscles do have an inspiratory function but a secondary one, and over time, the same was found for the sternocleidomastoid muscles, which become active during labored breathing. Shafer’s Textbook of Physiology in 1900 clearly identified them without specific attribution but also mentioned abdominal muscles for forced expiration. These basic biomechanics have remained the foundation of our understanding of respiratory mechanics (Figure 1.4). Later developments included the identification of the phrenic nerve motor neurons in the midcervical (C3–C5) spinal cord, “a straight, very discrete column of cells exactly paralleling the ventral longitudinal fissure”[Reference Hollinshead and Keswani27]. The phrenic nerve is a mixed sensory-motor nerve, and only recently, sensory-afferent neurons have been found to project to the cortex, explaining the perception of air hunger by patients[Reference Davenport and Vovk28]. However, the focus rapidly shifted back to characterization of the central respiratory drive, and interest in the workings of respiratory muscles waned. Further understanding of neuromuscular respiratory failure came with better understanding of the pathophysiology in acute neuromuscular disease, and clinicians led the way in explaining how diaphragm function and the upper airway could become affected. The rapid admission of a large number of dyspneic patients during the poliomyelitis epidemics put neuromuscular respiratory failure front and center.
A History of Clinical Observations
Clinicians who took care of acute neurologic disease did observe a vast number of changes in respiration. Excluding yawning, hiccups, and respiratory tics such as puffing, hissing, and snorting, attention nearly always focused on change in respiratory rates and rhythms. Already in the 1800s, studies linked brain injury to periodic abnormalities of its rhythm. Cheyne and later Stokes described the ascending and descending waxing-and-waning pattern so well known to clinicians[Reference Cheyne29, Reference Cheyne30].
The respiration is at first slow and heaving, then irregular and sometimes convulsive and lastly, interrupted. The patient from relaxation of the palate, snores loudly during inspiration, and sometimes during expiration the upper lip, from relaxation or palsy is loudly blown up from the teeth, as we often see it upon great exhaustion, as in the subsiding of an epileptic fit. Sometimes the breathing is soft. Immediately before death the respiration is irregular and is performed perhaps not more than three of four times in the minute. The irritability of the heart survives the respiration sting with my finger over the artery of a person who died of apoplexy. I distinctly felt the pulse beat after the last expiration. Interrupted respiration is justly considered as the most dangerous symptom. (Figure 1.5)
