


Introduction
The marine subsurface is a large microbial habitat, where the majority of biomass exists along continental margins (Kallmeyer et al., Reference Kallmeyer, Pockalny, Adhikari, Smith and D’Hondt2012) and microbial biomass tends to correlate with total organic carbon (Lipp et al., Reference Lipp, Morono, Inagaki and Hinrichs2008). Along the Peru Margin on Ocean Drilling Program Leg 201, cells reached abundances of up to 109 cells g–1 sediment and were particularly abundant in areas where geochemical gradients exist that can be used for microbial metabolism, such as sulfate/methane transition zones (SMTZs) (D’Hondt et al., Reference D’Hondt2004). Novel microbial lineages were discovered at the Peru Margin, which was the first location where metagenomics was used to study microbial populations (Biddle et al., Reference Biddle2006, Reference Biddle, Fitz-Gibbon, Schuster, Brenchley and House2008). Along the Costa Rica Margin, sampled during the Integrated Ocean Drilling Program (IODP) Expedition 338, novel lineages were also discovered using metagenomics (Farag et al., Reference Farag, Biddle, Zhao, Martino, House and León-Zayas2020, Reference Farag, Zhao and Biddle2021; Zhao and Biddle, Reference Zhao and Biddle2021). Among these novel lineages were multiple new branches of archaea, including the Asgard archaea within the lineages of Candidatus Lokiarchaeia (previously Lokiarchaeota) and Sifarchaeia (previously Sifarchaeota) (Farag et al., Reference Farag, Biddle, Zhao, Martino, House and León-Zayas2020, Reference Farag, Zhao and Biddle2021; Zhao and Biddle Reference Zhao and Biddle2021). Considering the rate at which novel lineages were discovered through the application of metagenomics methods, the deep subsurface of continental margins is an intriguing place to continue to examine microbial diversity via this method.
Advances in high-throughput sequencing have increased our ability to use metagenomic analyses to investigate the deep biosphere, although the low recovery of quality DNA from deep sediments creates difficulties. Some previous studies have had to rely on amplified DNA for sequencing (Biddle et al., Reference Biddle, Fitz-Gibbon, Schuster, Brenchley and House2008), while others have worked with only non-amplified DNA (Farag et al., Reference Farag, Biddle, Zhao, Martino, House and León-Zayas2020, Reference Farag, Zhao and Biddle2021; Zhao and Biddle, Reference Zhao and Biddle2021). In the Peru Margin study, amplification was performed using phi29-based multiple displacement amplification, which is not necessarily robust in terms of representational bias, and an internal control of amplified to non-amplified DNA was used (Biddle et al., Reference Biddle, Fitz-Gibbon, Schuster, Brenchley and House2008). More modern efforts in amplification have included the use of PCR, which tends to be less sensitive to DNA quality and more accurate in data representation (Arneson et al., Reference Arneson, Hughes, Houlston and Done2008; Martino et al., Reference Martino, Rhodes, Biddle, Brandt, Tomsho and House2012). PCR-based DNA amplification would allow for more accurate metagenomic analysis and should be considered when working with deep sediments that commonly have poor extraction yields and questionable DNA quality.
The IODP Expedition 339 to the Iberian Margin was an exciting opportunity to investigate the deep subsurface microbiome of a new continental margin. IODP site U1385 sits at 2578 m below sea level and is an important paleoclimatology location (Hodell et al., Reference Hodell, Lourens, Stow, Hernández-Molina and Alvarez Zarikian2013). Because of the consistent hemipelagic sediment depositional history at this site, with sedimentation rates averaging 10 cm ky–1, and excellent preservation, this sediment was used to correlate δ18O isotopes in Greenland ice cores with that of fossilised planktonic foraminifera in order to refine ice-volume estimates during millennial-scale events throughout the Pleistocene (de Abreu et al., Reference de Abreu, Shackleton, Schönfeld, Hall and Chapman2003; Abrantes et al., Reference Abrantes, Hodell, Carrara, Batista and Duarte2010). Although this site is well-preserved for climate history, the geochemical profile shows that microbial processing of hydrocarbons and sulfate should occur (Scientists, 2013), with sulfate completely removed from the sediment porewater by 50 m below seafloor (mbsf). At this depth, methane is also removed and indicates a subsurface SMTZ known to be an active growth zone for subsurface microbial life (Knab et al., Reference Knab, Cragg, Hornibrook, Holmkvist, Pancost, Borowski, Parkes and Jørgensen2009; Sørensen and Teske, Reference Sørensen and Teske2006; D’Hondt et al., Reference D’Hondt2004). Studies of sulfur and oxygen isotopes support a low rate of microbial sulfate reduction in the upper 7 m of sediment at this site, estimated to be ~6.6 × 10–9 mol cm–3 y–1, including a significant amount of intracellular sulfur reoxidation (Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016).
For this study, sediments from U1385 were examined using metagenomics methods to uncover subsurface microbial signatures. We found that nucleic acid recovery from this environment was challenging, and as such, utilised a PCR-based amplification method to amplify and sequence whole metagenomic DNA. From the analysis of the full metagenome and metagenome-assembled genomes (MAGs), details regarding microbial metabolism and taxonomy related to geochemical niches are presented.
Materials and methods
Samples of nannofossil-rich muds and clays, with varying proportions of biogenic carbonate and terrigenous sediment (Scientists, 2013), were collected from Site U1385A during the IODP Expedition 339 via the vessel the R/V JOIDES Resolution (37°34.0128′N, 10°7.6580′W; Scientists, 2013). Geochemistry was measured by standard IODP protocols and was retrieved from shipboard data (Scientists, 2013; Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016). Methane was measured by gas chromatography, sulfate was measured by ion chromatography, ammonium was measured by spectrophotometry and iron(II) was measured by inductively coupled plasma atomic emission spectroscopy (Scientists, 2013). Total organic carbon was low (<1%) (Scientists, 2013; Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016).
Immediately on collection, sediments were frozen at –80°C and were later transported to the shore-based laboratory via dry ice. Samples remained frozen at –80°C until use. Samples were gently thawed, and the core liner and contaminating outside sediment were removed. Sediment was collected via sterile spatula and added to DNA extraction tubes.
DNA was extracted using the MO BIO PowerMax Soil DNA Isolation Kit (MO BIO Laboratories Inc.) with a minor adjustment to the manufacturer’s protocol. Briefly, 10 g of sediment was processed according to the manufacturer’s instructions, with an additional step of incubation in a 65°C water bath for 15 min prior to step 4 (10-min vortex). A blank extraction was run alongside the samples with distilled water (DI) as the sample. The DNA concentrations of extracts were: 10.5 mbsf, 1.77 ng μL–1; 29.5 mbsf, 0.46 ng μL–1; 48.5 mbsf, 0.23 ng μL–1; 67.5 mbsf, 0.23 ng μL–1; 86.5 mbsf, 0.18 ng μL–1; 123 mbsf, 0.16 ng μL–1; blank, 0 ng μL–1.
Because a small amount of DNA was extracted from the samples, DNA was amplified via the Genomeplex Whole Genome Amplification Kit (Sigma-Aldrich Inc.). This kit uses random primers to perform whole-genome amplification (WGA) in a robust manner (Arneson et al., Reference Arneson, Hughes, Houlston and Done2008). For WGA, 2 ng of DNA was added to each reaction and 20 cycles were run using the manufacturer’s suggestions for denaturing and annealing temperatures. Triplicate reactions were pooled and products were purified using the Gen-Elute PCR Clean-Up Kit (Sigma-Aldrich Inc.). Yields for each sample were: 10.5 mbsf, 1.7 μg; 29.5 mbsf, 1.4 μg; 48.5 mbsf, 0.9 μg; 67.5 mbsf, 0.53 μg; 86.5 mbsf, 0.36 μg; 123 mbsf, 0.195 μg; blank, 0.1 μg. Samples were sent for Illumina library construction and sequencing on an Illumina HiSeq2500 machine at the University of Delaware Sequencing and Genotyping Center in the Delaware Biotechnology Institute.
