


The Janus kinase (JAK)-Signal Transducer and Activator of Transcription (STAT) signaling pathway is crucial for gene regulation in common and lineage-specific genetic programs (Suppl. Fig. 1) (Brooks & Putoczki, Reference Brooks and Putoczki2020; X. Hu et al., Reference Hu, Li, Fu, Zhao and Wang2021; Jankowski et al., Reference Jankowski, Lee, Wilflingseder and Hennighausen2021; Lee et al., Reference Lee, Jung and Hennighausen2021; Shillingford, Reference Shillingford2002; Xue et al., Reference Xue, Yao, Gu, Shi, Yuan, Chu, Bao, Lu and Li2023). Four known JAKs (JAK1, JAK2, JAK3, and TYK2) and seven STATs (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) collectively form the JAK and STAT gene families. The members of these gene families have crucial functions such as immune modulation, cell proliferation, and hematopoiesis (Brooks & Putoczki, Reference Brooks and Putoczki2020; X. Hu et al., Reference Hu, Li, Fu, Zhao and Wang2021; Lee et al., Reference Lee, Jung and Hennighausen2021; Xue et al., Reference Xue, Yao, Gu, Shi, Yuan, Chu, Bao, Lu and Li2023). The JAK and STAT genes carry mutations, such as single nucleotide polymorphisms (SNPs), representing the most prevalent form of genetic variation among individuals (L. X. Shen et al., Reference Shen, Basilion and Stanton1999). These variations are characterized based on their genomic location and/or their potential influence on gene expression or function (Chu & Wei, Reference Chu and Wei2019). In noncoding regions, such as promoters and enhancers, SNPs can play essential roles in gene regulation (Hecker et al., Reference Hecker, Lauber, Behjati Ardakani, Ashrafiyan, Manz, Kersting, Hoffmann, Schulz and List2023; Hoffmann et al., Reference Hoffmann, Trummer, Schwartz, Jankowski, Lee, Willruth, Lazareva, Yuan, Baumgarten, Schmidt, Baumbach, Schulz, Blumenthal, Hennighausen and List2023, Reference Hoffmann, Vaz, Chhatrala and Hennighausen2025; Lee et al., Reference Lee, Willi, Shin, Liu and Hennighausen2018; Peña-Martínez & Rodríguez-Martínez, Reference Peña-Martínez and Rodríguez-Martínez2024). Within coding regions, SNPs are further classified into synonymous variants, which preserve the amino acid sequence and are functionally silent and evolutionarily neutral, and nonsynonymous (missense) variants, which change amino acids and can influence protein structure and/or function (Chu & Wei, Reference Chu and Wei2019; Lio et al., Reference Lio, Düz, Hoffmann, Willruth, Baumbach, List and Tsoy2025; Tsoy et al., Reference Tsoy, Ameling, Franzenburg, Hoffmann, Liv-Willuth, Lee, Knabl, Furth, Voelker, Hennighausen, Baumbach, Kacprowski and List2024). It is well established that dysregulation of the JAK and STAT genes, through amino acid changes and altered regulatory element activity, can cause diverse pathophysiological outcomes such as autoimmune diseases, cancer, and infectious diseases (Erdogan et al., Reference Erdogan, Qadree, Radu, Orlova, de Araujo, Israelian, Valent, Mustjoki, Herling, Moriggl and Gunning2022; Hennighausen & Lee, Reference Hennighausen and Lee2020; Hoffmann, Willruth et al., Reference Hoffmann, Willruth, Dietrich, Lee, Knabl, Trummer, Baumbach, Furth, Hennighausen and List2024). Missense mutations drive functional changes of proteins by altering protein stability, disrupting protein-protein interactions, or compromising enzymatic functions, thus serving as potent drivers of disease (Teng et al., Reference Teng, Srivastava, Schwartz, Alexov and Wang2010).
Some mutations on JAK and STAT genes frequently lead to constitutive activation of the JAK-STAT pathway, a hallmark of various hematological malignancies. A well-known example is the JAK2Val617Phe/V617F mutation, which is highly prevalent in myeloproliferative neoplasms, occurring in approximately 90–95% of polycythemia vera (PV) cases and 50–60% of individuals with essential thrombocythemia and primary myelofibrosis (Perner et al., Reference Perner, Perner, Ernst and Heidel2019; Rampal et al., Reference Rampal, Al-Shahrour, Abdel-Wahab, Patel, Brunel, Mermel, Bass, Pretz, Ahn, Hricik, Kilpivaara, Wadleigh, Busque, Gilliland, Golub, Ebert and Levine2014). This gain-of-function mutation enhances JAK-STAT signaling even in the absence of cytokine stimulation, thereby driving uncontrolled cell proliferation and survival. Similarly, activating mutations in JAK1 and JAK3 have been identified in T-cell acute lymphoblastic leukemia (T-ALL), where they contribute to persistent JAK-STAT pathway activation (Girardi et al., Reference Girardi, Vereecke, Sulima, Khan, Fancello, Briggs, Schwab, de Beeck, Verbeeck, Royaert, Geerdens, Vicente, Bornschein, Harrison, Meijerink, Cools, Dinman, Kampen and De Keersmaecker2018; Waldmann, Reference Waldmann2017). Beyond hematological malignancies, dysregulation of the JAK-STAT pathway has been implicated in solid tumors and autoimmune diseases, underscoring its broader significance in human pathology (Łączak et al., Reference Łączak, Kuczyńska, Grygier, Andrzejewska, Grochowska, Gulaczyk and Lewandowski2022; O’Shea et al., Reference O’Shea, Schwartz, Villarino, Gadina, McInnes and Laurence2015). Mutations in STAT3, for instance, are linked to increased tumor invasiveness and poor clinical outcomes across multiple cancer types (Deng et al., Reference Deng, Li, Li, Mao, Ke, Liang, Lei, Lau and Mao2022; Klein et al., Reference Klein, Stoiber, Sexl and Witalisz-Siepracka2021).
