


Introduction
There are currently 33 species in fungal genus Pseudogymnoascus, with four species originating from the polar regions: Pseudogymnoascus antarcticus, Pseudogymnoascus australis, Pseudogymnoascus griseus and Pseudogymnoascus lanuginosus (Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021). The commonly studied Pseudogymnoascus destructans is a known fungal pathogen causing white nose syndrome in bats, and it was previously known as Geomyces destructans (Blehert et al. Reference Blehert, Hicks, Behr, Meteyer, Berlowski-Zier and Buckles2009, Gargas et al. Reference Gargas, Trest, Christensen, Volk and Blehert2009). Pseudogymnoascus spp. have been reported in a wide range of geographical locations with temperate temperatures (Minnis & Lindner Reference Minnis and Lindner2013, Drees et al. Reference Drees, Lorch, Puechmaille, Parise, Wibbelt and Hoyt2017, Zhang et al. Reference Zhang, Li, Chen, Liang and Han2023), but none has been reported from a tropical region to date. Generally, filamentous fungi in the subdivision Pezizomycotina (phylum Ascomycota) can produce various specialized metabolites (SMs) that have pharmaceutical utility, such as penicillin from Penicillium chrysogenum (Smith et al. Reference Smith, Burnham, Edwards, Earl and Turner1990) and lovastatin from Monascus spp. (Dikshit & Tallapragada Reference Dikshit and Tallapragada2015). As a filamentous fungus, polar Pseudogymnoascus produce various types of SM compounds such as antifungal amphiol (Fujita et al. Reference Fujita, Ikuta, Nishimura, Sugiyama, Yoshimura and Kakeya2021), antibacterial and antitumor macrosphelide A-B (Antipova et al. Reference Antipova, Zaitsev, Zhelifonova, Tarlachkov, Grishin, Kochkina and Vainshtein2023), cytotoxic ganodermaside A-D (Shi et al. Reference Shi, Li, Zheng, Zhang, Dai and Shang2021b) and antibacterial pseudophenone A (Shi et al. Reference Shi, Yu, Dai, Zhang, Hu, Zheng and Shi2021a). SMs have traditionally been called ‘secondary metabolites’, as they are generally not essential for life. However, the term ‘specialized metabolites’ is currently preferred as these metabolites have been shown to bestow very specific ecological or physiological advantages on the organism (Gavriilidou et al. Reference Gavriilidou, Kautsar, Zaburannyi, Krug, Müller, Medema and Ziemert2022).
In SM production, multiple genes are usually involved, which are arranged contiguously and called biosynthetic gene clusters (BGCs). The clusters generally have a gene-producing, chemically defining synthase (core gene) that modifies primary metabolites to form the core backbone of the SM compound. That backbone is then further modified by additional tailoring enzyme-encoding genes (Keller Reference Keller2019) to produce the final SM compound. To date, only two studies (Palmer et al. Reference Palmer, Drees, Foster and Lindner2018, Antipova et al. Reference Antipova, Zaitsev, Zhelifonova, Tarlachkov, Grishin, Kochkina and Vainshtein2023) have very briefly explored the BGCs of Pseudogymnoascus spp. in silico with antiSMASH, a database used for exploring and querying the BGCs of SMs in bacteria and fungi. However, only the results for the temperate fungus P. destructans are included in the antiSMASH repository (Blin et al. Reference Blin, Shaw, Medema and Weber2023a). The in silico prediction of BGCs using genomic data allows the assignment of known metabolites to their related BGCs, which could then be expressed for metabolite production in pharmaceutical applications. Additionally, genomic data have increased our understanding of the mechanistic link between genomic structure and metabolic output, enabling the systemic identification of BGCs. For instance, analysis of the Cyathus olla genome identified a specific BGC responsible for producing neuroactive cyathane diterpenes (Xie et al. Reference Xie, Zhao, Song, Qiao, Wang and Qi2024), and comparative genomics in Hericium rajendrae provided strong evidence regarding the biosynthetic pathway of erinacines (Wei et al. Reference Wei, Cheng, Zhu, Zhang, Cui, Wang and Qi2023). Furthermore, genomic analysis can reveal the evolutionary drivers of metabolic diversity. Studies on Auricularia species and C. olla show how events such as gene family expansion, transposon activity and whole-genome duplication shape a species’ unique BGC repertoire, tailoring its chemistry for ecological adaptation (Xie et al. Reference Xie, Zhao, Song, Qiao, Wang and Qi2024, Qi et al. Reference Qi, Kang, Zhang, Qi, Li and Vadim2025). Consequently, these genomic insights transform fungal bioprospecting from a discovery-based endeavour into a predictive science, enabling forms of targeted exploitation such as genetic manipulation of identified fungal BGCs to achieve an increased yield of desired compounds (Harvey et al. Reference Harvey, Tang, Schlecht, Horecka, Fischer and Lin2018, Roux et al. Reference Roux, Woodcraft, Hu, Wolters, Gilchrist and Chooi2020).