Reads were quality checked in the CLC-BIO Genomics Workbench (Qiagen). Poor-quality scores were detected over the first 30 bp of the majority of the reads as a result of an artifact of 30 bp identical nucleotides at the universal primer sites ligated to each read in the WGA step. Thus, the first 30 bp of each read was removed and assembly was performed on the resulting 120 bp paired-end reads. Sequences were assembled using IDBA-UD (Peng et al., Reference Peng, Leung, Yiu and Chin2012) up to 120 kmer length, run with the following flags called: --mink 40; --maxk 120; --step 20; --num_threads 18; --min_contig 300. Contigs were processed with PhyloSift (Darling et al., Reference Darling, Jospin, Lowe, Matsen, Bik and Eisen2014) for initial assessment of the taxonomic composition of the datasets. A high abundance of contigs in the blank and sediment samples was annotated as Delftia acidovorans, a species within the Betaproteobacteria that is a common contaminant of molecular biology reagents. This was deemed a contaminant and, as such, we removed reads associated with it. Reads were recruited from all datasets to the genome of Delftia acidovorans CCUG 15835 (NCBI Reference Sequence: NZ_KE150403.1) using FR-HIT (Niu et al., Reference Niu, Zhu, Fu, Wu and Li2011). Recruited reads were removed from the datasets using a custom perl script. These contaminant-cleaned metagenome data are deposited in the NCBI SRA Archive under BioProject PRJNA301144.
From the cleaned data, for each of the sampled depths, quality-controlled paired reads were assembled using SPAdes version 3.15.3 with default parameters, iterating through k-mers of 21, 29, 39, 59, 79 and 99 (Prjibelski et al., Reference Prjibelski, Antipov, Meleshko, Lapidus and Korobeynikov2020). The SPAdes assemblies were used to generate metabolic analyses for each depth. However, because metagenome binning using individual depth’s assembled SPADes contigs produced only few bins (0 to 3) at each depth, most with completeness percentages less than 40%, another assembly was generated by concatenating reads from all of the depths together, then using Megahit version 1.2.9, with presets meta-large and iterating through k-mers 27 and 117 in increments of 10 (Li et al., Reference Li, Liu, Luo, Sadakane and Lam2015). Assembled contigs were processed through bowtie2 version 2.5.1 (Langmead and Salzberg, Reference Langmead and Salzberg2012) and samtools version 0.1.19 (Li et al., Reference Li, Handsaker, Wysoker, Fennell, Ruan, Homer, Marth, Abecasis and Durbin2009) to generate mapped and sorted bam files, which were then used along with the assembled contigs as input to the metagenome binning tool Metabat2 (Kang et al., Reference Kang, Froula, Egan and Wang2015). Each metagenomic bin was then examined for completeness and contamination using CheckM version 1.2.2 (Parks et al., Reference Parks, Imelfort, Skennerton, Hugenholtz and Tyson2015). GTDB-Tk version 2.1.0 was used to assign taxonomic classifications to each of the bacterial and archaeal metagenome bins based on the Genome Taxonomy Database (GTDB) and names were updated based on release 10-RS226 (Chaumeil et al., Reference Chaumeil, Mussig, Hugenholtz and Parks2022). Efforts were made to reduce contamination levels in some of the metagenome bins to less than 10%. Vizbin (Laczny et al., Reference Laczny, Sternal, Plugaru, Gawron, Atashpendar, Margossian, Coronado, van der Maaten, Vlassis and Wilmes2015) was used to remove contamination, except for the Atribacterota bin. Binning was redone for this MAG using Semibin2 with default parameters (Pan et al., Reference Pan, Zhu, Zhao and Coelho2022) to reduce strain heterogeneity. In addition, taxonomic assignments, coverage data and GC content were used to exclude outlier contigs in this bin. Contamination levels using Semibin2 for all other bins were essentially unchanged. Taxonomy of genes inside each bin was predicted using usearch v11.0.667 (Edgar, Reference Edgar2010) using the UniRef100 database as of August 2022, and coverage data was produced by Metabat2 (Kang et al., Reference Kang, Froula, Egan and Wang2015).
Gene annotations and metabolic data for each depth sample and individual MAGs were generated using METABOLIC-G version 4.0, which includes CAZY and peptidase analyses (Zhou et al., Reference Zhou, Tran, Breister, Liu, Kieft, Cowley, Karaoz and Anantharaman2022). In addition, BlastKOALA (Kanehisa et al., Reference Kanehisa, Sato and Morishima2016) and the Kyoto Encyclopedia of Genes and Genomes website (https://www.kegg.jp/kegg/) were used to examine the completeness of metabolic pathways. Individual gene coverage information was determined by SPAdes k-mer coverage, summing all gene hits in METABOLIC-G for each depth and normalising each sample based on the number of reads in the deepest sample as the reference. Taxonomic classification of metabolic genes present in the metagenomes and MAGs was performed using usearch v11.0.667 (Edgar, Reference Edgar2010) against the BLAST-nr database, taking the first hit as the result. Additional coverage data were obtained using bbtools version 39.01 (BBMap, sourceforge.net/projects/bbmap/). 30S Ribosomal Protein S3 (rps3) sequences from the entire dataset and the MAGs were generated from annotation by Prokka version 1.14.5 (Seemann, Reference Seemann2014). The taxonomy of these sequences was then obtained via BLASTP searches of the non-redundant protein sequences database (nr) at NCBI (https://blast.ncbi.nlm.nih.gov). Coverage of each bin per depth was determined using gbtools (Seah and Gruber-Vodicka, Reference Seah and Gruber-Vodicka2015). Data including MAG sequences and metabolic analysis files are available at FigShare, https://doi.org/10.6084/m9.figshare.27633126.