In our previous work (Hoffmann & Hennighausen, Reference Hoffmann and Hennighausen2025), we conducted a large-scale survey of missense mutations within the JAK and STAT genes across two major repositories — the All of Us database (The All of Us Research Program Genomics Investigators et al., Reference Bick, Metcalf, Mayo, Lichtenstein, Rura, Carroll, Musick, Linder, Jordan, Nagar, Sharma, Meller, Basford, Boerwinkle, Cicek, Doheny, Eichler and Gabriel2024), which captures a broad range of genetic variation from the general but mostly healthy population in the United States, and the COSMIC (Bamford et al., Reference Bamford, Dawson, Forbes, Clements, Pettett, Dogan, Flanagan, Teague, Futreal, Stratton and Wooster2004; Sondka et al., Reference Sondka, Dhir, Carvalho-Silva, Jupe, McLaren, Starkey, Ward, Wilding, Ahmed, Argasinska, Beare, Chawla, Duke, Fasanella, Neogi, Haller, Hetenyi, Hodges and Teague2024) database, a leading resource for somatic mutations in cancer. Our investigation identified hundreds of unique amino acid–changing variants, some of which had been reported as disease-associated. This analysis provided a resource cataloging the breadth and frequency of missense mutations in JAK-STAT genes. Building on these findings, this study aims to focus explicitly on the identified disease-causing (by literature) SNPs and ClinVar (Landrum et al., Reference Landrum, Lee, Riley, Jang, Rubinstein, Church and Maglott2014) benign/likely benign SNPs and their distribution among specific functional domains of JAK and STAT proteins. We assess whether certain functional domains are more frequently affected and how these mutations correlate with disease. By mapping the structural distribution of disease-associated SNPs, we determine whether they predominantly occur on the protein surface or within its core. Additionally, we analyze specific target sequences, CRISPR target proximity, and amino acid composition patterns to identify shared vulnerabilities between disease-causing and ClinVar benign SNPs that may explain why specific regions are more mutation- or disease-prone.
Materials and Methods
All of Us Data Explorer
The All of Us Research Program gathers health and genomic data from participants residing in the U.S. We accessed the All of Us Controlled Tier Dataset v7 (All of Us Research Program Genomics Investigators et al., Reference Bick, Metcalf, Mayo, Lichtenstein, Rura, Carroll, Musick, Linder, Jordan, Nagar, Sharma, Meller, Basford, Boerwinkle, Cicek, Doheny, Eichler and Gabriel2024) (encompassing 413,000 participants) through the Data Browser to examine SNPs. We focused on missense mutations in the JAK-STAT gene families, specifically SNPs that alter amino acids, and we analyzed their frequency across various demographic groups. All data were anonymized according to program protocols. In accordance with All of Us guidelines, we included only missense mutations (excluding all other SNP types) identified in at least 20 participants and previously associated with a disease in the literature. The cut-off of 20 is dictated by the policy of All of Us due to privacy concerns. We focused only on missense mutations because they change one amino acid to another and therefore could have a significant impact on protein function (Pal & Moult, Reference Pal and Moult2015).
COSMIC (Catalogue of Somatic Mutations in Cancer)
The Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic; Bamford et al., Reference Bamford, Dawson, Forbes, Clements, Pettett, Dogan, Flanagan, Teague, Futreal, Stratton and Wooster2004; Sondka et al., Reference Sondka, Dhir, Carvalho-Silva, Jupe, McLaren, Starkey, Ward, Wilding, Ahmed, Argasinska, Beare, Chawla, Duke, Fasanella, Neogi, Haller, Hetenyi, Hodges and Teague2024) tracks somatic mutations identified in cancer. Using COSMIC v100 (>1,000,000 tumor samples), we examined missense mutations in the JAK-STAT pathway, focusing on those found in cancer samples. COSMIC details the mutational spectrum, tissue distribution, and associated cancers for each SNP, enabling direct comparison with findings from the All of Us cohort. We extracted mutations using COSMIC’s online tools, filtering by mutation type. For each SNP, we noted the number of samples carrying the mutation and referenced disease associations from existing literature. To align with the All of Us approach, we included only missense mutations (excluding all other SNP types) present in at least 20 tumor samples and that were previously associated with a disease in the literature. We used the cut-off of 20 as dictated by the policy of All of Us due to privacy concerns to have a consistent filter between the two databases.
Obtaining SNPs That are Disease-Associated in the Published Literature and SNPs Classified as Benign in ClinVar
We used the identified SNPs and literature from Hoffmann and Hennighausen (Reference Hoffmann and Hennighausen2025). We used the Athena – OHDSI Vocabularies Repository database (https://athena.ohdsi.org/search-terms/start) to generalize disease terms (Figure 1, Suppl. Table 1) (Reich et al., Reference Reich, Ostropolets, Ryan, Rijnbeek, Schuemie, Davydov, Dymshyts and Hripcsak2024).
Domain-specific distribution of missense mutations in JAK and STAT proteins and their associations with disease. The schematic representation of JAK (JAK2, JAK3, TYK2) and STAT (STAT1, STAT3, STAT4, STAT5B) proteins highlights the locations of missense mutations identified in the All of Us and COSMIC databases. Symbols indicate disease associations, including autoimmune diseases (turquoise circles), cancer/tumor (purple stars), infectious diseases (yellow triangles), blood disorders/hematopoietic system involvement (blue donuts), protective mutations against autoimmunity (green hexagons), and other genetic disorder order skin disorder (dark pink quarter of a circle). Mutations found in at least 20 individuals are labeled, with mutations found in All of Us (black font) or COSMIC (red font) and mutations found in All of Us and COSMIC are highlighted in bold red. This visualization provides insight into mutation clustering within functional domains. Protein domains are annotated as follows: STAT proteins include the N-terminal, coiled-coil, DNA-binding, linker, Src homology 2 (SH2), and transactivation (TAD) domains, while JAK proteins include the FERM (For protein 4.1, Ezrin, Radixin, and Moesin), SH2, pseudokinase, and kinase domains.

Figure 1 Long description
A diagram of the JAK and STAT proteins highlighting the locations of missense mutations identified in the All of Us and COSMIC databases. The diagram includes symbols indicating disease associations such as autoimmune diseases, cancer/tumor, infectious diseases, blood disorders/hematopoietic system involvement, protective mutations against autoimmunity, and other genetic disorders. Mutations found in at least 20 individuals are labeled, with mutations found in All of Us or COSMIC and mutations found in both databases highlighted. Protein domains are annotated for STAT proteins including the N-terminal, coiled-coil, DNA-binding, linker, Src homology 2 (SH2), and transactivation (TAD) domains, and for JAK proteins including the FERM, SH2, pseudokinase, and kinase domains.
We used the NCBI Entrez E-utilities API (https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi, with parameter “db”: “clinvar”) to filter all missense mutations from All of Us and COSMIC for only mutations categorized as benign by ClinVar.
The Python code can be found at GitHub: https://github.com/Firestar93/JAKSTAT_missenseSNPs_properties
Enzyme Cut Sites and CRISPR Sites on the Nucleotide Sequence and Amino Acid Composition Analysis on the Amino Acid Sequences
We downloaded the mRNA sequences from the CCDS database (Farrell et al., Reference Farrell, O’Leary, Harte, Loveland, Wilming, Wallin, Diekhans, Barrell, Searle, Aken, Hiatt, Frankish, Suner, Rajput, Steward, Brown, Bennett, Murphy, Wu and Pruitt2014; Harte et al., Reference Harte, Farrell, Loveland, Suner, Wilming, Aken, Barrell, Frankish, Wallin, Searle, Diekhans, Harrow and Pruitt2012; Pruitt et al., Reference Pruitt, Harrow, Harte, Wallin, Diekhans, Maglott, Searle, Farrell, Loveland, Ruef, Hart, Suner, Landrum, Aken, Ayling, Baertsch, Fernandez-Banet, Cherry, Curwen and Lipman2009; Pujar et al., Reference Pujar, O’Leary, Farrell, Loveland, Mudge, Wallin, Girón, Diekhans, Barnes, Bennett, Berry, Cox, Davidson, Goldfarb, Gonzalez, Hunt, Jackson, Joardar, Kay and Pruitt2018) for the following genes: STAT1 (CCDS2309.1), STAT3 (CCDS32656.1), STAT4 (CCDS2310.1), STAT5B (CCDS11423.1), JAK2 (CCDS6457.1), JAK3 (CCDS12366.1), and TYK2 (CCDS12236.1).