In order to explore the BGCs within the genomes of polar Pseudogymnoascus spp., their whole genome sequences (WGSs) need to be obtained. However, many of the genomes in GenBank for Pseudogymnoascus originate from temperate regions, where the pathogenic P. destructans is the most well studied. Although there are three polar Pseudogymnoascus spp. assemblies in GenBank (Leushkin et al. Reference Leushkin, Logacheva, Penin, Sutormin, Gerasimov and Kochkina2015), their species are unidentified and their sequencing depths are low, making them unsuitable for reference-based assembly. Therefore, based on these knowledge gaps, this study aims to explore the SM production potential of two polar Pseudogymnoascus species using computational prediction software. To achieve this, we constructed the WGSs de novo for both species. These produced WGSs will be particularly useful as reference genomes for studies aiming to investigate other polar strains, in which the predicted BGCs could help to bridge the gap between characterized SMs and their molecular counterparts.
Methodology
Fungal cultivation and genome sequencing
Two non-pathogenic, polar isolates of Pseudogymnoascus spp. deposited in the culture collection of the National Antarctic Research Centre (NARC), Universiti Malaya, Malaysia, were used in this study. Strain AKSP4 (AK07KGI 1202 R1-1 sp.4) was originally isolated from King George Island, Antarctica (Krishnan et al. Reference Krishnan, Alias, Wong, Pang and Convey2011), whereas strain HNDR4 (HND16 R4-1 sp.1) was from Hornsund in the Arctic (Ali et al. Reference Ali, Alias, Siang, Smykla, Pang, Guo and Convey2013). Each strain was revived by sub-cultivating it into five potato dextrose agar (PDA; Oxoid) plates and incubating at 15°C. The plate with the best growth was chosen for further cultivation in five 250 ml conical flasks of liquid medium containing 50 ml potato dextrose broth (PDB; Oxoid) and incubated at 15°C and shaken at 120 rpm for 8 days to obtain sufficient biomass.
Of the five flasks, only three flasks with sufficient biomass were selected, and each fungal culture in these three flasks along with their media were transferred into 50 ml centrifuge tubes. Tubes were centrifuged at 3000 rpm for 6 min at 15°C to obtain the fungal pellet. Genomic DNA extraction of fungal pellets, polymerase chain reaction (PCR) amplification, preliminary DNA sequencing for barcoding and whole-genome sequencing were outsourced to Apical Scientific Sdn. Bhd. Initial genomic DNA extraction for DNA barcoding tests was conducted using Presto™ Mini gDNA Yeast Kit (Geneaid, GBYB100). PCR amplification of the complete internal transcribed spacer (ITS) region was conducted using primers ITS1 (5´-TCCGTAGGTGAACCTGCG-3´; White et al. Reference White, Bruns, Lee, Taylor, Innis, Gelfand, Sninsky and White1990) and ITS4 (5´-TCCTCCGCTTATTGATATGC-3´; White et al. Reference White, Bruns, Lee, Taylor, Innis, Gelfand, Sninsky and White1990). The total reaction volume of 25 μl contained gDNA, 0.5 pmol of each primer, deoxynucleotides triphosphates (dNTPs; 200 μM each), 0.5 U thermostable DNA polymerase, supplied PCR buffer and water. The PCR was performed as follow: 1 cycle (98°C for 2 min) for initial denaturation, 25 cycles (98°C for 15 s; 60°C for 30 s; 72°C for 30 s) for annealing and extension and 1 cycle (72°C for 10 min) for final extension. The PCR products were purified using standard PCR clean-up methods. The purified PCR products were subjected to bidirectional sequencing with universal primers M13F (−20) and M13R-pUC (−26) using a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Once DNA barcoding testing had confirmed that fungal pellets were pure cultures of Pseudogymnoascus, the remainder pellets were extracted for their genomic DNA using SEPa Plant DNA Isolation Reagent Kit. A total of 0.2 μg DNA per sample was used as input material for the DNA library preparations using the NEBNext® Ultra™ IIDNA Library Prep Kit (Cat No. E7645). Briefly, the genomic DNA sample was fragmented by sonication to a size of 350 bp. Then, DNA fragments were end-polished, A-tailed and ligated with the full-length adapter for Illumina sequencing, followed by further size selection and PCR amplification. Afterwards, the PCR products were purified using an AMPure XP system (Beverly, MA, USA). Subsequently, library quality was assessed on an Agilent 5400 system (Agilent, USA) and quantified via quantitative PCR (1.5 nM). Whole-genome sequencing was performed in the 2 × 250 bp format on an Illumina NovaSeq 6000 system at 150× coverage. Raw data were cleaned using fastp (Chen Reference Chen2023) to remove paired reads that contained 1) adapter contamination, 2) uncertain nucleotides (N) constituting more than 10% of either read and/or 3) low nucleotide quality (Q < 5) constituting more than 50% of either read. Both raw and clean data were provided by Apical Sdn. Bhd.