The neurologic diseases that could lead to Cheyne–Stokes pattern of breathing were meningitis, encephalitis, cerebral hemorrhage, cerebral infarcts, cerebellar hemorrhage, and ruptured intracranial aneurysm but without emergence of a specific lesion, and it could be seen in both diffuse cortical and brainstem injury. Each of these disorders led to a markedly decreased level of consciousness, which led to Cheyne–Stokes. Many early clinicians interpreted this breathing abnormality as a signal of deterioration in clinical condition, an observation that is often still true to this day.
Title page.

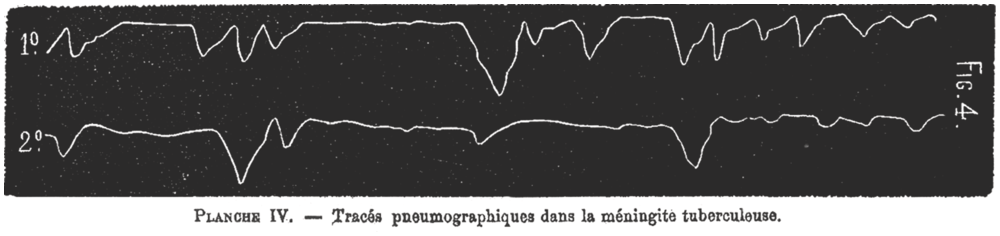
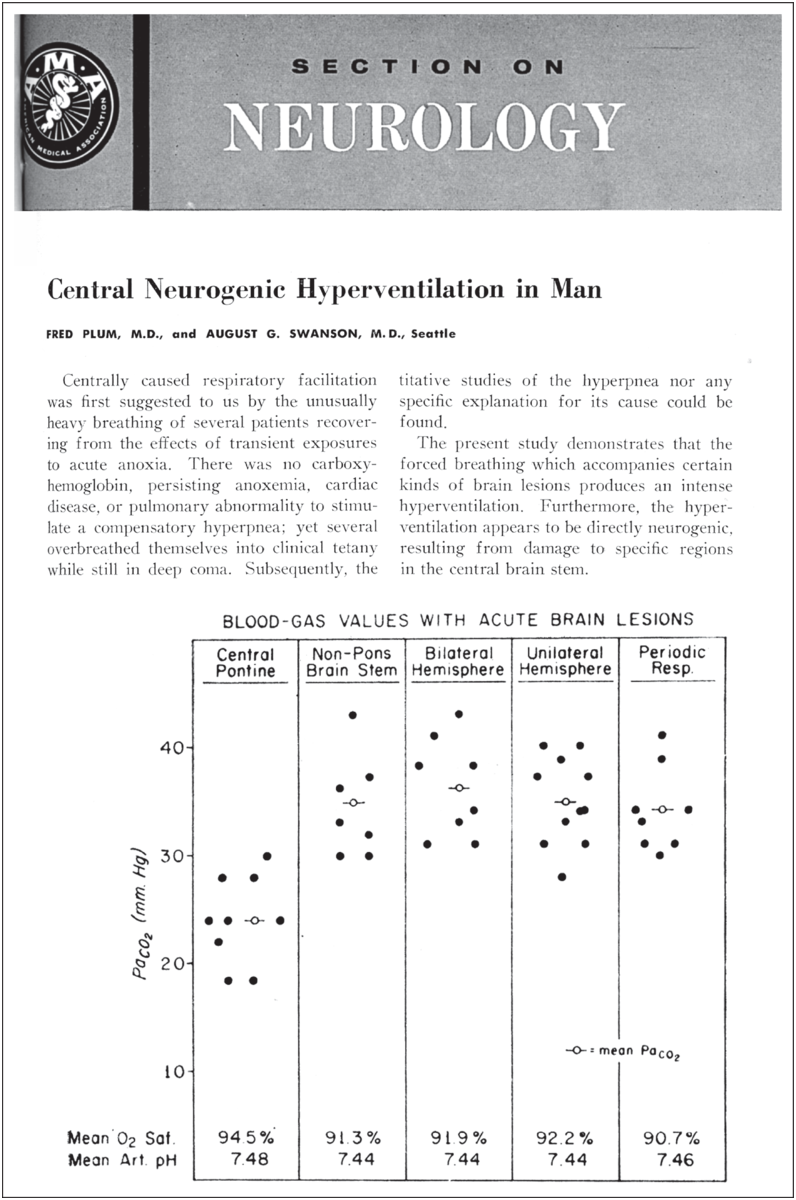
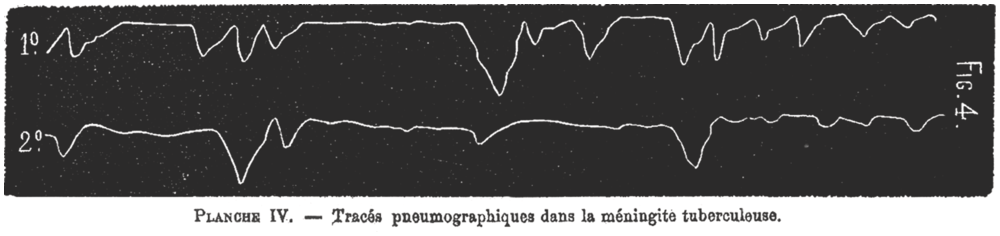
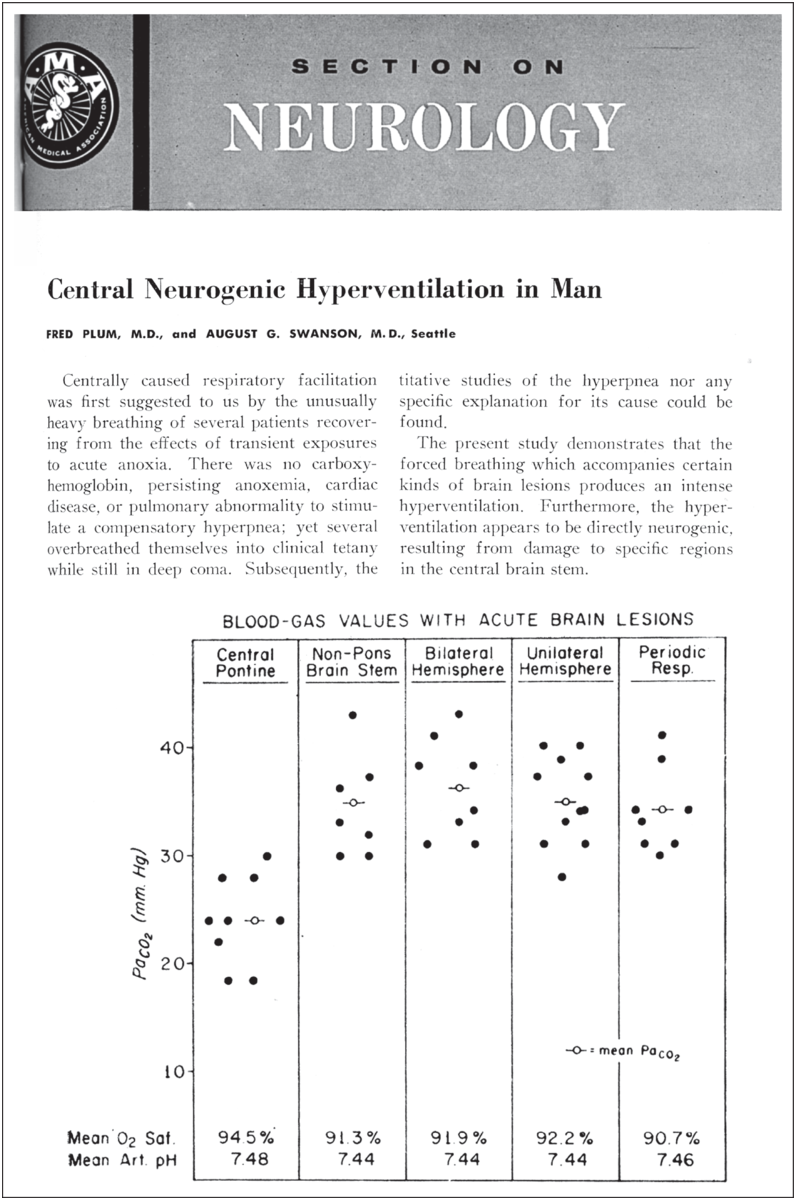
A related breathing abnormality was Biot’s breathing. The breathing pattern Biot described is irregular and rapid, with 10- to 30-second rhythmical pauses but sometimes with alternating periods of apnea and tachypnea. This breathing pattern has none of the crescendo-decrescendo cycles of Cheyne–Stokes breathing and is completely irregular with varying periods of apnea. Therefore, it was sometimes called ataxic breathing (Figure 1.6)[Reference Biot31, Reference Wijdicks32]. Biot connected this rhythme meningitique to tuberculosis but without claiming a possible connection with this (at that time) relatively common infection. In the German literature, the eponym appeared first in 1904 in Hofbauer’s book Semiologie und Differentialdiagnose der verschiedenen Arten von Kurzatmigkeit aufgrund der Atemkurve[Reference Hofbauer33]. In the English literature, it appeared in 1911 in the title of a review article by Connor who observed seven patients, six of whom had meningitis[Reference Connor34]. But perhaps the second most-known dysregulated respiratory rhythm identified as a neurogenic respiratory pattern was neurogenic hyperventilation. There was reluctance to attribute hyperventilation to a brain lesion rather than to a compensatory response in acute illness. Rapid hyperventilation was mostly seen as Kussmaul’s breathing in patients with a diabetic coma. Alveolar hyperventilation was typically clinically seen as a response to hypoxia or metabolic acidosis or was associated with a specific pulmonary disorder. (Hyperventilation with respiratory alkalosis was also incidentally noted in hepatic encephalopathy and patients in the last stages of deep coma.) Hyperventilation associated with a brain lesion was first carefully described by Plum and Swanson. The study compared nine patients with acute lesions in the pons, mostly caused by acute embolus to the basilar artery, to patients with brain lesions elsewhere and who lacked hyperventilation (Figure 1.7)[Reference Plum and Swanson35]. The authors hypothesized that structures in the medial pontine reticular formation were inhibitory to respiration, and thus, central neurogenic hyperventilation is the result of an uninhibited stimulation of the medullary respiratory centers by the lateral pontine reticular formation. We now know central neurogenic hyperventilation is indeed more common in pontine lesions (and not infrequently associated with primary brain tumor) but not exclusively so. It can be seen as part of sustained sympathetic hyperactivity in traumatic brain injury and in any severe global acute brain injury.