We used a house-made Python script to detect enzyme cut sites (using the Biopython package, specifically the Bio. Restriction module (Cock et al., Reference Cock, Antao, Chang, Chapman, Cox, Dalke, Friedberg, Hamelryck, Kauff, Wilczynski and de Hoon2009)) and the following CRISPR-Cas sites: SpCas9 recognizes the 5“-NGG-3” sequence, while SaCas9 targets 5“-NNGRRT-3”. NmCas9 requires the 5“-NNNNGATT-3” PAM, and St1Cas9 recognizes 5“-NNAGAAW-3”. Similarly, St3Cas9 targets 5“-NGGNG-3”, and CjCas9 recognizes the 5“-NNNNACA-3” sequence. FnCas9 operates with a 5“-YG-3” PAM, whereas TdCas9 exhibits specificity for 5“-NAAAW-3”. SpCas9-NG (SpG) has a relaxed PAM requirement of 5“-NG-3”, broadening its targeting scope. Lastly, SpRY functions as a near-PAM-less variant, with a preference for NRN sequences. To associate enzyme sites and CRISPR sites to a mutation, we stretch a window of 20 bps in each direction of the mutation since it was reported that 20 bps ensures that any potential restriction sites are adequately captured (Wang, Reference Wang2018). We used the UpSet Python library for visualizations (Lex et al., Reference Lex, Gehlenborg, Strobelt, Vuillemot and Pfister2014).
We used house-made Python scripts to detect amino acid combinations close (3 amino acids upstream and downstream) to ClinVar benign and disease-associated mutations using the Biopython package.
The code can be found at GitHub: https://github.com/Firestar93/JAKSTAT_missenseSNPs_properties
Visualization of the Protein Structures
We used the ChimeraX tool for the visualization of the protein structures (Goddard et al., Reference Goddard, Huang, Meng, Pettersen, Couch, Morris and Ferrin2018; Meng et al., Reference Meng, Goddard, Pettersen, Couch, Pearson, Morris and Ferrin2023; Ucsf Chimerax Pettersen et al., Reference Pettersen, Goddard, Huang, Meng, Couch, Croll, Morris and Ferrin2021). We used ALPHAFOLD MONOMER V2.0 (Jumper et al., Reference Jumper, Evans, Pritzel, Green, Figurnov, Ronneberger, Tunyasuvunakool, Bates, Žídek, Potapenko, Bridgland, Meyer, Kohl, Ballard, Cowie, Romera-Paredes, Nikolov, Jain, Adler and Hassabis2021) predicted structures for the following proteins: STAT1 (AF-P42224-F1-model_v4), STAT3 (AF-P40763-F1-model_v4), STAT4 (AF-Q14765-F1-model_v4), STAT5B (AF-P51692-F1-model_v4), JAK2 (AF-O60674-F1-model_v4), JAK3 (AF-P52333-F1-model_v4), and TYK2 (AF-P29597-F1-model_v4). ChimeraX files for interactive visualization can be found at figshare: https://doi.org/10.6084/m9.figshare.28597121.
Results
To better understand the characteristics of JAK and STAT mutations in terms of their location within the nucleotide and amino acid sequence, we focused on their distribution across protein domains and disease associations for STAT1, STAT3, STAT4, STAT5B, JAK2, JAK3, and TYK2. We could not identify any missense variants in STAT2, STAT5A, STAT6, and JAK1 that satisfied the All of Us requirements of at least 20 individuals harboring them. We identified domains particularly susceptible to disease-related alterations. We further assessed where they are located to evaluate their effects on protein structure and stability. Beyond structural localization, we analyzed the genomic context of these mutations, examining their proximity to specific feasible sequences and CRISPR target sites to assess potential regulatory influences and editing feasibility. Finally, we investigated conserved amino acid sequence patterns near disease-associated mutations to identify motifs contributing to mutation susceptibility.
Mutation Hotspots in JAK-STAT Proteins and Their Disease Relevance
We visualized mutations that were disease-associated with autoimmune disease, cancer, chronic disease, etc. (Figure 1, Suppl. Table 1) and ClinVar benign (Suppl. Fig. 2) from the All of Us and COSMIC database and investigated distinct patterns in the distribution of disease-associated SNPs across JAK and STAT proteins to highlight specific domains linked to various diseases. Autoimmune disorders were mainly associated with mutations in the coiled-coil domains of STAT1 (Uzel et al., Reference Uzel, Sampaio, Lawrence, Hsu, Hackett, Dorsey, Noel, Verbsky, Freeman, Janssen, Bonilla, Pechacek, Chandrasekaran, Browne, Agharahimi, Gharib, Mannurita, Yim, Gambineri and Holland2013) and STAT4 (Saevarsdottir et al., Reference Saevarsdottir, Stefansdottir, Sulem, Thorleifsson, Ferkingstad, Rutsdottir, Glintborg, Westerlind, Grondal, Loft, Sorensen, Lie, Brink, Ärlestig, Arnthorsson, Baecklund, Banasik, Bank and Bjorkman2022) critical for dimerization and transcriptional activity. In contrast, cancer-related mutations were predominantly found in the SH2 domains of STAT3 (Cheon et al., Reference Cheon, Xing, Moosic, Ung, Chan, Chung, Toro, Elghawy, Wang, Hamele, Hardison, Olson, Tan, Feith, Ratan and Loughran2022; D. Kim et al., Reference Kim, Park, Huuhtanen, Ghimire, Rajala, Moriggl, Chan, Kankainen, Myllymäki and Mustjoki2021; Kristensen et al., Reference Kristensen, Larsen, Rewes, Frederiksen, Thomassen and Møller2014; Olson et al., Reference Olson, Moosic, Jones, Larkin, Olson, Toro, Fox, Feith and Loughran2020; Ramsey et al., Reference Ramsey, Sabatini, Watson, Chawla, Ko and Sakhdari2023; Rivero et al., Reference Rivero, Mozas, Jiménez, López-Guerra, Colomer, Bataller, Correa, Rivas-Delgado, Bastidas, Baumann, Martínez-Trillos, Delgado, Giné, Campo, López-Guillermo, Villamor, Magnano and Matutes2021; M. Shen, Reference Shen2023; Yan et al., Reference Yan, Olson, Nyland, Feith and