De novo whole-genome assembly
The quality of clean data was assessed using FastQC (Andrew Reference Andrew2010). St. Petersburg genome assembler (SPAdes) v3.15.5 (Prjibelski et al. Reference Prjibelski, Antipov, Meleshko, Lapidus and Korobeynikov2020) was used to perform de novo assembly, resulting in the contig level assembly that was used for downstream analyses. funannotate v1.8.15 (Palmer & Stajich Reference Palmer and Stajich2020) was used to remove duplicated contigs and those of less than 500 bp. The National Centre for Biotechnology Information (NCBI) Foreign Contamination Screen (FCS) v0.5.4 (Astashyn et al. Reference Astashyn, Tvedte, Sweeney, Sapojnikov, Bouk and Joukov2024) was used to remove adaptor sequences and sequences from other foreign organisms within the assemblies. The contigs were subsequently sorted from shortest to longest in length. To assess genome sequence assembly quality, Quality Assessment Tool (QUAST) v5.2 (Gurevich et al. Reference Gurevich, Saveliev, Vyahhi and Tesler2013) and Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.5.0 (Simão et al. Reference Simão, Waterhouse, Ioannidis, Kriventseva and Zdobnov2015) were used, with the leotiomycetes_odb10 lineage database created on 8 January 2024 as the reference class for BUSCO.
Phylogenetic analysis for species identification
In order to confirm the species of strains AKSP4 and HNDR4, phylogenetic analysis was conducted based on ITS, nuclear large subunit (LSU) rDNA, DNA replication licensing factor (MCM7), RNA polymerase II second largest subunit (RPB2) and translation elongation factor (TEF1α) sequences that were obtained from the whole-genome assemblies of this study. For the ITS, LSU and MCM7 sequences, Wong et al. (Reference Wong, Mohamad-Fauzi, Rizman-Idid, Convey, Smykla and Alias2022) had previously identified these sequences for both strains AKSP4 and HNDR4 used in this study. Basic Local Alignment Tool (BLAST) was used to confirm the presence of these sequences within the whole-genome assemblies of this study. Furthermore, the RPB2 and TEF1α sequences were searched within the whole-genome assemblies of this study with BLAST, referring to the sequences in Zhang et al. (Reference Zhang, Li, Chen, Liang and Han2023) (Table S1). Hits were filtered using E-value, similarity percentage and number of gaps. SAMtools v1.18 (Danecek et al. Reference Danecek, Bonfield, Liddle, Marshall, Ohan and Pollard2021) was used to extract the sequences from the whole genomes.