Biot’s breathing.
Finally, an extremely rare neurogenic breathing pattern was named after the original German fable “the curse of Ondine” by Giraudoux[Reference Severinghaus and Mitchell36]. (The curse was a punishment for unfaithfulness when Ondine took away her husband’s automatic functions. It included loss of all automatic functions – seeing, hearing, moving, and breathing.) The syndrome, described in 1962, belongs to the primary alveolar hypoventilation syndromes. Next to a genetic defect, acquired forms of Ondine’s curse have always involved lesions in the respiratory centers of the medulla oblongata. Later, isolated cases of central hypoventilation were described with acute stroke, tumors, infections, and multiple sclerosis, but most authors struggled to link it with a specific condition[Reference Sugar37].
When reading these original descriptions, there is no evading the point that circumstances have changed greatly. In our current world, clinicians in ICUs of major hospitals are less concerned with breathing patterns because patients are intubated, sedated, and mechanically ventilated, and each of these patterns may disappear once a patient is liberated from the ventilator. However, breathing patterns are important if only to show the presence of brain disease. If observed and particularly when sustained, MRI studies may be needed. Moreover, often these breathing abnormalities are seen in more or less stable patients, which allow detailed observations. In the more dramatic presentations, clinicians past and present recognize that respiratory difficulty in neurologic disease as an agonal phenomenon. In the past, of course, it was irreversible because artificial ventilation could not be provided. Some recognized (neurogenic) pulmonary edema when coma deepened, and the pupils became fixed. There was tachypnea with bloody sputum and tachycardia until death, which was explained by a major sympathetic storm causing massive vasodilation in the pulmonary vascular bed. Mouth-to-mouth resuscitation came into practice over the course of nineteenth-century resuscitation. Some might have suggested using bellows for ventilation, but this was potentially harmful because the pressure in the inflated lung could blow up the bronchiole and cause a fatal tension pneumothorax. In comatose patients with a major “apoplexy,” breathing stopped and was considered part of the dying process – with no need or obligation to intervene.