Results
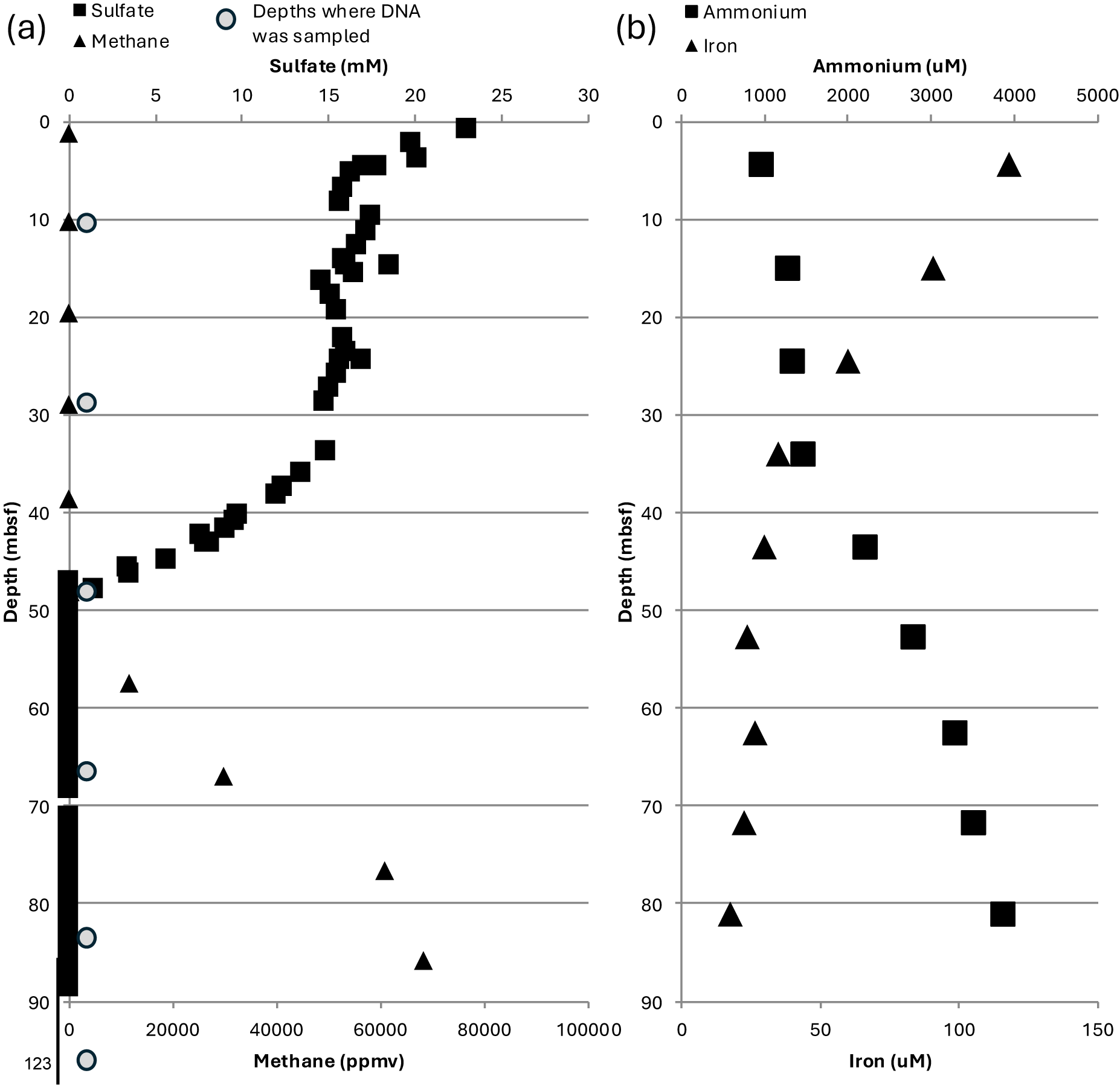
The samples from IODP Site U1385 came from depths that span zones of sulfate reduction, methane production, iron removal and ammonium buildup (Fig. 1). The SMTZ is traditionally considered a signature of microbial metabolism, whereas iron and ammonium shifts, while potentially microbially induced, can also be potential signatures of general diagenesis of organic matter. Considering the small amount of DNA retrieved from samples, WGA, having been deemed quantitatively robust (Arneson et al., Reference Arneson, Hughes, Houlston and Done2008), was used to increase the amount of DNA for sequencing. Therefore, although we treated the data as quantitative in a relative depth to depth manner for our study, the data cannot be viewed as comparably quantitative to samples from any other sites handled in a different manner.
Geochemistry and depths sampled for DNA at Site U1385. (a) Methane and ammonium increase with depth and (b) sulfate and iron shift with depth. Data from IODP Database (Scientists, 2013).

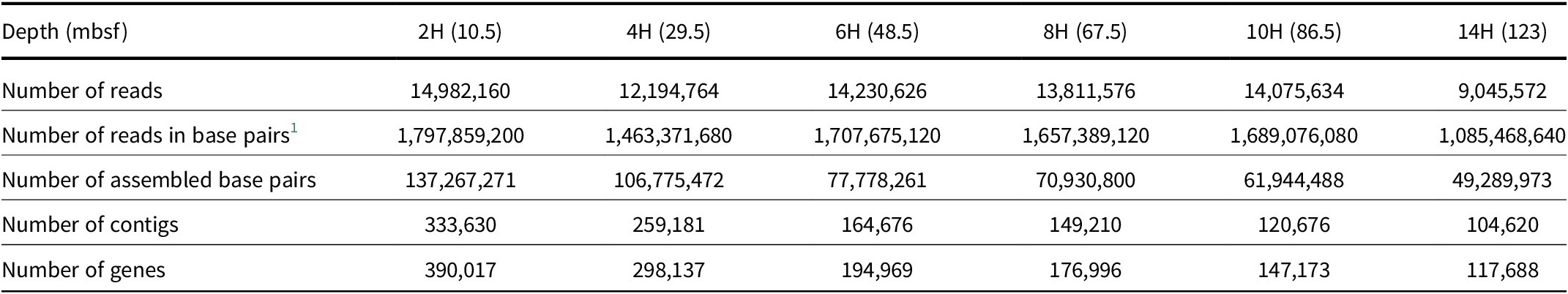
In general, the number of assembled contigs declined with depth, with the number of associated genes ranging from 390,017 at 10.5 mbsf to 117,688 at 123 mbsf (Table 1). The MAGs were binned using a co-assembly of all depths and six MAGs were created with completeness greater than 40% and contamination less than 8.5%. Because of the amount of data that did not bin into a MAG, we present the data as the bulk metagenome first, followed by individual MAG results. The MAGs in this study should be considered examples of a community metabolism, as they were assembled from multiple sediment depths.
Metagenome data summary

1 The number of reads in base pairs is based on 120 bp per read.
Complete metagenome: taxonomy by depth
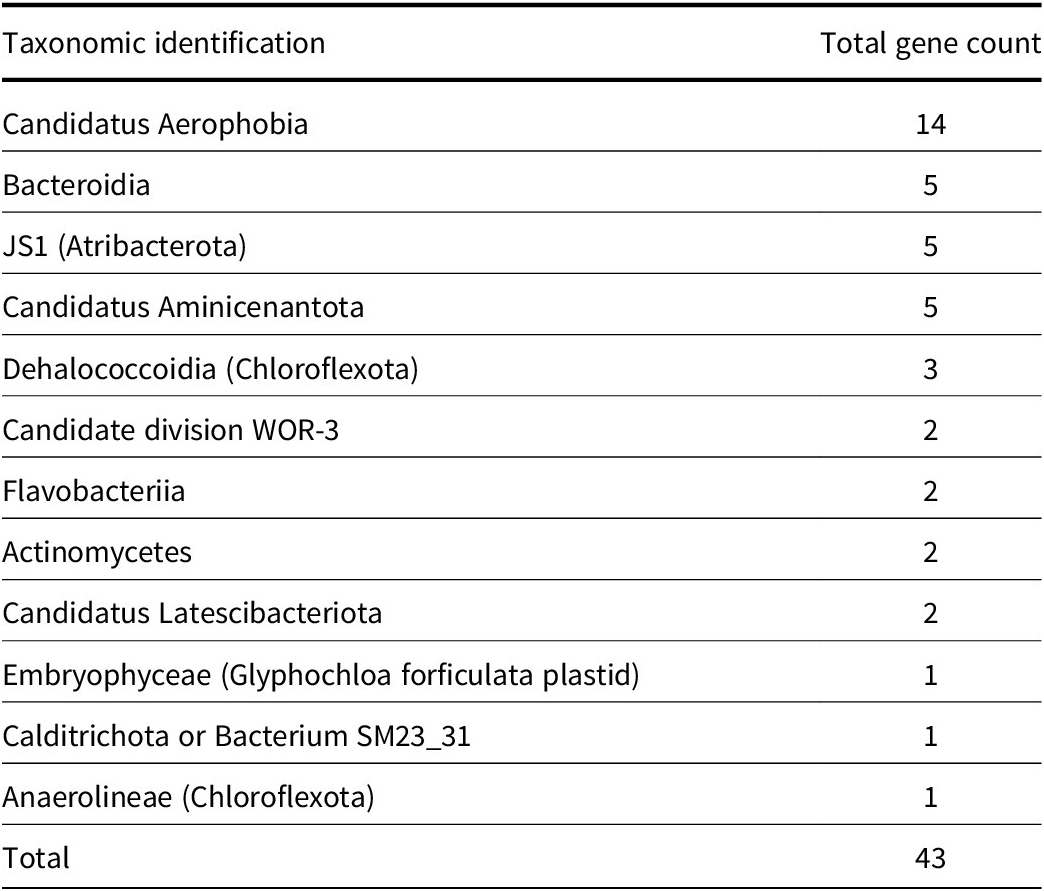
Because of the limited detection of 16S rRNA genes, the rps3 gene was used to detect taxonomic genes in the entire metagenome. Overall, 43 rps3 genes were identified, with the majority of genes coming from Candidatus Aerophobia (14), Bacteriodia (five), Candidatus Aminicenantota (five) and Atribacterota (five) (Table 2). Notably, no archaeal rps3 genes were identified in the bulk analysis.