Molecular Evolutionary Genetics Analysis (MEGA) 11 (Tamura et al. Reference Tamura, Stecher and Kumar2021) was used to trim and concatenate the ITS, LSU, MCM7, RPB2 and TEF1α sequences of strains AKSP4 and HNDR4. For each of the genetic marker sequences, multiple sequence alignment was conducted using ClustalW on the two strains of this study and on other reference sequences (Pseudogymnoascus spp. and Pseudogeomyces spp.) obtained from GenBank. Each genetic marker sequence was then concatenated to produce a final alignment file containing 67 taxa that was then used for downstream analyses. The maximum likelihood (ML) tree for species identification was constructed using IQ-TREE v2.3.4 (Minh et al. Reference Minh, Schmidt, Chernomor, Schrempf, Woodhams, Von Haeseler and Lanfear2020) with 10 000 ultrafast bootstrap values (Hoang et al. Reference Hoang, Chernomor, Von Haeseler, Minh and Vinh2017), where the best-fit nucleotide substitution model was calculated using ModelFinder (Kalyaanamoorthy et al. Reference Kalyaanamoorthy, Minh, Wong, Von Haeseler and Jermiin2017) provided within the same software. Bayesian inference (BI) analysis was performed using Mr. Bayes v3.2.7 (Ronquist et al. Reference Ronquist, Teslenko, Van der Mark, Ayres, Darling and Höhna2012), where Markov chain Monte Carlo (MCMC) simulations were run for 5 × 107 generations with a sampling frequency of every 103 generations. The substitution model GTR + I + G was used, and a burn-in of 25% was selected, whereby the remaining trees were used to construct a 50% majority rule consensus tree. Pseudogeomyces spp. were used as the outgroup for both analyses. figtree v1.4.4 (Rambaut Reference Rambaut2016) was used to visualize the tree.
Genome annotation
Prior to genome annotation, custom repeat databases specific to the AKSP4 and HNDR4 assemblies constructed in this study were generated using RepeatModeler v2.05 (Smit et al. Reference Smit, Hubley and Green2013) and then processed with RepeatMasker v4.1.5 (Smit et al. Reference Smit, Hubley and Green2013) for soft masking. Gene predictions were conducted with funannotate v1.8.15 (Palmer & Stajich Reference Palmer and Stajich2020) using the masked assemblies. Expressed sequence tags (ESTs) belonging to the class Leotiomycetes from NCBI and protein sequences of Pseudogymnoascus from UniProtKb were downloaded on 7 April 2024 as evidence for homology-based functional annotation. The gene prediction analysis produced an annotation file in GFF3 format and a FASTA file of predicted amino sequences. The latter was used within InterProScan v5.70-102.0 (Jones et al. Reference Jones, Binns, Chang, Fraser, Li and McAnulla2014), eggNOG v2.1.12 (Huerta-Cepas et al. Reference Huerta-Cepas, Szklarczyk, Heller, Hernández-Plaza, Forslund and Cook2018, Cantalapiedra et al. Reference Cantalapiedra, Hernández-Plaza, Letunic, Bork and Huerta-Cepas2021), dbCAN2 (Zhang et al. Reference Zhang, Yohe, Huang, Entwistle, Wu and Yang2018) and BlastKOALA (Kanehisa et al. Reference Kanehisa, Sato and Morishima2016). InterProScan v5.70-102.0 mapped the proteins to their corresponding gene ontology (GO) terms, and resulting charts of the 10 highest GO counts for each biological process (BP), cellular component (CC) and molecular function (MF) were produced using SRPlot (Tang et al. Reference Tang, Chen, Huang, Zhang, Zeng and Zhang2023). Output from eggNOG v2.1.12 and dbCAN2 was also incorporated into funannotate v1.8.15 to annotate clusters of orthologous groups (COGs) and carbohydrate-activating enzymes (CAZymes). BlastKOALA was used to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations.
Identification of specialized metabolite biosynthetic gene clusters
The fungiSMASH analysis provided within the antiSMASH v7.1.0 web server (Blin et al. Reference Blin, Shaw, Augustijn, Reitz, Biermann and Alanjary2023b) was used to search for gene clusters involved in SM production. The assembled whole-genome FASTA file was uploaded as an input file to antiSMASH. The respective annotation files in GFF3 format were also uploaded. A detection strictness setting of ‘Relaxed’ was applied, and all available features provided by the web server were chosen, such as ‘Cluster Blast’ and ‘Known Cluster Blast’. These features provide information on similar BGCs identified in other organisms that were available in the antiSMASH repository. Information regarding experimentally validated BGCs and the respective natural products were also retrieved from the Minimum Information about a Biosynthetic Gene Cluster (MIBiG) database. The antiSMASH results were filtered based on two criteria: 1) reference BGC from MIBiG shared at least 60% of genes and 2) the amino acid (AA) sequence of the core gene showed at least 60% similarity to that of the reference BGC.