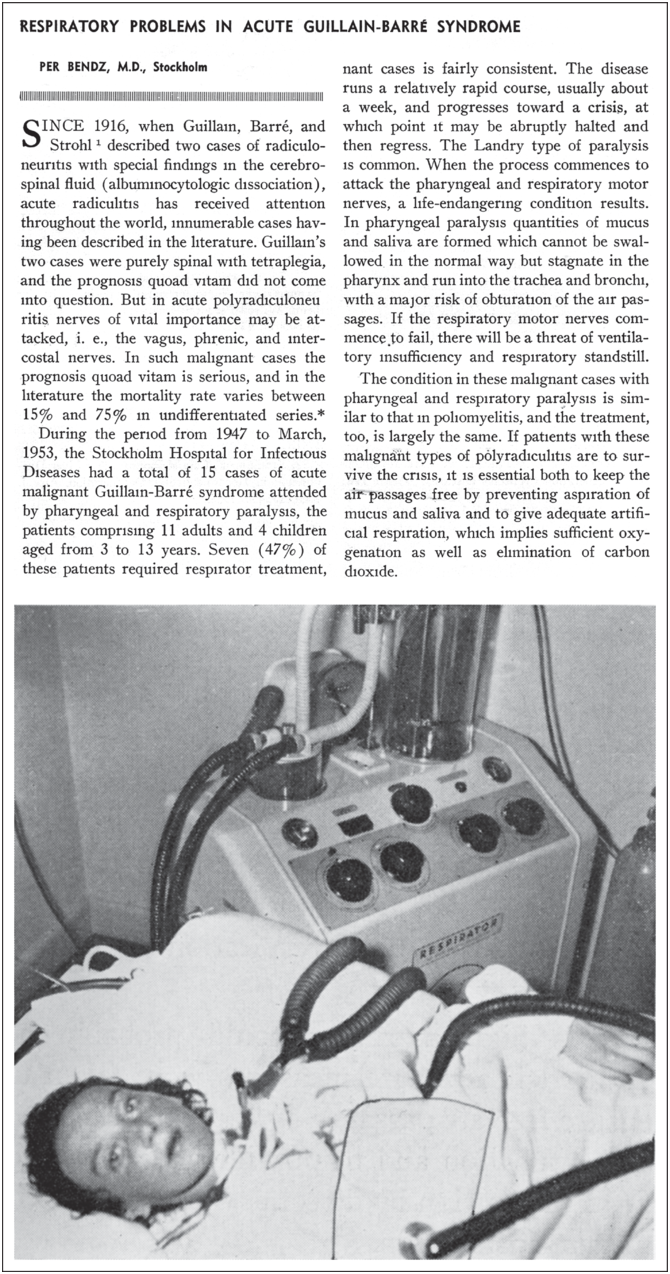
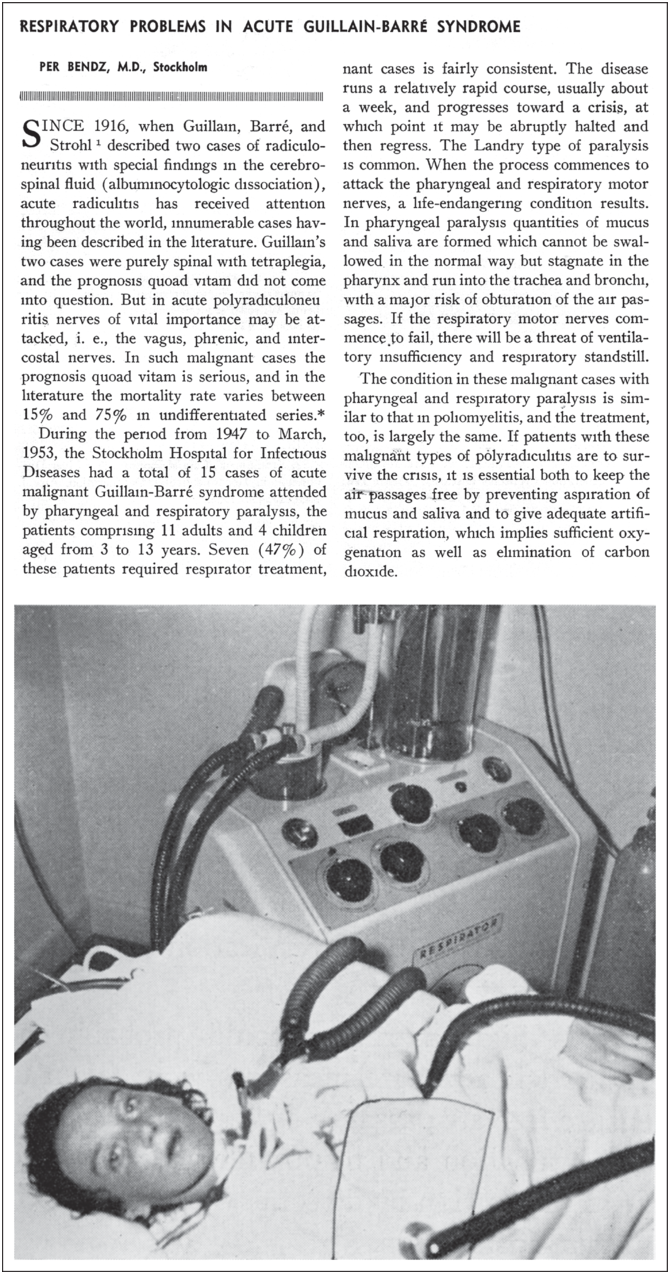
In the premechanical ventilation era, it was far more difficult for clinicians to do anything other than to observe certain acute neuromuscular disorders that induced dying from cessation of respiration. Patients unable to move air in and out despite the will to do so were under a great amount of distress but could not be helped. One of the first observations came from the disorder now known as Guillain–Barré syndrome (GBS). In 1859, Landry reported a 43-year-old man with fatal progressive weakness starting in all four limbs and difficulty walking, followed by worsening weakness involving the diaphragm, facial, laryngeal, and jaw muscles[Reference Landry38]. Gradually, more severe cases of GBS were reported, but until the 1950s, these patients died from respiratory arrest. A case record from Massachusetts General Hospital in 1951 described a 13-year-old girl with GBS with “tired” breathing and “pharyngeal” mucus,” requiring both suction and bronchoscopy and complicated by fatal aspiration pneumonia on the sixth hospital day. In this report, a “respirator” was used, most probably, a tank ventilator[39]. Bendz’s important article on respiratory care in GBS begins with three patients placed in a cuirass who “died with gurgling mucus in the pharynx”[Reference Bendz40]. A fourth case, that of a 28-year-old woman, rapidly progressing, again with a combination of dysphagia and respiratory distress leading to cyanosis and absent diaphragmatic breathing, was salvaged because a tracheostomy was performed promptly; the patient was connected to an Engström respirator and successfully weaned. This patient might have been one of the first published cases of successful respiratory care and positive-pressure mechanical ventilation in severe GBS (Figure 1.8).
Mechanical ventilation with tracheostomy in Guillain–Barré syndrome.