Count of 30S ribosomal protein S3 (rps3) from all depths

Complete metagenome: metabolism by depth
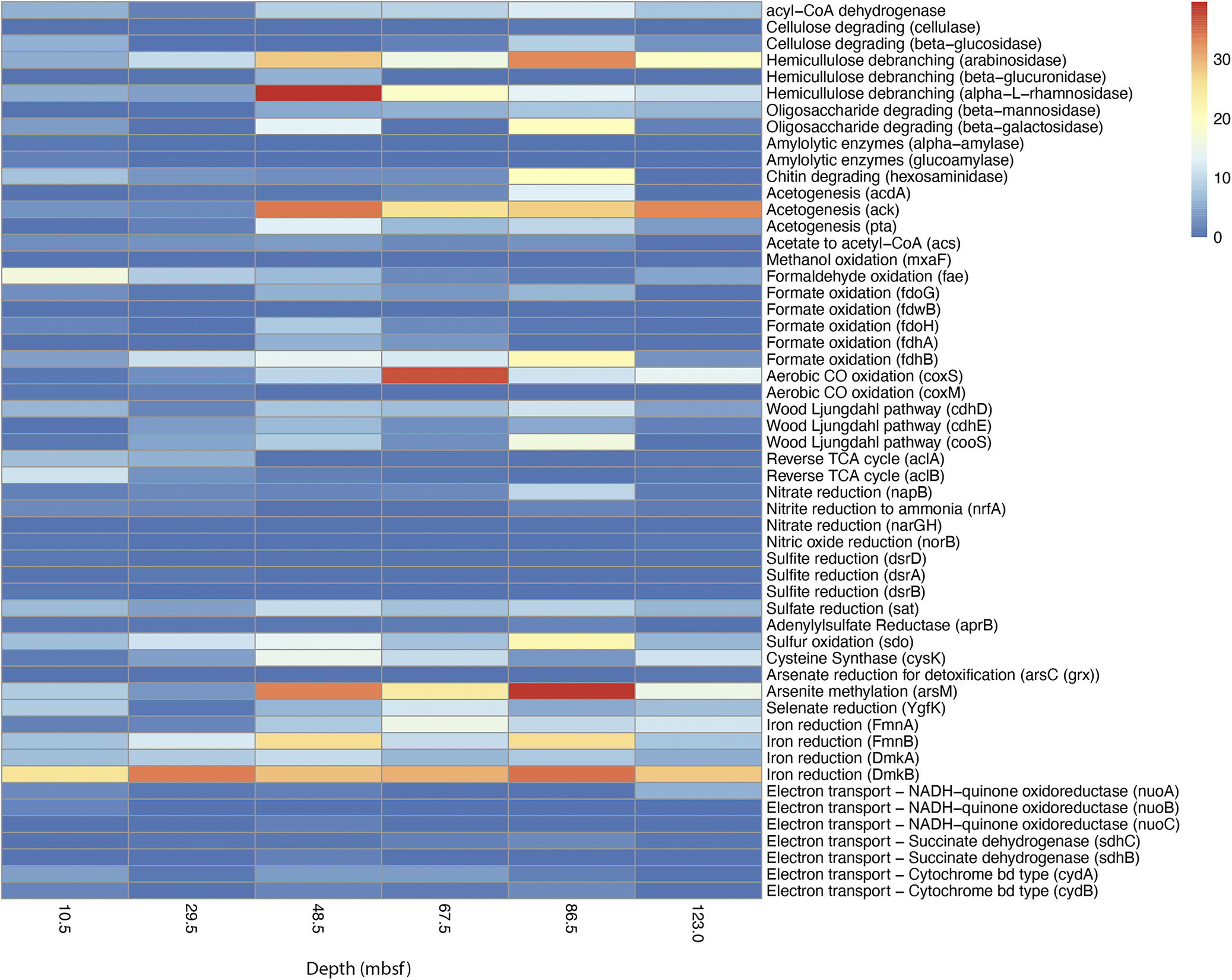
Genes for key central metabolic pathways were present at all depths. These pathways included glycolysis, gluconeogenesis, pentose phosphate pathway, phosphoribosyl pyrophosphate biosynthesis and the tricarboxylic acid cycle (TCA) cycle (Supplementary Table S1). Genes attributed to amino acid, nucleotide and fatty acid biosynthesis were also present across all depths. Despite the signature of methane removal from porewater (Fig. 1a), no methane coenzyme reductase A (mcrA) genes were found, which would be indicative of methanogenesis and anaerobic methane oxidation (Supplementary Table S1). Genes indicative of acetogenesis and carbon fixation via the Wood–Ljungdahl pathway were widespread (Fig. 2).
Selected metabolic pathway gene counts by depth. Gene counts are calculated by first summarising the average coverage for each gene, and then normalising the result by the number of reads at each depth, using the deepest sample as the standard.

Genes associated with other metabolic pathways appear to be more depth-specific. For example, many assimilatory sulfur-processing genes were present across all depths, including sulfate adenylyltransferase (sat) and cysteine synthase (cysK) (Fig. 2). In addition, adenylylsulfate reductase subunit B (aprB), typically used in the dissimilatory sulfate reduction pathway, was recovered at all depths except 123 mbsf. However, genes used in the complete dissimilatory sulfate reduction pathway were limited to shallower depths (Fig. 2), which are probably reflective of the geochemistry (Fig. 1a). The key step for sulfate reduction is performed by dissimilatory sulfite reductase (dsrAB). Genes for dsrB (annotated as from a Chloroflexota) were found only at 10.5 mbsf, and dsrA genes (annotated as from a Myxococcia) were found only at 29.5 mbsf (Fig. 2).
Dissimilatory nitrate reduction (DNR) was indicated from periplasmic nitrate reductase (nap) genes found at all depths and one subunit of the membrane-bound nitrate reductase (narI) gene at 123 mbsf (Fig. 2). Genes for nitrite reductase (nrf), the second step in DNR, were found at all depths except 48.5 and 67.5 mbsf. The nap genes were annotated as belonging to Candidatus Aerophobia and Chloroflexota, and narI was annotated to the class Betaproteobacteria. Genes for nrf were also found in these groups and for Thorarchaea at 123 mbsf.
Iron reduction genes, primarily flavin mononucleotide transferase (fmn) and dimethylmenequinone biosynthesis protein (dmk), were very common at all depths (Fig. 2), and were again reflective of the geochemistry (Fig. 1b). The genes were taxonomically assigned to a wide range of organisms. Arsenate, selenate and dimethyl sulfoxide (DMSO) processing genes were also detected, but no dissimilatory arsenate, selenate or DMSO reductases were found (Supplementary File S1).
Genes used for electron transport chains appeared to be present, perhaps coupled with the reduction processes noted above. Many nicotinamide adenine dinucleotide (NADH) dehydrogenase subunits were present, most of which were for the NADH:ubiquinone oxidoreductase respiratory complex 1 (nuoA-N) (Fig. 2). The numbers of subunits detected out of 14 with depth were: nine at 10.5 mbsf; five at 29.5 mbsf; 12 at 48.5 mbsf; two at 67.5 mbsf; four at 86.5 mbsf; one at 123.0 mbsf. In addition, genes for different subunits of succinate dehydrogenase (sdh) were found at all depths, with all four subunits being detected at 48.5 mbsf. At shallower depths, these genes were annotated as belonging to Chloroflexota, whereas genes annotated as belonging to Bacteroidota were predominant at deeper depths.