Results
Fungal culture and whole-genome sequence data
After 10 days of incubation on PDA plates, AKSP4 showed a reddish-pink, fluffy and slightly raised colony with a filiform margin. Exudates were absent from the plate, and the medium was red. The strain HNDR4 colony was whitish-yellow with a powdery surface, irregular and slightly raised aerial mycelium with a filiform margin. Exudates were absent from the plate, and the underside of the colony was yellow (Fig. 1). In the PDB flasks, strain AKSP4 showed red supernatant, whereas that of HNDR4 was yellow. All three PDB flasks chosen for each strain produced averages of 2.63 and 9.04 g pellets for AKSP4 and HNDR4, respectively.
Culture plates of a. red Pseudogymnoascus griseus (AK07KGI 1202 R1-1 sp. 4, strain AKSP4) and yellow Pseudogymnoascus australis (HND16 R4-1 sp. 1, strain HNDR4) after 10 days of incubation at 15°C on potato dextrose agar.

Preliminary DNA barcoding tests confirmed that these strains belong to the genus Pseudogymnoascus. For genome sequencing, a total of 6 742 252 000 bp of raw sequencing data were obtained for AKSP4, which were cleaned and trimmed to produce a final length of 5 943 265 707 bp. In contrast, HNDR4 generated 5 943 265 707 bp of raw data and 3 805 023 098 bp of clean reads. Both strains have ~90% clean reads reaching Q30 (Table I) that are acceptable for downstream analyses.
Summary of the clean data quality as well as the metrics of the final whole-genome assemblies for Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4).

Table I. Long description
See table for full data. Table 1_long-desc.doc
De novo whole-genome assembly statistics
The final assembly of AKSP4 consisted of 2172 contigs, whereas HNDR4 had 586 contigs, with assembly sizes of 35.60 and 32.00 Mb, respectively (Table I). The N50 for AKSP4 and HNDR4 were 80 040 and 214 294 bp, respectively. AKSP4 and HNDR4 showed a BUSCO completeness value of more than 95%, with GC contents of 50.34% and 50.53%, respectively. Approximately 8.05% and 6.67% of repeats such as retroelements, DNA transposons and simple repeats were found in the AKSP4 and HNDR4 assemblies, respectively.
Phylogenetic analyses
BLAST searches for the ITS, LSU and MCM7 sequences from the whole-genome assemblies in the present study were found to have near-perfect similarity to the reference markers used based on E-values, sequence similarities and number of gaps (Table S2). ITS was found in contig AKSP4_768 (nucleotide position: 2926–3496) and contig HNDR4_329 (nucleotide position: 2621–3196). LSU markers were located in contig AKSP4_768 (nucleotide position: 3539–4511) and contig HNDR4_329 (nucleotide position: 3240–4212). Additionally, MCM7 was located in contig AKSP4_20 (nucleotide position: 110 460–111 073) and contig HNDR4_8 (nucleotide position: 376 945–377 575). Both RPB2 and TEF1α sequences were only found in one location in the assemblies of AKSP4 and HNDR4 (Table S3). In AKSP4, RPB2 was found in contig AKSP4_2 (nucleotide position: 221 315–222 278) and TEF1α was found in contig AKSP4_1 (nucleotide position: 323 128–324 061). Both RPB2 and TEF1α were reverse complemented prior to downstream analyses. For HNDR4, RPB2 was found in contig HNDR4_1 (nucleotide position: 547 496–548 459) and TEF1α was found in contig HNDR4_6 (nucleotide position: 263 032–263 967). These identified sequences were used for phylogenetic analyses.
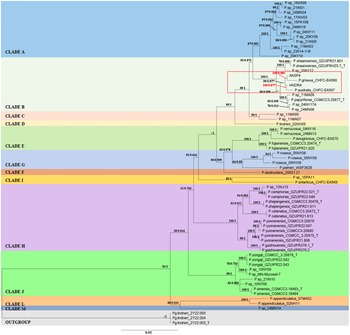
The concatenated alignment of the ITS + LSU + MCM7 + RPB2 + TEF1α dataset from 64 Pseudogymnoascus and 3 Pseudogeomyces taxa produced a final alignment length of 3775 bp (ITS: 370 bp, LSU: 1576 bp, MCM7: 496 bp, RPB2: 526 bp and TEF1α: 807 bp). The best-fit evolutionary model obtained by ML analysis for the concatenated dataset was TPM2 + I + G4 (I = 0.7311, G = 0.5386). Both BI and ML analyses showed similar tree topologies; therefore, the BI phylogram is shown with the support values of ML and BI analyses (Fig. 2). Both analyses identified strain AKSP4 as P. griseus and strain HNDR4 as P. australis (Fig. 2). Strain AKSP4 and P. griseus showed high support values with 100% ML bootstrap with 0.985 PP, whereas HNDR4 and P. australis showed 100% ML support with 0.877 posterior probability (PP) values (Fig. 2). Clade B also contains Pseudogymnoascus shaanxiensis, Pseudogymnoascus papyriferae and other unidentified strains (Pseudogymnoascus sp. 20KY12, Pseudogymnoascus sp. 11MA08, Pseudogymnoascus sp. 04NY17A and Pseudogymnoascus sp. 24MN06).