Similarly, deaths were portended in myasthenia gravis with choking, fluid regurgitation through the nose, diminished palatal reflex, and a lowered tone of voice suggesting weakness of the vocal cord adductors. Additionally, patients were unable to thrust out the cheek and avoid protrusion of the tongue. When chest movements were measured circumferentially, the difference between inspiration and expiration did not exceed a quarter of an inch. Dyspnea with exertion was common as well as “unaccountable attacks of breathlessness, during which the patient is in danger”[Reference Viets41]. Oral sputum accumulation accompanied these dyspneic attacks with patients unable to swallow or to spit it out. (“The tongue appears to sink back into the mouth.”) Pulling back the tongue markedly improved the symptoms. The fatal cases were a result of choking, “dyspneic attacks,” or “dyspneic attacks with pneumonia.” It was concluded that “involvement of respiratory muscles with consequent attacks of dyspnea was a symptom of gravest significance,” and the authors warned that death could occur during one of these attacks. Later, in a notable article, Rowland reported 39 fatal cases that he observed between 1930 and 1955. Presciently, he urged the physician to watch for premonitory attacks of respiratory distress, use sedatives cautiously, initiate early mechanical ventilation, and control for infection[Reference Viets41–Reference Viets44]
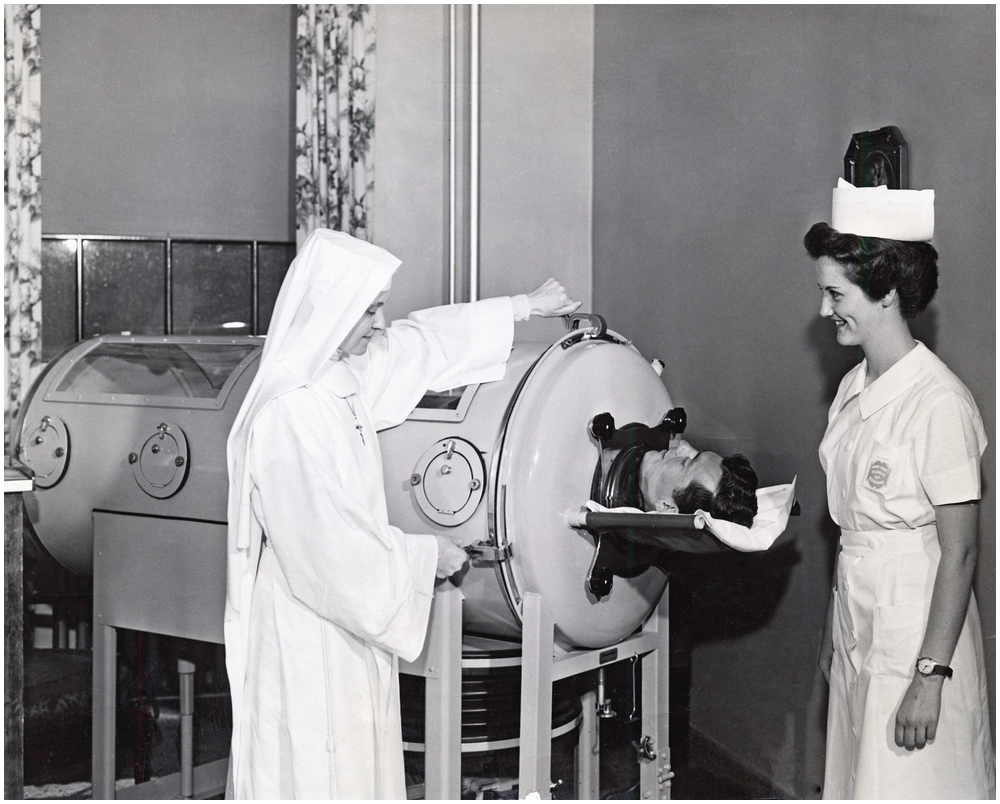
Engraved into our collective memory are the poliomyelitis epidemics that changed the healthcare landscape. The most severe summer outbreaks of poliomyelitis occurred in the United States and Europe in the 1950s. There were great challenges in managing large numbers of patients with poliomyelitis who presented in a brief time period and overwhelmed resources. Support of patients was initially rudimentary with little specific treatment. When the brainstem became involved, patients had difficulty clearing secretions from oropharyngeal weakness, and respiration became compromised. Many of these patients had back stiffness and severe pain from hypertonicity worsening respiratory excursions. These patients, in an early paralytic stage of anterior poliomyelitis, developed weakness of the respiratory muscle pump involving both intercostal and diaphragm muscles. In some, the neck muscles were able to create sufficient respiratory movements and compensate for diaphragm weakness. Critical shortages of ventilatory support in patients with the bulbar type of poliomyelitis resulted in serious accumulation of oropharyngeal secretions and diaphragmatic weakness and required both old (tracheostomy and negative-pressure ventilation) and new (positive pressure-ventilation) interventions. Specialized hospital units were necessary to provide this comprehensive care for these patients presenting with a new set of clinical problems. The worst cases needed prolonged placement in an “iron lung.” Within the chamber – sealing the patient at the neck – a negative pressure caused the abdomen and thorax to expand with air flowing in. A cycle was produced by returning to atmospheric pressure. (Patients in the iron lung had their chest expanded every four seconds.) Many patients could be liberated from the device or transitioned to a cuirass ventilator, but some survivors remained permanently dependent (Figure 1.9)[Reference Lassen45].
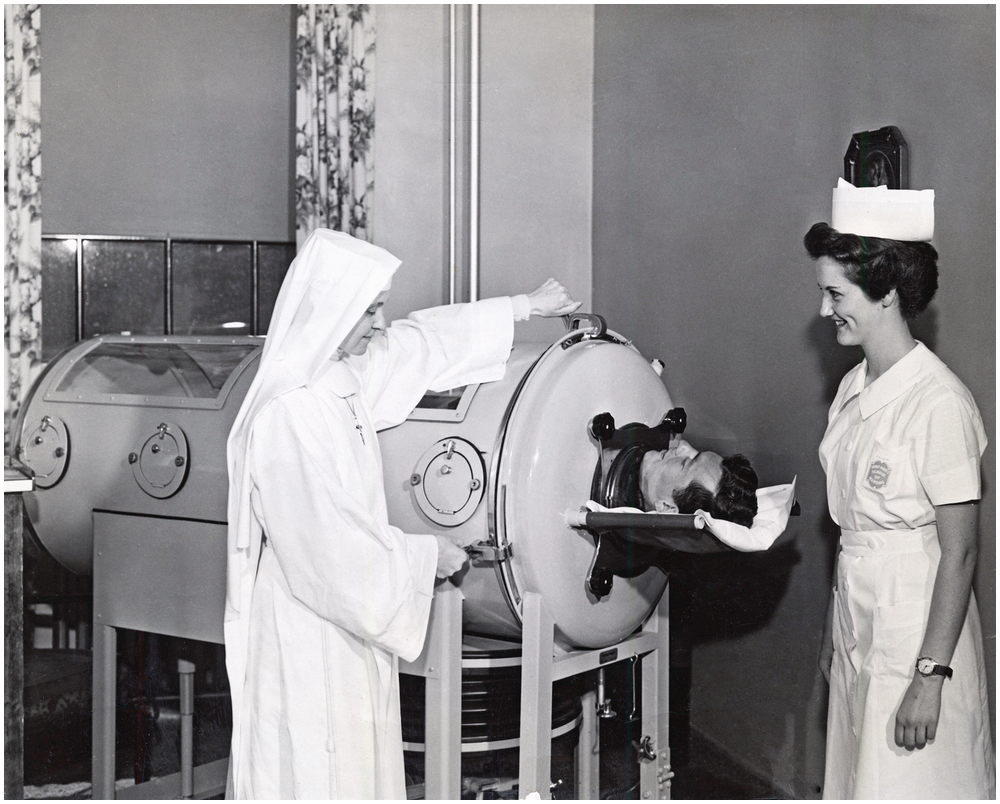
Neurologists have been struck by the fact that admission to intensive care is often a result of a neurologic disease, but the acute care of poliomyelitis was rarely the responsibility of neurologists, who delegated management to anesthesiologists and later to orthopedic surgeons, pediatricians, and physical therapists. There were, major exceptions[Reference Wijdicks46]. Ritchie Russell at the University of Oxford became interested in respiratory insufficiency in poliomyelitis. He summarized his clinical work and innovations in poliomyelitis in an important, comprehensive, multifaceted book, which he wrote to address perceived gaps in knowledge because “it seems to be no one’s task to study the disease as a whole”[Reference Russell47]. Ritchie Russell’s articles were instrumental in identifying multiple types of poliomyelitis defined by their respiratory care exigencies. He divided cases into three categories: (1) patients with difficulty in swallowing but no respiratory weakness; (2) patients with weakness of respiratory muscles but no bulbar weakness; and (3) patients with paralysis of both respiratory muscles and oropharyngeal muscles. He included the need to examine the respiratory muscles and emphasized that the abdominal muscles played a key role in coughing up secretions. He mentioned inability to count beyond 10 in one breath as a sign of reduced vital capacity[Reference Wijdicks46–Reference Russell48]. Care included postural drainage to avoid aspiration in patients without respiratory failure and the use of