Loughran2015) and STAT5B (Freiche et al., Reference Freiche, Couronné, Bruneau and Hermine2022; Z. Hu et al., Reference Hu, Medeiros, Xu, Yuan, Peker, Shao, Tang, Mai, Thakral, Rios, Hu and Wang2023; Yin et al., Reference Yin, Tam, Walker, Kaur, Ouseph, Xie, Weinberg, Li, Zuo, Routbort, Chen, Medeiros, George, Orazi, Arber, Bagg, Hasserjian and Wang2023), suggesting that alterations in these key signaling interfaces contribute to tumorigenesis. Hematological malignancies and blood disorders are strongly associated with SNPs in the Pseudokinase (Arai et al., Reference Arai, Yoshimitsu, Otsuka, Ito, Miyazono, Nakano, Obama, Nakashima, Hanada, Owatari, Nakamura, Tokunaga, Kamada, Utsunomiya, Haraguchi, Hayashida, Fujino, Odawara, Tabuchi and Ishitsuka2023; Delio et al., Reference Delio, Bryke, Mendez, Joseph and Jassim2023; Eichstaedt et al., Reference Eichstaedt, Verweyen, Halank, Benjamin, Fischer, Mayer, Guth, Wiedenroth, Egenlauf, Harutyunova, Xanthouli, Marra, Wilkens, Ewert, Hinderhofer and Grünig2020; Haji Paiman et al., Reference Haji Paiman, Mat Nasir, Miptah, Saidon and Abdul Monir2024; Krah et al., Reference Krah, Miotke, Li, Patel, Bowen, Pomicter and Patel2023; Panovska-Stavridis et al., Reference Panovska-Stavridis, Eftimov, Ivanovski, Pivkova-Veljanovska, Cevreska, Hermouet and Dimovski2016) and kinase (Kapralova et al., Reference Kapralova, Horvathova, Pecquet, Fialova Kucerova, Pospisilova, Leroy, Kralova, Milosevic Feenstra, Schischlik, Kralovics, Constantinescu and Divoky2016; Maaziz et al., Reference Maaziz, Garrec, Airaud, Bobée, Contentin, Cayssials, Rimbert, Aral, Bézieau, Gardie and Girodon2023; Tun et al., Reference Tun, Buka, Graham and Dyer2022) domains of JAK2, regions essential for modulating JAK-STAT signaling. We can also observe a plethora of literature associating the JAK2 Pseudokinase (Bahar et al., Reference Bahar, Barton and Kini2016; Bourrienne et al., Reference Bourrienne, Loyau, Faille, Gay, Akhenak, Farkh, Ollivier, Solonomenjanahary, Dupont, Choqueux, Villeval, Plo, Edmond, Ho-Tin-Noé, Ajzenberg and Mazighi2024; Carreño-Tarragona et al., Reference Carreño-Tarragona, Varghese, Sebastián, Gálvez, Marín-Sánchez, López-Muñoz, Nam-Cha, Martínez-López, Constantinescu, Sevilla and Ayala2021; Choi et al., Reference Choi, Messali, Uda, Abu-Zeinah, Kermani, Yabut, Lischer, Castillo Tokumori, Erdos, Lehmann, Sobas, Rao and Scandura2024; Gupta, Varma, Kumar et al., Reference Gupta, Varma, Kumar, Naseem, Sachdeva, Sreedharanunni, Binota, Bose, Khadwal, Malhotra and Varma2023; Gupta, Varma, Sreedharanunni et al., Reference Gupta, Varma, Sreedharanunni, Abdulkadir, Naseem, Sachdeva, Binota, Bose, Malhotra, Khadwal and Varma2023; Hassan et al., Reference Hassan, Abdellateif, Radwan, Hameed, Desouky, Kamel and Gameel2022; Krah et al., Reference Krah, Miotke, Li, Patel, Bowen, Pomicter and Patel2023; Lin et al., Reference Lin, Nebral, Gertzen, Ganmore, Haas, Bhatia, Fischer, Kuhlen, Gohlke, Izraeli, Trka, Hu, Borkhardt, Hauer and Auer2019; Mambet et al., Reference Mambet, Babosova, Defour, Leroy, Necula, Stanca, Tatic, Berbec, Coriu, Belickova, Kralova, Lanikova, Vesela, Pecquet, Saussoy, Havelange, Diaconu, Divoky and Constantinescu2018; Pace et al., Reference Pace, Guadagno, Russo, Gencarelli, Carlea, Di Spiezio, Bertuzzi, Mascolo, Grimaldi and Insabato2023; Patchell et al., Reference Patchell, Keohane, O’Shea and Langabeer2024; Puli’uvea et al., Reference Puli’uvea, Immanuel, Green, Tsai, Shepherd and Kalev-Zylinska2024; Roncero et al., Reference Roncero, López-Nieva, Cobos-Fernández, Villa-Morales, González-Sánchez, López-Lorenzo, Llamas, Ayuso, Rodríguez-Pinilla, Arriba, Piris, Fernández-Navarro, Fernández, Fraga, Santos and Fernández-Piqueras2016; Schulze et al., Reference Schulze, Stengel, Jaekel, Wang, Franke, Roskos, Schneider, Niederwieser and Al-Ali2019; Skoczen et al., Reference Skoczen, Stepien, Mlynarski, Centkowski, Kwiecinska, Korostynski, Piechota, Wyrobek, Moryl-Bujakowska, Strojny, Rej, Kowalczyk and Balwierz2020; Veitia & Innan, Reference Veitia and Innan2022; R. Z. Xu et al., Reference Xu, Karsan, Xu and Berry2022; Yongchao Zhang et al., Reference Zhang, Zhao, Liu, Zhang and Zhang2024) and kinase (Benton et al., Reference Benton, Boddu, DiNardo, Bose, Wang, Assi, Pemmaraju, Kc, Pierce, Patel, Konopleva, Ravandi, Garcia-Manero, Kadia, Cortes, Kantarjian, Andreeff and Verstovsek2019; Kapralova et al., Reference Kapralova, Horvathova, Pecquet, Fialova Kucerova, Pospisilova, Leroy, Kralova, Milosevic Feenstra, Schischlik, Kralovics, Constantinescu and Divoky2016; Mambet et al., Reference Mambet, Babosova, Defour, Leroy, Necula, Stanca, Tatic, Berbec, Coriu, Belickova, Kralova, Lanikova, Vesela, Pecquet, Saussoy, Havelange, Diaconu, Divoky and Constantinescu2018; Schulze et al., Reference Schulze, Stengel, Jaekel, Wang, Franke, Roskos, Schneider, Niederwieser and Al-Ali2019) domains to cancer. Similarly, cancer-related mutations were frequently observed in the Pseudokinase domain of JAK3 (Agarwal et al., Reference Agarwal, MacKenzie, Eide, Davare, Watanabe-Smith, Tognon, Mongoue-Tchokote, Park, Braziel, Tyner and Druker2015; Bergmann et al., Reference Bergmann, Schneppenheim, Seifert, Betts, Haake, Lopez, Maria Murga Penas, Vater, Jayne, Dyer, Schrappe, Dührsen, Ammerpohl, Russell, Küppers, Dürig and Siebert2014; Bouchekioua et al., Reference Bouchekioua, Scourzic, de Wever, Zhang, Cervera, Aline-Fardin, Mercher, Gaulard, Nyga, Jeziorowska, Douay, Vainchenker, Louache, Gespach, Solary