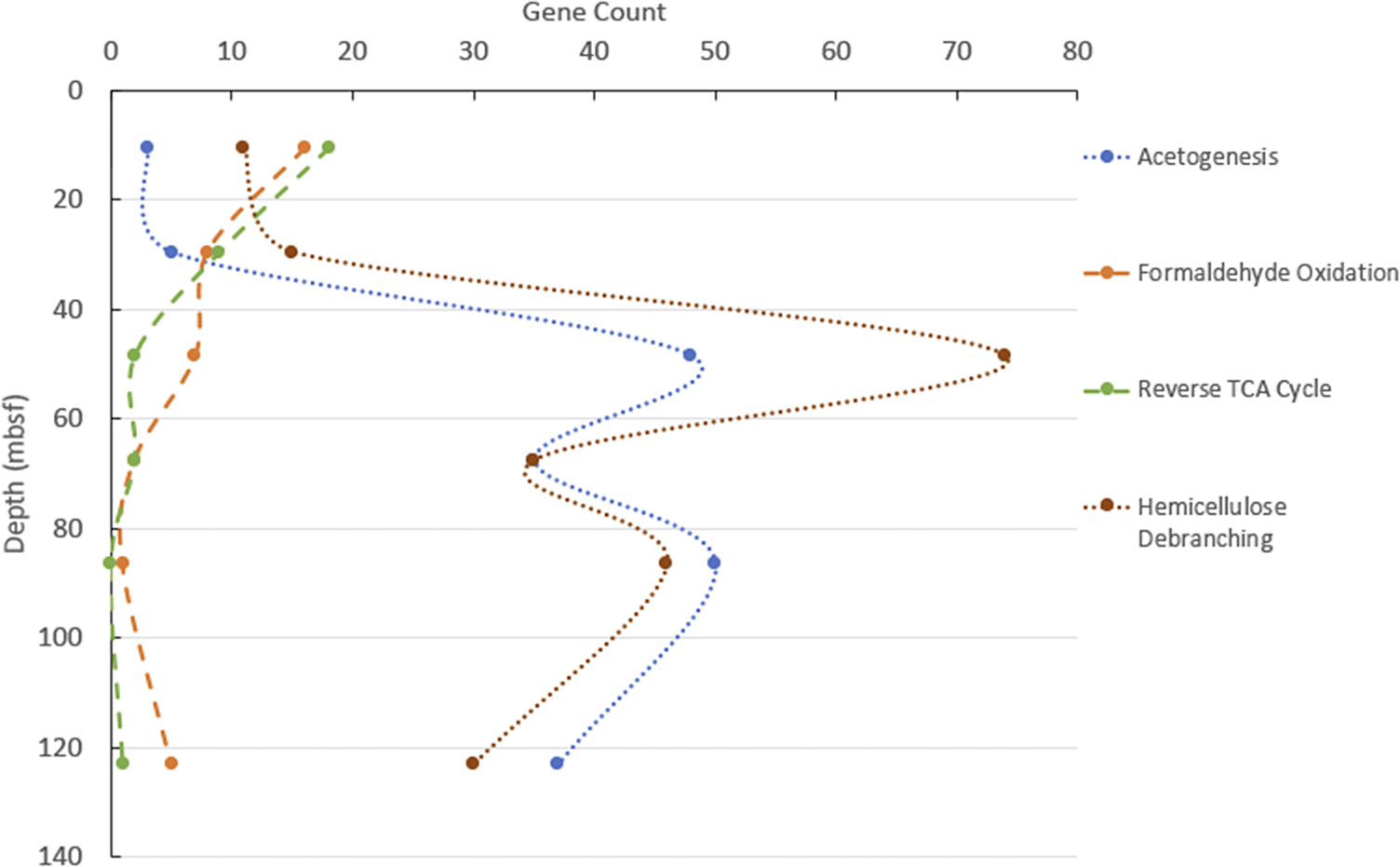
Some metabolic genes had distinct increases or decreases with depth. Formaldehyde oxidation and reverse TCA cycle genes were present in the shallow depth, then decreased (Fig. 3). These gene sequences were most commonly assigned to taxonomic lineages within the Archaea, such as Promethearchaeia, Thorarchaeia, Heimdallarchaeia and Candidatus Bathyarcaeia. Other genes, such as those for acetogenesis, were most commonly annotated as Atribacterota, Aerophobia, Bacteroidia and Clostridia, and increased at depth (Fig. 3), peaking at 48.5 and 86.5 mbsf (Fig. 3). Also following this trend of subsurface peaks were genes for hemicellulose debranching, which was found across a wide range of organisms (Figs 2 and 3). Surprisingly, the gene for aerobic carbon monoxide (CO) oxidation (coxS), annotated as Atribacterota, was most abundant at 67.5 mbsf, where geochemistry suggested no oxygen was present.
Metabolisms that vary greatly by depth. Shown are the categories of acetogenesis: formaldehyde oxidation, reverse tricarboxylic acid cycle (TCA) cycle and hemicellulose debranching. Gene counts are calculated by first summarising the average coverage for each gene, and then normalising the result by the number of reads at each depth, using the deepest sample as the standard.

Across all depths, 1195 carbohydrate genes were identified (Supplementary Table S2), with the greatest number (444) at 48.5 mbsf. The five most abundant categories included GH 4 (including glucosidase, galactosidase), GH13 (alpha-amylase and others), GH 29 (fucosidase), GH 38 (mannosidase) and GH109 (acetylgalactosaminidase). In addition, 12,773 peptidase genes were found (Supplementary Table S3), with the greatest number at the shallow 10.5 mbsf, and gradual declines at lower depths. The most common peptidases were C26, a gamma-glutamyl hydrolase, and M38, a beta-aspartyl dipeptidase.