inverted V and tilted bed. Ritchie Russell discussed intermittent positive pressure ventilation, tracheostomy, bronchoscopy, and methods of chest physiotherapy[Reference Russell49]. He advised treatment that varied from continuing postural drainage to aspiration of secretions and tube feeding and use of tank respirators, later followed by the development of the Oxford respiratory pump for poliomyelitis. He pioneered, together with technicians and anesthesiologists, a positive-pressure respiration facility that was also later used for other conditions. He criticized Lassen’s method of using medical students’ bag ventilation and devised the Radcliffe respiration pump in 1953 with Edgar Schuster, which could more accurately control amount of pressure, rate, and volume but also create a waveform with rapid flow rate and rise of pressure at inspiration to force air through the tracheostomy tube[Reference Russell and Schuster50, Reference Russell, Schuster, Smith and Spalding51].
In the United States, A. B. Baker’s contributions were also important in describing the essential involvement of the vagal nerve, which impaired swallowing and caused faulty innervation to the larynx leading to obstruction of the airway. He later noted that “we advocate a ‘total push’ form of physiologic support and understanding of respiratory failure in bulbar poliomyelitis and prevention of secondary systemic complications.” Baker was instrumental in determining the essence of recognition of pooling of secretions and impaired speech, which indicated impending obstruction of the airway. Anxiety and restlessness associated with hypoxemia appeared to clear after adequate oxygenation. Baker’s contribution was to emphasize that delay in performing a tracheostomy subjected the patient to severe hypoxemia and increased danger of pulmonary edema. Laryngeal stridor and dyspnea (despite adequate chest excursions), cyanosis, and encephalopathic symptoms were indicators of severe obstruction of the airway requiring emergency intubation or tracheostomy. Baker recognized that involvement of vital centers (without cranial nerve involvement) leads to irregular, shallow respiration in some patients and, in others, marked dysautonomic features (flushed skin, cherry red lips, tachycardia, and reduced pulse pressure with cold and clammy skin). He showed, in patients who came to autopsy, necrosis of areas below the floor of the fourth ventricle. A subgroup was found with both central respiratory centers and peripheral involvement requiring a respirator. To monitor for early anoxia, Baker used an oximeter or calorimeter attached to the earlobe (forerunner of the pulse oximeter[Reference Ross and Baker52–Reference Monke, Shapiro and Baker55].
Plum described the effects of cuirass-type ventilators, the effects of the rapid rocking bed on ventilation, carbon dioxide retention with normal oxygenation but a damaged controlling respiratory center from bulbar poliomyelitis and the presence of hypertensive urgencies in patients requiring ventilation in tank respirators. He clearly described carbon dioxide retention and its significance in determining adequate management. In patients with slowly progressive poliomyelitis, small degrees of failure of carbon dioxide elimination occurred in a considerable proportion of patients. He identified not only the effect of hypoxemia and hypercapnia but also observations on measurements of vital capacity. In addition, he emphasized that the respiratory center is depressed by high carbon dioxide tensions, and only the carotid bodies could provide chemical stimulation of respiration after the respiratory center is impaired and stimulated by hypoxemia. In 1952, he reported patterns of changes in pulmonary function in patients recovering from poliomyelitis[Reference Plum and Lukas56–Reference Plum and Swanson61].
Later, Plum studied patients with acute central respiratory failure in the wards of the King County Hospital, Seattle, Washington, and the Providence Hospital, Anchorage, Alaska. Patients with respiratory insufficiency convalescing from poliomyelitis were studied in the Northwest Respiratory and Rehabilitation Center. He developed a technique to minimize trauma while suctioning the patient after tracheostomy. Faulty suctioning could lead to severe mucous membrane damage and potentially asphyxiating tracheal bronchitis. Furthermore, in a clinical and pathologic study, Plum found that central respiratory failure in poliomyelitis was medullary in origin. He noted that permanent damage to chemoreceptive neurons in the respiratory center could explain low CO2 responsiveness after poliomyelitis. Each recognized that airway obstruction and respiratory compromise in a neurologic disease shared some similarities with acute neuromuscular disease (myasthenia gravis and GBS), such as pooling secretions and predictive signs of early hypoxemia, but other observations, such as bulbar dysrhythmic respiratory failure, were new and original.