and Coppo2014; de Martino et al., Reference de Martino, Gigante, Cormio, Prattichizzo, Cavalcanti, Gigante, Ariano, Netti, Montemurno, Mancini, Battaglia, Gesualdo, Carrieri and Ranieri2013; Ehrentraut et al., Reference Ehrentraut, Schneider, Nagel, Pommerenke, Quentmeier, Geffers, Feist, Kaufmann, Meyer, Kadin, Drexler and MacLeod2016; Koo et al., Reference Koo, Tan, Tang, Poon, Allen, Tan, Chong, Ong, Tay, Tao, Quek, Loong, Yeoh, Yap, Lee, Lim, Tan, Goh, Cutcutache and Lim2012; Rivera-Munoz et al., Reference Rivera-Munoz, Laurent, Siret, Lopez, Ignacimouttou, Cornejo, Bawa, Rameau, Bernard, Dessen, Gilliland, Mercher and Malinge2018; Sato et al., Reference Sato, Toki, Kanezaki, Xu, Terui, Kanegane, Miura, Adachi, Migita, Morinaga, Nakano, Endo, Kojima, Kiyoi, Mano and Ito2008; Sim et al., Reference Sim, Kim, Kim, Jeon, Nam, Ahn, Keam, Park, Kim, Kim and Heo2017; L. Xu et al., Reference Xu, Wilson, Laetsch, Oliver, Spunt, Hawkins and Skapek2016), reinforcing its role in malignant transformation. Infectious diseases, on the other hand, appeared to be linked to mutations in the FERM domain of JAK3 (Zhong et al., Reference Zhong, Wang, Ma, Gou, Tang and Song2017), the DNA binding domain of STAT1 (Uzel et al., Reference Uzel, Sampaio, Lawrence, Hsu, Hackett, Dorsey, Noel, Verbsky, Freeman, Janssen, Bonilla, Pechacek, Chandrasekaran, Browne, Agharahimi, Gharib, Mannurita, Yim, Gambineri and Holland2013), and all over TYK2 (Kerner et al., Reference Kerner, Laval, Patin, Boisson-Dupuis, Abel, Casanova and Quintana-Murci2021, Reference Kerner, Ramirez-Alejo, Seeleuthner, Yang, Ogishi, Cobat, Patin, Quintana-Murci, Boisson-Dupuis, Casanova and Abel2019; Ogishi et al., Reference Ogishi, Arias, Yang, Han, Zhang, Rinchai, Halpern, Mulwa, Keating, Chrabieh, Lainé, Seeleuthner, Ramírez-Alejo, Nekooie-Marnany, Guennoun, Muller-Fleckenstein, Fleckenstein, Kilic, Minegishi and Boisson-Dupuis2022), which play a crucial role in cytokine receptor binding and immune response regulation. Interestingly, mutations were associated with autoimmunity (Li et al., Reference Li, Gakovic, Ragimbeau, Eloranta, Rönnblom, Michel and Pellegrini2013; López-Isac et al., Reference López-Isac, Campillo-Davo, Bossini-Castillo, Guerra, Assassi, Simeón, Carreira, Ortego-Centeno, García de la Peña, Beretta, Santaniello, Bellocchi, Lunardi, Moroncini, Gabrielli, Riemekasten, Witte, Hunzelmann and Martín2016; Motegi et al., Reference Motegi, Kochi, Matsuda, Kubo, Yamamoto and Momozawa2019) and protective against autoimmunity (Diogo et al., Reference Diogo, Bastarache, Liao, Graham, Fulton, Greenberg, Eyre, Bowes, Cui, Lee, Pappas, Kremer, Barton, Coenen, Franke, Kiemeney, Mariette, Richard-Miceli, Canhão and Plenge2015; Enerbäck et al., Reference Enerbäck, Sandin, Lambert, Zawistowski, Stuart, Verma, Tsoi, Nair, Johnston and Elder2018; Jensen et al., Reference Jensen, Attfield, Feldmann and Fugger2023; Motegi et al., Reference Motegi, Kochi, Matsuda, Kubo, Yamamoto and Momozawa2019) in TYK2, suggesting that variations in these regions may provide resilience against autoimmune diseases, which is an ongoing field of study (Molitor et al., Reference Molitor, Hayashi, Lin, Dunn, Peterson, Poston, Kurnellas, Traver, Patel, Akgungor, Leonardi, Lewis, Segales, Bennett, Truong, Dani, Naphade, Wong, McDermott and Rassoulpour2025; Syed et al., Reference Syed, Ballew, Lee, Rana, Krishnan, Castela, Weaver, Chalasani, Thomaidou, Demine, Chang, Coomans de Brachène, Alvelos, Vazquez, Marselli, Orr, Felton, Liu, Kaddis and Evans-Molina2025).
Examining the Relationship Between Structural Alterations in JAK-STAT Proteins and the Pathogenic Potential of Mutations
Next, we investigated the protein structure using the AlphaFold3 AI-predicted model to determine where the mutations impact structural alterations. We observed that disease-associated SNPs were more frequently found in linker regions connecting secondary structural elements, such as between alpha helices and beta sheets. When these mutations occurred within an alpha helix or beta sheet, they were predominantly located at the boundary of the structure, with only rare occurrences in the middle (Figure 2, Figure 3). This suggests that mutations in transition regions may have a more significant impact on protein dynamics and function, potentially altering folding, stability, or interactions with other molecules. In contrast, benign mutations were more often embedded within well-defined secondary structures, but rarely in linker regions. Furthermore, we observed that disease-associated mutations were predominantly located within the interior of the protein’s 3D structure, suggesting that these variants may impact structural integrity or disrupt key protein-protein interactions. Conversely, benign mutations were more commonly found on surface-exposed regions of the protein (i.e., an amino acid that faces the outside of the protein) (Suppl. Fig. 3).
Structural analysis of disease-associated and ClinVar benign missense variants in the STAT proteins. The panel illustrates the secondary structure localization of disease-associated (left) and benign/likely benign (right) mutations mapped onto AlphaFold predicted protein structures.

Structural analysis of disease-associated and ClinVar benign missense variants in the JAK proteins. The panel illustrates the secondary structure localization of disease-associated (left) and benign/likely benign (right) mutations mapped onto AlphaFold predicted protein structures.