MAGs: taxonomy by depth
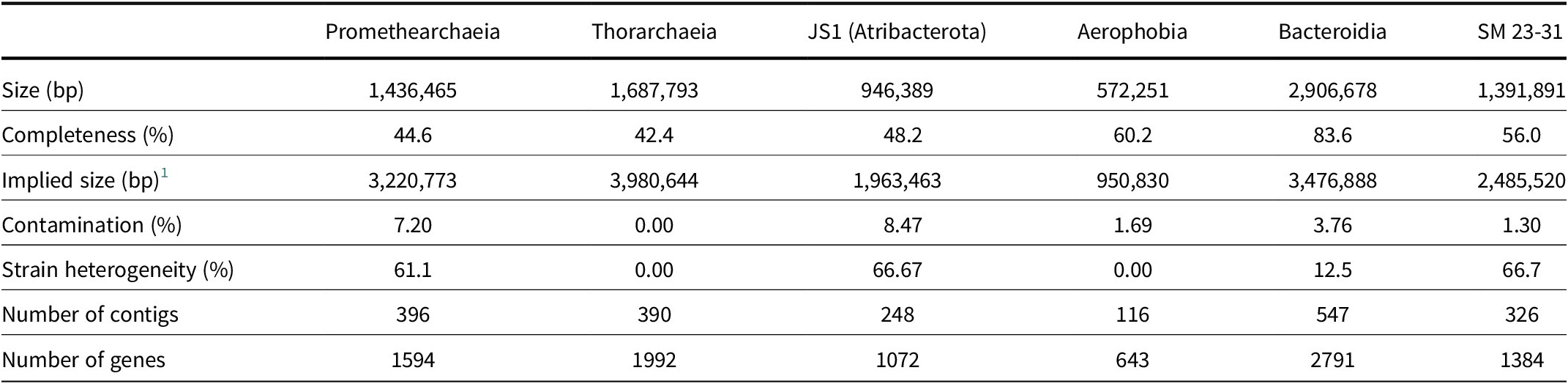
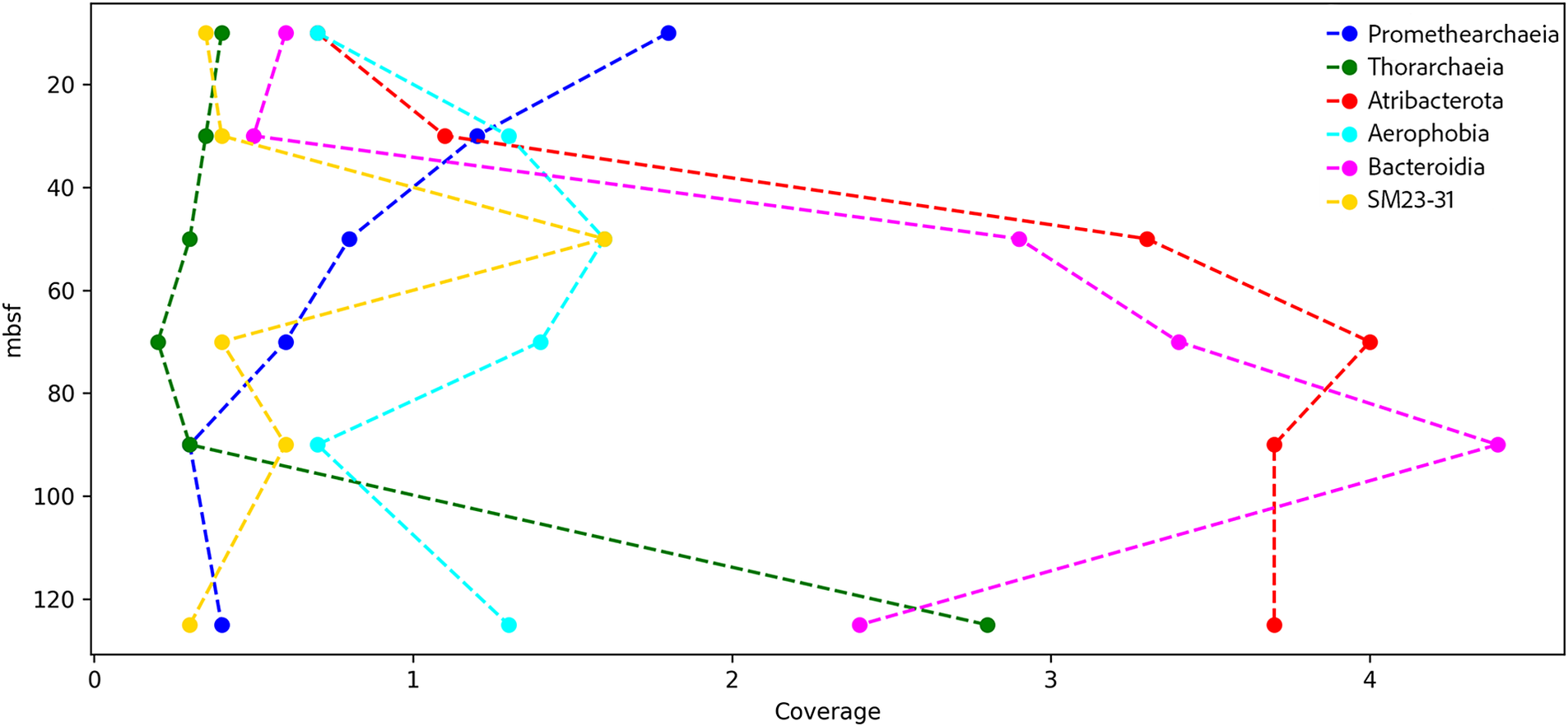
Two MAGs were assembled from Archaea, Promethearchaeia and Thorarchaeia (Table 3). Based on coverage data, gene sequences from Promethearchaeia were more abundant at shallow depths, with abundance declining gradually at deeper depths (Fig. 4). Thorarchaeia gene sequences were present at shallower depths, with maximum abundance in the deepest sample, 123 mbsf (Fig. 4).
Metagenome-assembled genome (MAG) statistics

1 Implied genome size equals MAG size divided by completeness.
Coverage of metagenome-assembled genomes (MAGs) across each depth. Each point represents the average coverage for each MAG at a specific depth. Coverage was calculated as the mean sequencing depth across all contigs within each MAG.

Four bacterial MAGs were assembled: Atribacterota, Aerophobia, Bacteriodia and SM 23-31 (Table 3). The gene sequence abundance for Atribacteria and Bacteroidetes increased with depth, with peaks at 67.5 and 86.5 mbsf, respectively, and were the most represented MAGs by coverage across all depths (Fig. 4). Sequences from Aerophobia and SM23-31 were present at all depths, with both peaking at 48.5 mbsf (Fig. 4).
MAGs: metabolism by MAG
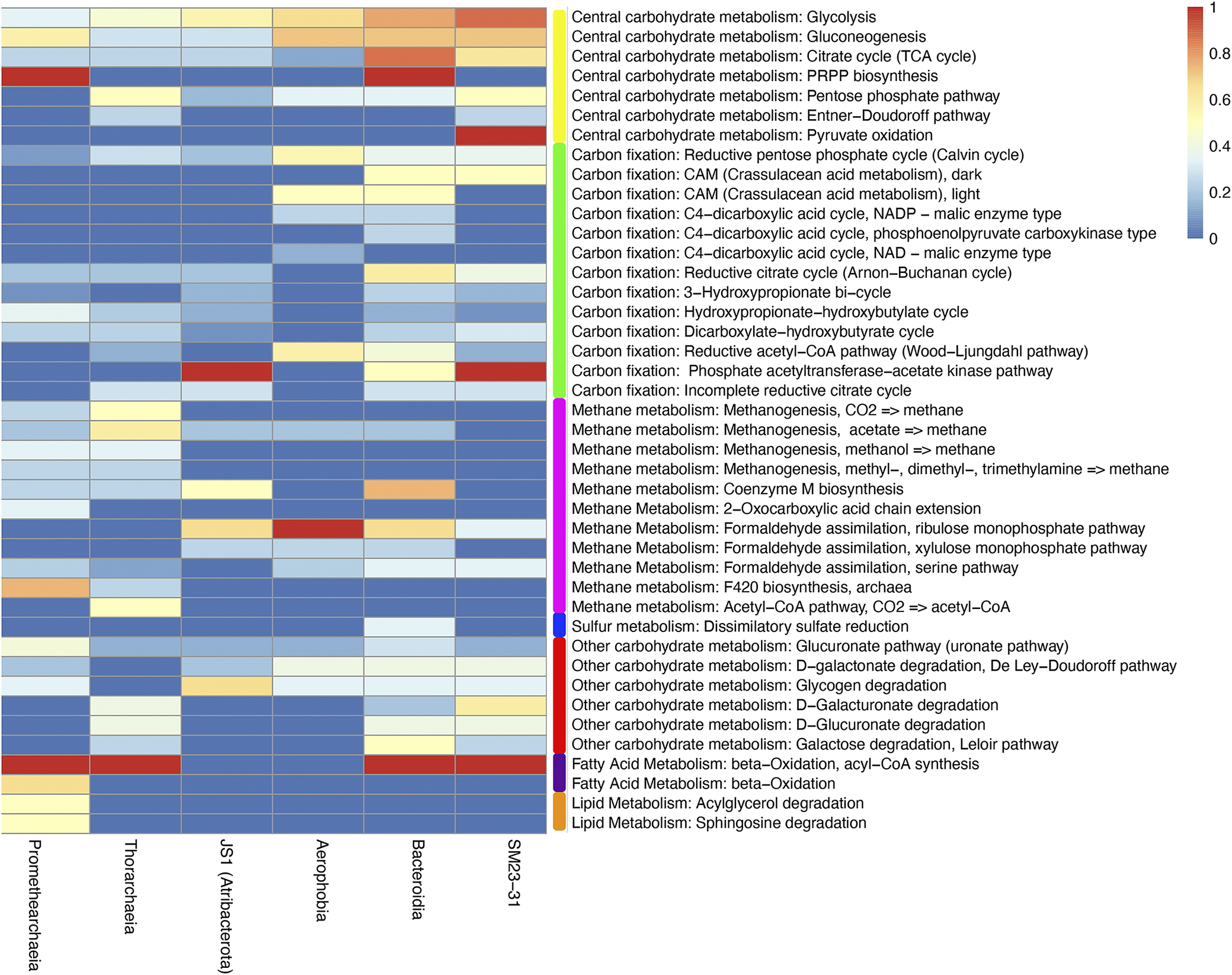
Because of the high strain heterogeneity created by binning MAGs across all depths of sediment samples (Table 3), we present the metabolic features of each MAG as a class-level community analysis. As a result of low completion percentages in all of the MAGs, complete metabolic pathways were rare. However, all of the MAGs appeared to be capable of a heterotrophic lifestyle utilising sugars or carbohydrates, primarily via fermentative pathways (Fig. 5).