Another major development in understanding the neurology of breathing were the observations in neurosurgical trauma units. Neurosurgeons, ostensively more concerned with expanding contusions or cerebral edema, did not typically focus on abnormal rhythms of respiration, but the more astute ones would follow up after being alerted by neuroscience nursing staff that the breathing of the patient had changed.
Most known were periodic breathing surges during the rise and fall of ICP with irregular respiration and hyperventilation at the top of a plateau wave. Hoff and Breckenridge studied many clinical breathing patterns[Reference Hoff and Breckenridge62]. In a number of experiments, they found that an intrinsically slow rhythm of the brainstem, normally expressed in deep, “all-or-nothing,” or gasping breathing, accounted for periodicity of breathing. In 1972, North and Jennett stated in the Lancet: “Abnormal breathing is often regarded as an incidental feature of brain damage, and little clinical attention is paid to it”[Reference North and Jennett63].
Conventional wisdom prior to the 1970s held that respiratory patterns were of localizing value in neurological diagnosis and could identify specific lesions. Cheyne–Stokes breathing with localized deep hemispheric and basal ganglia lesions and central neurogenic hyperventilation with lesions of the upper brainstem but with lower lesions of the pontine tegmentum could lead to apneustic breathing, cluster breathing, and short-cycle Cheyne–Stokes respiration. Ataxic breathing was linked to lesions in either the medulla or lower portion of the pons. Others claimed that periodic breathing in humans was a result of supramedullary dysfunction[Reference Plum, Brown and Snoep64].
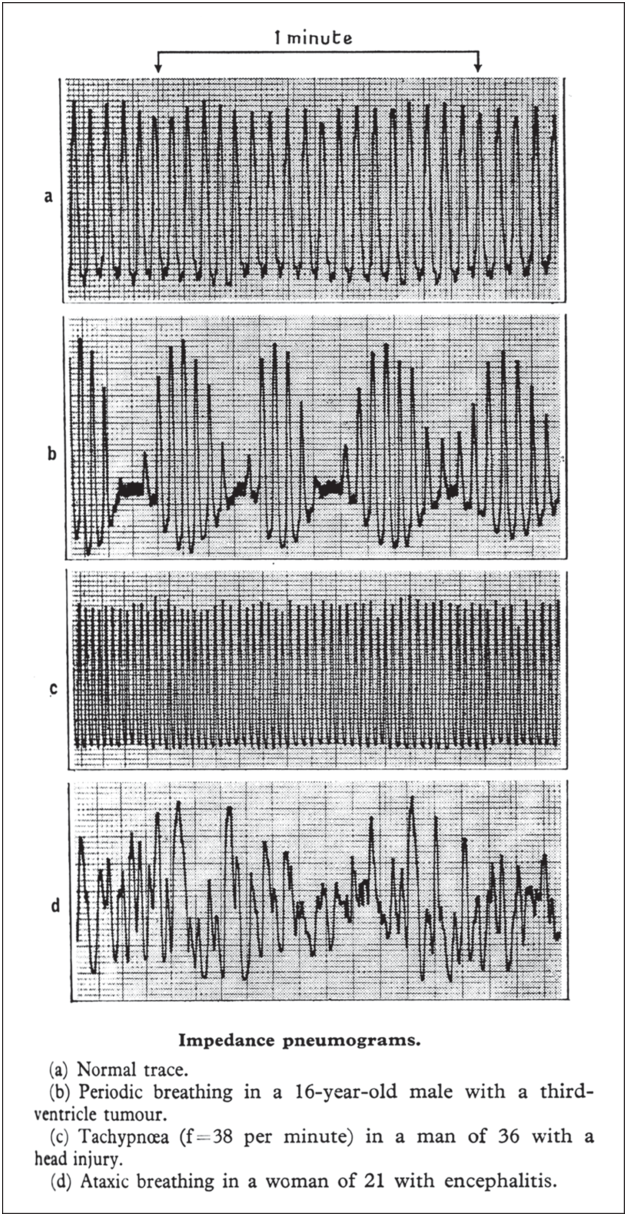
However, a series of studies by Jennett and North carefully examined the respiratory responses in clinical cases of brain damage and demonstrated several variants for the first time. Most notably, they simplified (and denied) the claim that breathing patterns correlated with specific lesions in the brain and brainstem. They examined this localization paradigm in the neurosurgical ICU, hoping for clinical confirmation “if these associations could be applied not only to chronic neurological disorders but also to acute brain damage, then recognition of particular breathing patterns could be very useful.” An early study in neurosurgical patients allowed recording of patterns and identified three main abnormalities A follow-up study in neurocritical care patients investigated 90 patients with traumatic brain injury and 43 patients with aneurysmal subarachnoid hemorrhage. The breathing pattern was recorded using an impedance pneumograph. Their definition of breathing patterns was as follows: periodic breathing, regularly recurring cycles of changing ventilation, the smallest tidal volume being not more than half that at the peak of the cycle; tachypnea, respiratory frequency over 25 breaths per minute; irregular breathing, an erratic variation in frequency and in tidal volumes, the smallest breaths being less than half the volume of the largest; and sighing, an occasional deep but prolonged breath, at least twice the usual tidal volume for the patient.
Jennett and North found no abnormal breathing patterns in a substantial minority (40%) of their patients. Periodic respiration was found in 41% of patients but with a preponderance of periods shorter than the typical one minute of Cheyne–Stokes respiration (Figure 1.10). Upper-airway obstruction caused the regularly recurring periods of apnea, and elevating the mandible restored a normal breathing pattern in some patients. Tachypnea was noted in over 25% of their patients, but many had normal PaCO2 with abnormalities other than tachypnea. Tachypneic patients with hyperpnea showed a significant negative correlation between minute volume and PaCO2, although the scatter was wide[Reference Jennett and North65]. Irregular breathing was seen in a third of their patients, of whom only three showed irregularities of the degree and persistence to merit the term ataxic breathing. Thirty patients showed two different abnormal patterns during the recording, and two patients showed all three major abnormalities. Slow respiratory frequencies (7 to 10 breaths per minute) were recorded in 12 patients[Reference Ashbridge, Jennett and North66, Reference Jennett, Ashbridge and North67]. Sighing and gasping was seen in a few patients.