Figure 3 Long description
Panel A: A diagram of JAK2 protein structure showing disease-associated variants. The diagram includes labels for specific amino acid changes such as Asn1108Ser, Arg1063His, Ile724Thr, Glu846Asp, Val617Phe/Ile, Arg564Leu, Arg683Gly/Ser, and Gly571Ser. Panel B: A diagram of JAK2 protein structure showing ClinVar benign/likely benign variants. The diagram includes labels for specific amino acid changes such as Leu892Val, Cys480Phe, Leu113Val, Asn337Asp, Val392Met, Lys244Arg, and Gly127Asp. Panel C: A diagram of JAK3 protein structure showing disease-associated variants. The diagram includes labels for specific amino acid changes such as Arg925Ser, Arg840Cys, Ala573Val, Met511Ile, Ala572Val, Pro132Thr, Arg222His, Val722Ile, and Arg657Gln. Panel D: A diagram of JAK3 protein structure showing ClinVar benign/likely benign variants. The diagram includes labels for specific amino acid changes such as Val217Met, Arg121His, Pro396Leu, Ile1003Val, Arg451Gln, His897Arg, and Ala1090Thr. Panel E: A diagram of TYK2 protein structure showing disease-associated variants. The diagram includes labels for specific amino acid changes such as Arg231Trp, Ala928Val, Val362Phe, Gly634Glu, Pro1104Ala, and Gly761Val. Panel F: A diagram of TYK2 protein structure showing ClinVar benign/likely benign variants. The diagram includes labels for specific amino acid changes such as Val15Ala, Ala53Thr, Pro820His, Gly744Trp, Arg719His, His993Tyr, Ala928Val, Glu1163Gly, Val362Phe, and Pro1104Ala.
Conserved Amino Acid Patterns in Proximity to Disease-Associated and ClinVar Benign Variants
We further compared amino acid patterns surrounding the ClinVar benign and disease-associated mutations (three amino acids upstream and three amino acids downstream of the mutated site (see Materials and Methods, Figure 4). This comparative analysis of amino acid patterns in benign and disease-associated variants revealed distinct compositional differences across disease-associated and ClinVar benign variants. In the single-residue analysis (top panel), certain amino acids, such as valine (Val), glutamic acid (Glu), and methionine (Met), exhibited higher frequencies in proximity to disease-associated variants compared to benign variants, while others, such as serine (Ser), alanine (Ala), tyrosine (Tyr), and arginine (Arg), appeared more frequently in benign variants.
Comparative analysis of amino acid patterns (one amino acid, two amino acid combinations, and three amino acid combinations out of three upstream and three downstream of the variant in All of Us or COSMIC) in benign and disease-associated variants. Amino acid compositions for benign variants are blue, and disease variants are red.

At the dipeptide level (second panel), several combinations were more prominent in proximity to disease-associated mutations such as (1) leucine and glutamic acid (Leu+Glu), (2) leucine and leucine (Leu+Leu), (3) aspartic acid and leucine (Asp, Leu), and (4) arginine and glutamic acid (Arg+Glu). Some combinations were uniquely present close to disease-associated variants: (1) phenylalanine and methionine (Phe+Met), (2) arginine and valine (Arg+Val), and (3) glutamic acid and methionine (Glu+Met).
Expanding to combinations of three amino acids out of six surrounding amino acids (third panel), we observed some combinations that are uniquely in proximity to disease-associated variants: (1) leucine, serine, leucine (Leu+Ser+Leu), (2) aspartic acid, leucine, leucine (Asp+Leu+Leu), (3) aspartic acid, serine, leucine (Asp, Ser, Leu), (4) leucine, leucine, glutamic acid (Leu+Leu+Glu), (5) leucine, isoleucine, glutamic acid (Leu+Ile+Glu), and (6) leucine, glutamic acid, aspartic acid (Leu+Glu+Asp). On the other hand, we detected the following combinations only close to benign variants (1) lysine, proline, glycine (Lys+Pro+Gly), (2) glycine, serine, tyrosine (Gly+Ser+Tyr), (3) arginine, arginine, arginine (Arg+Arg+Arg), and (4) arginine, threonine, arginine (Arg+Thr+Arg).
The observed differences in amino acid compositions surrounding disease-associated and benign variants suggest underlying structural and functional constraints that contribute to pathogenicity. A key trend is the enrichment of hydrophobic residues, particularly leucine (Leu), isoleucine (Ile), methionine (Met), and phenylalanine (Phe), in proximity to disease-associated variants. The frequent occurrence of Leu+Leu, Leu+Glu, and Leu+Glu+Asp combinations suggests that these mutations often occur within hydrophobic cores, where they may disrupt protein stability or alter packing interactions. Similarly, the presence of methionine (Met) in disease-associated motifs, such as Glu+Met and Phe+Met, points to potential disruptions in hydrophobic regions, particularly in proteins involved in enzymatic activity or membrane function. Conversely, benign variants appear to favor polar and flexible residues, with an overrepresentation of serine (Ser), glycine (Gly), and threonine (Thr). The exclusive presence of Gly+Ser+Tyr and Lys+Pro+Gly in benign variants suggests that these substitutions predominantly occur in solvent-exposed loops or linker regions, where flexibility and structural adaptability mitigate the effects of mutation. Additionally, the recurrent occurrence of arginine-rich motifs (Arg+Arg+Arg and Arg+Thr+Arg) in benign variants indicates that these substitutions are likely involved in electrostatic interactions or protein-protein binding sites that can accommodate mutational changes without significant functional consequences. A second striking pattern is the enrichment of negatively charged residues (glutamic acid, Glu, and aspartic acid, Asp) near disease-associated variants, suggesting potential disruptions in salt-bridge interactions and protein stability. The presence of Arg+Glu, Asp+Leu+Leu, and Leu+Glu+Asp in disease-associated variants indicates that these mutations may destabilize electrostatic interactions or affect protein folding. In contrast, the benign variants tend to retain positively charged arginine (Arg), which is commonly involved in stabilizing protein structures or mediating protein-protein interactions. Taken together, these findings suggest that disease-associated variants frequently occur in structurally constrained regions, where mutations disrupt core hydrophobic interactions, electrostatic balance, or functional interfaces. In contrast, benign variants are more likely to appear in flexible or surface-exposed regions, where mutations are better tolerated due to the preservation of local structural dynamics. The distinct differences in amino acid preferences between disease and benign variants provide insights into the physicochemical constraints that contribute to pathogenicity and may aid in refining predictive models for variant classification.
Analyzing the Nucleotide Sequence Near Benign and Disease-Associated Variants
To explore the genomic context of disease-associated and ClinVar benign mutations, we analyzed the nucleotide sequences surrounding these variants, focusing on a 20 bp region around each mutation. The first part of our investigation aimed to identify patterns of nucleotide sequences and assess whether specific sequences are preferentially found near disease-associated or benign mutations (Figure 5, Suppl. Fig. 4). Our analysis identified nucleotide sequences of 625 restriction enzyme sites out of a total of 1,088 within the examined regions. Notably, several nucleotide sequences were exclusively present near disease-associated mutations but absent in ClinVar benign variants. These included the nucleotide sequences of DpnI, Asi256I, DpnII, MalI, Lcr047I, NdeII, Bsp143I, Sau3AI, FaiI, MspJI, and BssMI. We found that the nucleotide sequence ‘GATC’ was predominantly present near disease-associated mutations but was entirely absent in the vicinity of ClinVar benign mutations.