A phylogram constructed using internal transcribed spacer (ITS), nuclear large subunit (LSU) rDNA, DNA replication licensing factor (MCM7), RNA polymerase II second largest subunit (RPB2) and translation elongation factor (TEF1α) gene regions for 67 strains of Pseudogymnoascus, with Pseudogeomyces spp. as the outgroup. Maximum likelihood (ML) bootstrap value and Bayesian posterior probability (PP) are indicated at each branch (ML/PP). The branch support values for strains AKSP4 (100/0.985) and HNDR4 (100/0.877) are labelled in red. Nomenclature of clades is labelled according to Minnis & Lindner (Reference Minnis and Lindner2013). ‘T’ indicates sequences derived from a type specimen.

Genome annotation
Annotation statistics (Fig. 3) showed similar values for both P. griseus (strain AKSP4) and P. australis (strain HNDR4), with an average percentage difference of ~4.10% between the two genomes. Among CAZymes, both strains have the highest number for glycoside hydrolases (GHs; P. griseus (strain AKSP4): 577 genes; P. australis (strain HNDR4): 570 genes; Fig. 4). Similar trends were observed in the COGs (Fig. 5), where contigs with ‘[S] unknown functions’ were the highest (P. griseus (strain AKSP4): 2733 genes; P. australis (strain HNDR4): 2552 genes) and none were annotated for category ‘(R) general function prediction’.
The number of functional annotations in assemblies of Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4). BGC = biosynthetic gene cluster; CAZyme = carbohydrate-activating enzyme.

Bar chart summarizing the types of carbohydrate-activating enzymes (CAZymes) found in Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4).

Figure 4 Long description
The types of CAZymes on the x-axis are Glycoside Hydrolases, Auxilliary Activities, Carbohydrate-Binding Activities, Carbohydrate Esterases, GlycosylTransferases, Polysaccharide Lyases.
The number of contigs annotated with their respective cluster of orthologous groups (COG) classification for Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4) using the eggNOG mapper.

Figure 5 Long description
See list for the full types of cluster of orthologous annotations on x-axis. Figure 5_long-desc.doc
Figure 6 shows that the distributions of gene functions in both strains are similar, where most genes were linked to functions such as protein families: genetic information processing (P. griseus (strain AKSP4): 806 genes; P. australis (strain HNDR4): 800 genes), genetic information processing (P. griseus (strain AKSP4): 759 genes; P. australis (strain HNDR4): 755 genes) and carbohydrate metabolism (P. griseus (strain AKSP4): 399 genes; P. australis (strain HNDR4): 395 genes). Figure 7 shows that most genes annotated under the BP of GO category are involved in membrane transport (P. griseus (strain AKSP4): 567 genes, P. australis (strain HNDR4): 563 genes), regulation of DNA-templated transcription (P. griseus (strain AKSP4): 349 genes, P. australis (strain HNDR4): 366 genes) and carbohydrate metabolic processes (P. griseus (strain AKSP4): 314 genes, P. australis (strain HNDR4): 310 genes). As for CC, the three highest categories are genes having a role in the membrane layer (P. griseus (strain AKSP4): 861 genes, P. australis (strain HNDR4): 842 genes), the cytoplasm (P. griseus (strain AKSP4): 774 genes, P. australis (strain HNDR4): 750 genes) and the nucleus (P. griseus (strain AKSP4): 768 genes, P. australis (strain HNDR4): 747 genes). Finally, for the MF category, most genes were involved in protein binding (P. griseus (strain AKSP4): 726 genes, P. australis (strain HNDR4): 694 genes), adenosine triphosphate (ATP) binding (P. griseus (strain AKSP4): 609 genes, P. australis (strain HNDR4): 586 genes) and oxidoreductase activity for P. griseus (strain AKSP4; 448 genes), whereas for P. australis (strain HNDR4) more genes were involved in zinc ion binding (451 genes). Genes involved in nucleolus-related functions in CC were not included in the chart for P. australis (strain HNDR4) as it ranked 11th with a gene count of 93. Similarly, genes involved in DNA repair under the BP category were also ranked 11th with a gene counts of 99 for P. griseus (strain AKSP4), and therefore this also was not included in the chart.