The percentage completeness of selected metabolic pathways among metagenome-assembled genomes (MAGs). The completeness of each pathway was determined per KEGG BlastKOALA. Coloured bars are added to delineate metabolic categories.

Archaeal MAGs
The Promethearchaeia and Thorarchaeia MAGs were 44.6% and 42.4% complete, respectively (Table 3); however, from what genes were present, annotations suggested that both should have the ability to perform glycolysis and gluconeogenesis, and degrade fatty acids, carbohydrates and peptides (Fig. 5; Supplementary Table S4). Both archaeal MAGs had full beta oxidation, acyl-coA synthesis pathways (Fig. 5). Although both MAGs have some genes involved in methanogenesis pathways, the key gene of methyl coenzyme M reductase was not detected (Supplementary Table S4). For carbon fixation, the genes for the rTCA cycle (aclA and aclB) were found in the Promethearchaeia MAG, whereas genes for the Wood–Ljungdahl pathway were found in the Thorarchaeia MAG. The Promethearchaeia MAG contained 16 genes found among nine carbohydrate-active enzymes (CAZY) categories, encoding the ability to degrade carbohydrates, including cellulose, chitin and hexose sugars (Supplementary Table S5). This MAG additionally had 17 peptidase genes that were found in categories that included cysteine, metallo, serine and threonine peptidase (Supplementary Table S6). The Thorarchaeia MAG had only one carbohydrate degradation gene (CAZY GH133), an amylo-alpha-1,6-glucosidase, and had 20 peptidase genes also across all peptidase categories (Supplementary Tables S5 and S6). For energy generation, the Promethearchaeia MAG had two iron reduction genes (ndh2 and dmkB), but oxidative phosphorylation pathways were not detected. In the Thorarchaeia MAG, iron reduction genes were also detected (fmnA, dmkA and dmkB). Fragments of an electron transport chain were found, including some NADH dehydrogenase gene subunits (two of 14 for nuo; two of 16 for ndh). In addition, two of nine subunits for the V/A type ATPase gene and one of four fumarate reductase genes were detected.
Bacterial MAGs
The Atribacterota MAG, at 48.2% complete (Table 3), included partially complete glycolysis (five of nine genes in the pathway), gluconeogenesis (two of seven) and TCA cycle (two of eight) genes (Fig. 5). Two acetogenesis genes (ack and pta) were present, along with coxS, involved in CO oxidation, and the sdo gene involved in sulfur oxidation. These findings are consistent with published genomes of Atribacterota that suggest they utilise a fermentative acetogenic metabolism involving carbohydrates or organic acids (Vuillemin et al., Reference Vuillemin, Vargas, Coskun, Pockalny, Murray, Smith, D’Hondt and Orsi2020). Only three carbohydrate degradation genes were detected, including potential amylases or rhamnosidases, and 20 peptidase genes were present (Supplementary Tables S5 and S6). Seven of nine subunits of a V/A type ATPase were detected, but only one subunit (nuoF) of NADH dehydrogenase. No subunits of succinate dehydrogenase, cytochromes nor any Fe–Fe or Ni–Fe hydrogenases were detected (Supplementary Table S4).
The bacterial group SM23-31 has not been well described, but other MAGs in Candidate Division LCP-89 are probably fermentative and suggest carbohydrate and amino acid degradation (Youssef et al., Reference Youssef2019). Our SM23-31 MAG was similar to these previously described groups, as it had eight of nine possible glycolysis genes and five of eight genes in the TCA cycle (Fig. 5). Genes in acetogenesis (pta and ack) were present as well, but key Wood–Ljungdahl genes were not detected. Ten carbohydrate degradation genes were found, involving hexose sugars such as rhamnose, galactose and mannose (Supplementary Table S5), and 16 peptidase genes were also identified, including cysteine, metallo and serine varieties (Supplementary Table S6). Two genes potentially involved in iron reduction were detected (fmnB and dmkB). Many respiratory electron transport genes were also present, including 13 of 14 possible units of NADH dehydrogenase gene nuo, a complete succinate dehydrogenase gene (sdh), three ATPase subunits and two subunits of cytochrome b/d complex (Supplementary Table S4). Based on the annotations, it is unclear what the terminal electron acceptor might be.
The Aerophobota MAG was 60.2% complete (Table 3), with six of nine glycolysis genes and one of eight TCA cycle genes detected (Fig. 5). In addition, gluconeogenesis (five of seven genes), the pentose phosphate pathway (two of six genes) and the Wood–Ljungdahl pathway (four of seven, including cdhD and cdhE, genes) were detected (Fig. 5). Formaldehyde assimilation via the ribulose monophosphate pathway was complete, although many other pathways were incomplete. A V/A-type ATPase, with eight of nine subunits, was annotated, but only one NADH dehydrogenase subunit (nuoF) was detected. Carbohydrate degradation genes were mostly undetected, except in CAZY family GH109, which included acetylgalactosaminidases and acetylhexosaminidases (Supplementary Table S5). Thirteen peptidases were found, including cysteine, metallo, serine and threonine varieties (Supplementary Table S6).
The Bacteroidia MAG was 83.6% complete, having its highest coverage at 89.5 mbsf (Table 3; Fig. 4). Many genes involved in central carbon metabolism were present, but many pathways were not complete, including glycolysis (seven of nine genes), gluconeogenesis (five of seven), the TCA cycle (seven of eight) and the pentose phosphate pathway (two of six) (Fig. 5). Bacteroidia are thought to specialise in the degradation of polysaccharides, such as cellulose, xylan and starch, originating with algae (Zheng et al., Reference Zheng, Cai, Liu, Liu and Sun2020). Our data suggested similar specialisation. In the MAG, 64 carbohydrate degradation genes were detected, including those involving cellulose, hemicellulose and chitin (Supplementary Table S5). Additionally, 38 peptidase genes, involving the cysteine, metallo and serine varieties, were detected (Supplementary Table S6). Some acetogenesis (ack and pta) and Wood–Ljungdahl genes (three of seven) were present (Fig. 5). Only one potential gene (nrfA) in the DNR pathway was detected. Similarly, only aprA was found for the sulfate reduction pathway. Two genes suggesting iron reduction were also present (fmnB and dmkB). Some electron transport or oxidative phosphorylation genes were detected, although only two of 14 NADH dehydrogenase B/A subunits were detected. However, three of four succinate dehydrogenase genes, two of three cytochrome bd complex subunit genes and three of four F-type ATPases subunits were present (Supplementary Table S4).