Enzyme restriction site analysis in proximity to disease-associated and ClinVar benign variants in JAK and STAT genes. (a,b) The top 25 restriction enzymes identified near disease-associated and benign variants, respectively. (c) Venn diagram illustrating the overlap of restriction sites found near disease-associated variants (red) and ClinVar benign variants (blue).

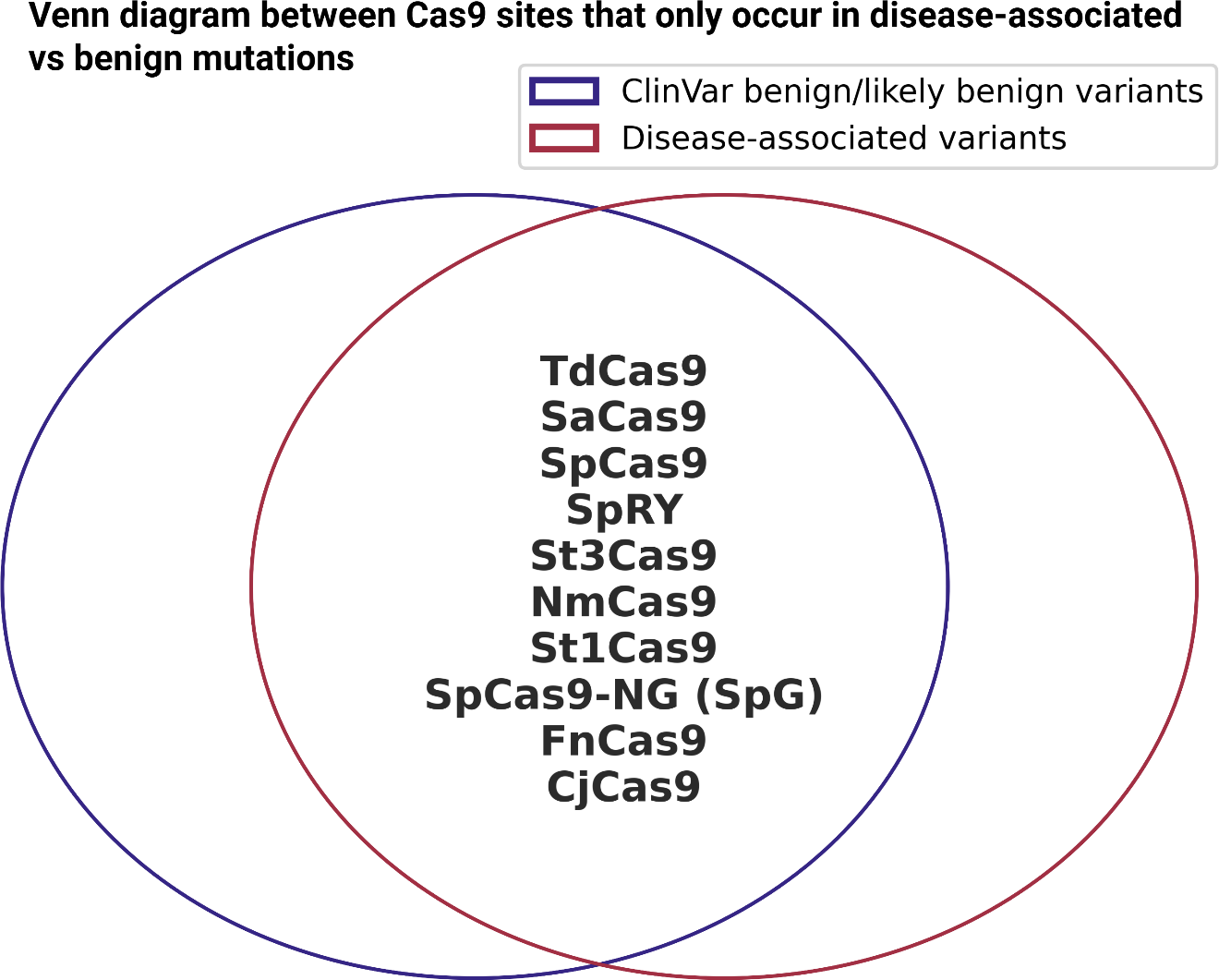
The second part of this analysis is to assess whether multiple Cas9 enzymes preferentially target sequences near disease-associated or ClinVar benign mutations; we analyzed the presence of Cas9 recognition sites within a 20 bp region surrounding these mutations (Figure 6, Suppl. Fig. 5). We examined a range of Cas9 enzymes, including SpCas9, SaCas9, NmCas9, St1Cas9, St3Cas9, CjCas9, FnCas9, TdCas9, xCas9, SpCas9-NG (SpG), SpRY, HiFi Cas9 (HF1), eSpCas9 (1.1), and HypaCas9 that recognized different PAM sequences (Suppl. Table 2) (Acharya et al., Reference Acharya, Ansari, Kumar Das, Hirano, Aich, Rauthan, Mahato, Maddileti, Sarkar, Kumar, Phutela, Gulati, Rahman, Goel, Afzal, Paul, Agrawal, Pulimamidi, Jalali and Chakraborty2024; Du et al., Reference Du, Zhu, Qian, Xue, Zheng and Huang2023; Guo et al., Reference Guo, Ren, Zhu, Tang, Wang, Zhang and Huang2019; Hibshman et al., Reference Hibshman, Bravo, Hooper, Dangerfield, Zhang, Finkelstein, Johnson and Taylor2024; Hou et al., Reference Hou, Zhang, Propson, Howden, Chu, Sontheimer and Thomson2013; Ikeda et al., Reference Ikeda, Fujii, Sugiura and Naito2019; H. K. Kim et al., Reference Kim, Lee, Kim, Park, Min, Choi, Huang, Yoon, Liu and Kim2020; Liang et al., Reference Liang, Zhang, Li, Yang, Fei, Liu and Qin2022; Müller et al., Reference Müller, Lee, Gasiunas, Davis, Cradick, Siksnys, Bao, Cathomen and Mussolino2016; Schmidheini et al., Reference Schmidheini, Mathis, Marquart, Rothgangl, Kissling, Böck, Chanez, Wang, Jinek and Schwank2024; Slaymaker et al., Reference Slaymaker, Gao, Zetsche, Scott, Yan and Zhang2016; Vakulskas et al., Reference Vakulskas, Dever, Rettig, Turk, Jacobi, Collingwood, Bode, McNeill, Yan, Camarena, Lee, Park, Wiebking, Bak, Gomez-Ospina, Pavel-Dinu, Sun, Bao, Porteus and Behlke2018; Wu et al., Reference Wu, Tang and Tang2020; Yifei Zhang et al., Reference Zhang, Zhang, Xu, Wang, Chen, Wang, Wu, Tang, Wang, Zhao, Gan and Ji2020). Our analysis did not reveal a strong preference for any Cas9 protein’s cut site being predominantly located near disease-associated mutations, but absent in ClinVar benign mutations. These findings suggest that while CRISPR/Cas9 target sites are present around the variants, there is no clear enrichment in disease-associated sites that would indicate preferential targetability.