Kyoto Encyclopedia of Genes and Genomes (KEGG) functional annotation of genes in Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4) using BlastKOALA.

Figure 6 Long description
Each color in the legend represents a different annotation type. See list for all the annotation types. Figure 6_long-desc.doc
The top 10 highest gene ontology annotation counts for each biological process (BP), cellular component (CC) and molecular function (MF) subgroup for a. Pseudogymnoascus griseus (strain AKSP4) and b. Pseudogymnoascus australis (strain HNDR4). ATP = adenosine triphosphate.

Figure 7 Long description
X-axis is represented by the ontology annotation terms for each subgroups and y-axis is represented by the gene count for each terms for P. griseus (strain AKSP4) and P. australis (strain HNDR4). Biological process subgroup is in green, cellular component in orange and molecular function in purple. See list for the full ontology terms in each subgroup on the x-axis. Figure 7_long-desc.doc
Putative biosynthetic gene clusters and potential specialized metabolites
Totals of 58 and 51 BGCs were predicted in P. griseus (strain AKSP4) and P. australis (strain HNDR4), respectively (Tables S4 & S5). Most of these BGCs were for polyketides (23 and 18 BGCs, respectively; Fig. 8). Based on the selection criteria, several putative BGCs were identified with the potential to produce compounds similar to choline, swainsonine, trichobrasilenol and geodin (Figs 9 & 10 & Tables S6 & S7). Among the predicted BGCs that showed similarities to MIBiG entries, 18 and 15 BGCs (for P. griseus (strain AKSP4) and P. australis (strain HNDR4), respectively) did not fit the two selection criteria.
Bar chart summarizing the types of specialized metabolites (SMs) predicted to be produced by the putative biosynthetic gene clusters (BGCs) predicted for Pseudogymnoascus griseus (strain AKSP4) and Pseudogymnoascus australis (strain HNDR4) using antiSMASH.

Figure 8 Long description
The types of specialised metabolites predicted to be produced by the BGCs on the x-axis are NRPS, NRPS-like, Terpene, PK, indole, Hybrid (NRP/PK), Hybrid(terpene/PK), fungal ribosomally synthesized and post translationally modified peptide-like.
Putative biosynthetic gene clusters (BGCs) for Pseudogymnoascus griseus (strain AKSP4) that fulfilled the selection criteria. The compounds potentially produced are a. choline, b. swainsonine, c. trichobrasilenol and d. geodin. Core genes are labelled with their respective IDs. The proportion of genes in each reference BGC (choline, swainsonine, trichobrasilenol and geodin) that is shared with the predicted BGC regions of P. griseus (strain AKSP4) are indicated within parentheses. Genes with matching colours between the reference BGCs and predicted BGCs of P. griseus (strain AKSP4) indicate that they share sequence similarity between each other.

Putative biosynthetic gene clusters (BGCs) for Pseudogymnoascus australis (strain HNDR4) that fulfilled the selection criteria. The compounds potentially produced are a. choline, b. geodin and c. trichobrasilenol. Core genes are labelled with their respective IDs. The proportion of genes in each reference BGC (choline, geodin and trichobrasilenol) that is shared with the predicted BGC regions of P. australis (strain HNDR4) are indicated within parentheses. Genes with matching colours between the reference BGCs and predicted BGCs of P. australis (strain HNDR4) indicate that they share sequence similarity between each other.

Figure 9 shows that, in P. griseus (strain AKSP4), 100% of the BGCs from Aspergillus nidulans FGSC A4, which produces choline, have a 66% AA similarity to the core gene of BGC region 3.2. The BGC from Alternaria oxytropis, which produces swainsonine, has 83% of its genes shared with that of the BGC in region 53.1, where the core genes have a 67% AA similarity. Approximately 60% of the genes in the BGC of Annulohypoxylon truncatum that produces trichobrasilenol were shared with the BGC in region 96.1, showing 63% AA similarity in the core genes that matched. For BGC in Aspergillus terreus NIH2624 that produces geodin, 75% of its genes are matched to that of region 110.1, where their core genes show 64% AA similarity. Further details regarding AA similarity for the other matched genes can be found in Table S6.