They concluded:
The only breathing pattern that could be used reliably as a localizing sign in patients with acute brain damage was the gross and persistent irregularity associated with hypercapnia, which points to a lesion in the medulla. Otherwise, breathing abnormalities were common but diverse. Central neurogenic hyperventilation did not relate periodic breathing either to the site or the prognosis of acute brain damage. All patients with lesions in the medulla had abnormal patterns; medullary and pontine lesions were frequently associated with irregular breathing.
There were other observations by neurologists in the ICU. In 1962, Plum et al. studied the effect of brief hyperventilation (five deep breaths), which lowered the PaCO2 by at least 6 mm Hg, in large groups of both normal subjects and patients with brain damage. They concluded that apnea greater than 12 seconds was sufficiently abnormal to be diagnostic of bilateral forebrain damage[Reference Plum, Brown and Snoep64]. When apnea followed hyperventilation, its incidence and duration presumably correlated with the magnitude of the reduction in PaCO2. Follow-up studies showed discrepancies in 100 abnormal patients from the neurosurgical wards of the Institute of Neurological Sciences. All had intracranial lesions but were sufficiently alert to cooperate in the test; most were suffering from traumatic brain injury, brain tumor, or recent subarachnoid hemorrhage. In one or more of the three tests, over half of them showed apnea more than five seconds longer than their greatest spontaneous interval. They found that the incidence of apnea correlated to drowsiness or disorientation. However, neither the value to which end tidal PaCO2 was reduced nor the amount by which it was reduced bore any relation to the duration of apnea. This study confirmed that brief voluntary hyperventilation commonly causes apnea in patients with supramedullary lesions. Apnea was related to drowsiness rather than to the extent of the lesion; it was unrelated to the measured reduction in end-tidal carbon dioxide tension. There was no association between responsiveness to PaCO2 and either bilateral forebrain damage or periodic breathing. (No mention was made of sympathetic storming, which is typically associated with paroxysms of tachypnea, sweating, and tachycardia.) Looking back, we can only conclude that the specific localizations attributed to certain breathing patterns were inconsistent or perhaps incorrect. We can additionally conclude that extrapolation of laboratory experiments to the bedside is again potentially problematic.
The Neurology of Apnea
Apnea is less common than hypoventilation and often short lived. When apnea is prolonged, it may be permanent. Historically, when did we discover that the respiratory drive can be permanently lost? For sure, any clinicians summoned to the bedside of a cyanotic patient, with or without a pulse, would not hesitate to resuscitate – just because these efforts can be successful. It is not exactly known how long we can “tolerate” apnea (within minutes followed by circulatory arrest), and thus, attempts continue with some patients breathing again. The apnea time intervals – from recovery to irretrievability – have been nothing but an educated guess, and the permanence of apnea has never been downright accepted despite expansive definitions.
The most apposite moment in the history of the neurology of breathing has been the discovery of permanent apnea in mechanically ventilated patients with acutely catastrophic brain and brainstem injury. This would occur only in brain death and only when all brainstem reflexes became lost. In 1959, the neurosurgeons Pierre Wertheimer and Jacques Descotes with the neurologist Michel Jouvet were among the first to propose criteria for these new clinical states (A propos du diagnostic de la mort système nerveux). They not only observed apnea but stopped the ventilator to stimulate the respiratory centers with increasing respiratory acidosis, all to no avail[Reference Wertheimer, Jouvet and Descotes68]. The same condition was later called coma dépassé and said to be characterized by immobility of the eyes in a neutral position, mydriasis and absent light reflex, absent blinking with stimuli, absence of swallowing reflexes, drooping of the jaw, absence of motor responses to any stimuli, muscle hypotonia, tendon areflexia, equivocal plantar reflexes, absence of spontaneous respiration after discontinuation of ventilation, and also immediate cardiovascular collapse as soon as vasopressors were stopped. A disturbance of thermoregulation with core temperature depends on the environmental temperature[Reference Mollaret and Goulon69]. Empirically, neurologists then found that respiratory drive stopped as one of the last acts of rostrocaudal damage to the brainstem commonly involving the bordering lower medulla oblongata pressure centers, which led to apnea and hypotension simultaneously. This presentation had the signature of a terminally dying person. It also became quickly understood that once apnea was found in a calamitous brain injury with no elicitable brainstem reflexes (not confounded by major toxin or other mimicker), breathing would never return. No medical or surgical intervention could reverse it. This comatose state therefore stood apart from other degrees of coma and was “beyond” (dépassé) what had been seen before, irrevocable and a neurologic definition of death. Finally, medicine found a solid criterion for apnea and added a key milestone.
Conclusions
This chapter has surveyed the experimental and clinical progress in understanding the neurology of breathing through the centuries but with a focus on the last century. Quickly recognized as vital for mammalian life, the regulation of breathing was beyond understanding for many investigators. The discovery of the respiratory centers in the lower brainstem opened a field of inquiry, and there is ongoing experimental research in defining its physiologic responses to changes.
The history of clinical observations was different and resulted from acute and astute observers but with fragmented attention. The localizing value of breathing patterns was limited, and more generally, neurogenic breathing patterns were exceedingly difficult to differentiate from lung injury or respiratory compensation of an acid-base disbalance. Any change in respiratory rhythm tells the clinician only that the patient is potentially deteriorating neurologically and that oxygenation may become marginal requiring endotracheal intubation and mechanical ventilation. The experimental localization paradigm could not always be confirmed in a clinical setting nor was there any indication that certain breathing patterns related to outcome. That is not to say that outcome was determinatively fatal once the respiratory mechanics failed in acute neuromuscular diseases. The development of the iron lung and other respirators[Reference Drinker and McKhann70, Reference Goerig, Filos, Ayisi, Atkinson and Boulton71] and, eventually, tracheostomy and positive pressure ventilation increased survival, a chance to heal, and even recover fully.