Discussion
The Iberian Margin was visited by the drilling program because of its value for paleo-history and paleoclimate proxies (Seah and Gruber-Vodicka, Reference Seah and Gruber-Vodicka2015; Scientists, 2013). Its location and preservation allowed for an opportunity to link marine, ice and terrestrial climate records (Tzedakis et al., Reference Tzedakis, Margari and Hodell2015). However, amongst the well-preserved nannofossils and planktonic foraminifera (Girone et al., Reference Girone, De Astis, Sierro, Hernández-Almeida, Alonso Garcia, Sánchez Goñi, Maiorano, Marino, Trotta and Hodell2023), microbial signatures appear to be widespread but usable DNA was extremely difficult to recover from this location, probably because of the large amount of clay (Oliveira et al., Reference Oliveira, Rocha, Rodrigues, Jouanneau, Dias, Weber and Gomes2002). As such, a method using PCR-adapter WGA for DNA was applied to overcome small amounts of DNA with poor quality. This WGA has been shown to have little representational bias. However, we do acknowledge that bias is possible and interpret our results with caution. Previous attempts to use PCR-based amplification of deep sediment used a technique, random amplification metagenomic PCR (RAMP), built for a specific sequencing technology with longer primers for amplification (Martino et al., Reference Martino, Rhodes, Biddle, Brandt, Tomsho and House2012). The WGA technique used in this current study is sequencing method-independent and uses shorter primers and a more robust polymerase and therefore, we suggest, is better suited to sediment samples compared to the previously developed RAMP method.
We examined the metagenome across depths of Site U1385, going as deep as 123 mbsf where sediment was dated at roughly 1.2 million years of age (Hodell et al., Reference Hodell, Lourens, Crowhurst, Konijnendijk, Tjallignii, Jimenez-Espejo, Skinner and Tzedakis2015). However, direct comparisons of potentially modern versus preserved genomic molecules were not performed in this study and would require experimental methods that were strongly inhibited by the sedimentary material. Yet, because of the variability annotated metabolisms that we see throughout the differing depths of the metagenome, we interpret these data as potential indicators of active subsurface life.
Examining the total metagenome, some genetic signatures reflected porewater geochemistry, such as related to the sulfur cycle, but others did not, such as for the iron cycle. For instance, sulfate concentrations were low from porewater from the surface to approximately 48 mbsf, and the only complete pathways for sulfate reduction were from the shallow sediments. These results agree with what had been predicted through isotope analysis, that microbial sulfate reduction would be strongest within the top ~10 m and then stimulated again at the SMTZ (Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016). The presence of genes for assimilatory sulfur processes also agreed with the isotopic-based estimate that there is a high percentage of intracellular recycling of sulfur species (Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016). In contrast, genes for iron reduction were present across the majority of the sediment column, although iron reduction has not been described as a significant metabolism in deep sediments, potentially because iron is commonly a limiting nutrient for the ocean. However, the site’s location on a narrow continental shelf and the delivery of material via the Tagus River provide terrestrial sediment that has significantly more iron than most marine-sourced sediment (Tzedakis et al., Reference Tzedakis, Drysdale, Margari, Skinner, Menviel, Rhodes and Taschetto2018). We hypothesise that the signature of iron reduction was stronger across genomes as a result of this riverine discharge and burial. Additionally, isotopic evidence from this site indicated the potential for a deep portion of pyrite formation that microbial iron reduction may enable (Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016). The signatures for acetogenesis increased with depth, which was expected as a metabolism in a deep sediment environment (Lever, Reference Lever2012). Counterintuitively, hemicellulose degradation also increased at depth. This may be as a result of to the stimulation of organisms at the SMTZ and be a byproduct of increased microbial metabolism. However, despite the SMTZ geochemical signature of anaerobic methane oxidation at ~48 mbsf and the increase of methane at greater depths, no genomic signatures for these metabolisms were detected. These results were similar to other continental margin studies (Biddle et al., Reference Biddle, Fitz-Gibbon, Schuster, Brenchley and House2008; Carr et al., Reference Carr, Orcutt, Mandernack and Spear2015). The microbes performing anaerobic methane oxidation and methanogenesis in the deep biosphere are still elusive.
The binning of the metagenomes and analysis of coverage showed that microbes may have metabolic niches in the subsurface. The increase of Atribacteria and Bacteroidia in deep sediment reflects the well-known anaerobic and fermentative abilities of these groups (Vuillemin et al., Reference Vuillemin, Vargas, Coskun, Pockalny, Murray, Smith, D’Hondt and Orsi2020; Zheng et al., Reference Zheng, Cai, Liu, Liu and Sun2020). SM23-31 is commonly identified in anaerobic marine sediments and by genomic appearances seems to be a powerhouse fermenter (Youssef et al., 2019). The noticeable increase of SM23-31 signal at the SMTZ may be indicative that there is more usable substrate activated in this area, but we have no geochemical support for this hypothesis. The Aerophobia, Promethearchaeia and Thorarchaeia are also common inhabitants of anaerobic marine sediments, but their distribution and metabolic profiling in this study suggest their main feature is fermentation (Zhang et al., Reference Zhang, Fang, Yin, Lai, Kuang, Zhang, Xu, Wegener, Wang and Dong2023). However, our genome analysis does not show much if any variation from what is known of these groups from other locations around the globe, suggesting that the major group activities are phylogenetically well preserved.
The variations seen in coverage and genome function across depths suggest that the metagenome is representative of an active microbial signature and not a remnant or depositional signature because the correlations of genomic signatures match best to geochemistry and not any paleodeposition signal. The ability to separate functions into genomic bins allows for the clearer signature that there is variation across sediment depths. Our genetic data cannot assess any rate of microbial activity. However, researchers have used the porewater geochemistry, complemented with isotopic analysis, to indicate that slow processes are occurring over multiple depths and sediment ages (Turchyn et al., Reference Turchyn, Antler, Byrne, Miller and Hodell2016). This microbial activity does not damper the ability to use this site for nannofossil or pollen analysis because these signatures are probably well preserved as a result of anoxic sediment and large clay amounts at this site (Yang et al., Reference Yang2019; Sanchez Goñi et al., Reference Sanchez Goñi, Eynaud, Turon and Shackleton1999). Hence we assume, bolstered by the previously calculated sulfate reduction rates in the shallow sediments, that the metabolic rate of any of the microbes described in this study is quite low and acknowledge our hypotheses would need a direct study of microbial rates in the subsurface to provide proof of microbial activity. With this metagenomic examination of sediment from the Iberian Margin, we demonstrate a method to amplify low abundance and poor-quality DNA with metagenomics, and describe the potential microbial community of IODP Site U1385. Our analysis is surely an insufficient first pass in examining this area of the deep biosphere. With its valued climate history, perhaps this area of sediment will be retrieved again in the future and experiments can be designed to analyse its hypothetically slow but potentially active subsurface life.
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1180/gbi.2026.10007.
Acknowledgements
This project was funded by NSF OCE grant 1333104 to JFB. JR was also supported by the IODP Schlanger Drilling Fellowship. IF and GC were supported by EMRE. We thank all shipboard scientists and crew on IODP Expedition 339. Support from the University of Delaware Bioinformatics Data Science Core Facility (RRID:SCR_017696), including use of the BIOMIX and BioStore computational resources, was made possible through funding from Delaware INBRE (NIGMS P20GM103446), NIH Shared Instrumentation Grant (NIH S10OD028725), the State of Delaware, and the Delaware Biotechnology Institute.
Competing interests
The authors declare no competing interests with this work.



 Open access
Open access