CRISPR cut site analysis in proximity to disease-associated and benign mutations. Venn diagram illustrating the overlap of Cas9 cut sites uniquely occurring in either disease-associated (red) or benign (blue) mutations.

Discussion
This study provides a comprehensive analysis of missense mutations in the JAK-STAT pathway, highlighting differences between disease-associated and ClinVar benign variants regarding structural localization, biochemical properties, and genomic context. By integrating mutation data from the All of Us and COSMIC databases, we identified properties that may help explain why specific variants contribute to disease while others remain functionally neutral. Our findings suggest that disease-associated mutations frequently disrupt core hydrophobic interactions, electrostatic balance, or functional interfaces, whereas benign variants are more commonly found within secondary structures like helices and sheets and also in surface-exposed regions, where structural constraints are less restrictive. These observations reinforce the notion that pathogenicity is not merely a function of amino acid substitution alone but is heavily influenced by protein architecture and nearby context on the nucleotide and amino acid level.
Our current understanding of disease-associated mutations is largely based on the analysis of individual SNPs, whereas real-world genetic variation often involves combinations of mutations that may interact in yet unknown ways. The lack of experimental data on epistatic interactions is a major limitation (Suppl. Text 1: Limitations and considerations) in mutation interpretation, as the functional impact of a given variant may depend on the presence of additional mutations within the same gene or pathway (Blumenthal et al., Reference Blumenthal, Baumbach, Hoffmann, Kacprowski and List2020; Hernández-Lorenzo et al., Reference Hernández-Lorenzo, Hoffmann, Scheibling, List, Matías-Guiu and Ayala2022; Hoffmann, Poschenrieder et al., Reference Hoffmann, Poschenrieder, Incudini, Baier, Fritz, Maier, Hartung, Hoffmann, Trummer, Adamowicz, Picciani, Scheibling, Harl, Lesch, Frey, Kayser, Wissenberg, Schwartz, Hafner and Blumenthal2024). While our stringent inclusion criteria (minimum 20 occurrences due to All of Us policy) allowed for robust analysis of more prevalent variants, it is important to acknowledge that rare yet highly pathogenic mutations, especially those associated with rare inherited disorders, might not be captured by this approach.
The GATC sequence is well known in bacteria as a recognition site for DNA adenine methyltransferase (Dam) and plays key roles in DNA repair, replication timing, and gene regulation (Flusberg et al., Reference Flusberg, Webster, Lee, Travers, Olivares, Clark, Korlach and Turner2010). In E. coli, for example, GATC sites guide mismatch repair machinery to correct errors on the newly synthesized DNA strand (Horton et al., Reference Horton, Zhang, Blumenthal and Cheng2015). Although human cells do not use the same bacterial repair system, recent work shows that sequence context, including short motifs like GATC, can influence where replication errors occur and how repair systems handle them (Hasenauer et al., Reference Hasenauer, Barreto, Lotton and Matic2025). GATC-like sequences can also be recognized by certain transcription factors or occur in open chromatin regions, meaning changes nearby could alter gene expression (Mardenborough et al., Reference Mardenborough, Nitsenko, Laffeber, Duboc, Sahin, Quessada-Vial, Winterwerp, Sixma, Kanaar, Friedhoff, Strick and Lebbink2019).
Future work could explore whether selected missense variants influence STAT dimer formation by combining state-aware structural hypotheses with independent computational estimates of interface perturbation and focused experimental validation, recognizing that dimerization is conformation-dependent and not fully captured by static structural predictions alone.
Understanding genetic variation requires considering not only individual mutations but also their broader structural and functional context. As data and experimental tools improve, distinguishing ClinVar benign from disease-associated variants will become more precise. Addressing gaps such as epistatic interactions and underreported mutations will clarify how JAK-STAT variants affect health. Integrating computational models of protein–protein interactions with high-throughput experiments could systematically investigate multi-variant and epistatic effects. In vivo studies introducing predicted single or combined SNPs into homozygous mouse lines, then crossbreeding and applying RNA-seq, ChIP-seq, and allergenic challenge assays (Gad, Reference Gad1994), could reveal how complex mutation patterns shape pathway function. Clinical sample analysis will be critical to assess whether variants labeled as benign may contribute to disease in certain contexts, with our curated references offering a starting point for prioritization. Examining noncoding regulatory regions for pathogenic mutations may uncover additional mechanisms, but current datasets (All of Us, COSMIC) lack matched transcriptomic or epigenomic data, and patient samples were unavailable for this study. Future integration of genomic variation with matched expression and chromatin profiles will be essential for linking regulatory mutations to function. Combining large-scale genomics with advanced experimental systems offers a path to further investigate JAK-STAT variation and translate these insights into future therapeutic advances.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/thg.2026.10054.
Data availability
This study used data from the All of Us Research Program’s controlled Tier Dataset v.7, available to authorized users on the Researcher Workbench (https://databrowser.researchallofus.org/). Data from COSMIC v100 is available at (https://cancer.sanger.ac.uk/cosmic). The Python code can be found at: https://github.com/Firestar93/JAKSTAT_missenseSNPs_properties. Intermediate results and files can be found at: https://doi.org/10.6084/m9.figshare.28597121.
Acknowledgments
The authors want to thank Jakub Jankowski, Lothar Hennighausen, Priscilla A. Furth, and the members of the Laboratory of Cell & Molecular Biology (LCMB), NIDDK, NIH, for their valuable input. The figures were created with Biorender.com. Parts of the figures include icons from Flaticon.com under a paid license. The text was partly rephrased using ChatGPT version 4, Grammarly, and scite.ai under a paid license. Paperpile, under a paid license, was used to collect references in the correct format. We gratefully acknowledge All of Us participants for their contributions, without whom this research would not have been possible. We also thank the National Institutes of Health’s All of Us Research Program for making available the participant data, samples, and cohort examined in this study.
Author contributions
M.H. planned the project, executed the analysis, and wrote the manuscript. H.K. conceptualized the project and revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This research was supported by the Intramural Research Programs (IRPs) of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) within the National Institutes of Health (NIH). The contributions of the NIH author(s) were made as part of their official duties as NIH federal employees, are in compliance with agency policy requirements, and are considered Works of the United States Government. However, the findings and conclusions presented in this paper are those of the author(s) and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services. Funding support is in part from Georgetown University Medical Center.
Competing interests
The authors declare no competing interests.








 Open access
Open access