Figure 10 shows that, for P. australis (strain HNDR4), a similar choline-producing BGC from A. nidulans FGSC A4 is shared with the core gene of region 1.1, with a 67% AA similarity. The geodin BGC from A. terreus NIH2624 has 75% of its genes shared with region 11.1, and the core genes have 64% AA similarity. Lastly, 60% of genes in the trichobrasilenol-producing BGC from A. truncatum are shared with that of region 40.1, showing 70% AA similarity in the matched core genes. Further details regarding AA similarity for the other matched genes can be found in Table S7.
Discussion
The genome assemblies generated here are the first for P. griseus and P. australis with sequencing depths of 150×. These are much higher quality than the current genome assemblies of unidentified polar Pseudogymnoascus (GCA_000750925.1, GCA_000750935.1 and GCA_000750995.1) in GenBank (Leushkin et al. Reference Leushkin, Logacheva, Penin, Sutormin, Gerasimov and Kochkina2015); therefore, the genome assemblies of the present two species produced will serve as better reference genomes for future studies. The identification of strains AKSP4 and HNDR4 as P. griseus and P. australis, respectively, is consistent with their placement in Clade B in phylogenies (Wong Reference Wong2019, Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021, Zhang et al. Reference Zhang, Li, Chen, Liang and Han2023), which have at least included ITS, LSU and MCM7 markers. Both strains showed phylogenetic affinity to Clade B (as reported by Wong Reference Wong2019). Similarly, the placement of other Pseudogymnoascus spp. within their respective clades is also consistent with the results from previous reports (Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021, Zhang et al. Reference Zhang, Li, Chen, Liang and Han2023). Interestingly, Clade B also contained other Pseudogymnoascus spp. from locations outside of polar regions, such as P. shaanxiensis (Zhang et al. Reference Zhang, Dong, Chen, Mou, Lu and Han2020) and P. papyriferae (Zhang et al. Reference Zhang, Li, Chen, Liang and Han2023) from China and other unidentified strains from the bat hibernaculum in eastern North America (Lorch et al. Reference Lorch, Lindner, Gargas, Muller, Minnis and Blehert2013). Although these species from China and polar regions share the same clade despite their different geographical locations, each of the species still formed a distinct lineage with strong phylogenetic support values (Zhang et al. Reference Zhang, Dong, Chen, Mou, Lu and Han2020, Reference Zhang, Li, Chen, Liang and Han2023, Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021). Additionally, morphological studies of P. griseus and P. australis (Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021) showed distinct differences in the size and shape of their conidia when compared to other Pseudogymnoascus species from other geographical locations within Clade B (Zhang et al. Reference Zhang, Li, Chen, Liang and Han2023). Moreover, P. griseus (strain AKSP4) and P. australis (strain HNDR4) from the present study showed different culture characteristics and colours when grown on PDA instead of other media types (Villanueva et al. Reference Villanueva, Vásquez, Gil-Durán, Oliva, Díaz and Henríquez2021). Composition of nutrient medium and culture conditions are known to influence the colony morphology and metabolic pathways of fungi (Trivedi et al. Reference Trivedi, Vishwakarma, Saawarn, Mahanty and Hait2022).
Although antiSMASH generated many putative BGCs, metrics such as the proportion and types of genes that are shared and their AA similarities are important factors to be considered. In P. griseus (strain AKSP4) BGC region 3.2 (Fig. 9), the final compound may have a more complex structure than choline, as there are multiple genes that are not found in the reference BGC0002276. Nonetheless, choline is a precursor for phosphatidylcholine (Phillips Reference Phillips, Caballero, Finglas and Toldrá2016) that plays an important role in membrane fluidity regulation (Kanno et al. Reference Kanno, Wu, Scapa, Roderick and Cohen2007), which is crucial for cell survival in cold temperatures. The production of a choline-like compound may serve as a precursor for other phospholipids, potentially helping polar Pseudogymnoascus to adapt to the cold environment that reduces membrane fluidity (Johnson Reference Johnson2018).
Another potential compound is the indolizidine alkaloid swainsonine (also known as ‘locoweed’). It can modify glycoproteins and causes lysosomal storage disease by inhibiting α-mannosidase (Colegate et al. Reference Colegate, Dorling and Huxtable1979), causing severe toxicosis in various livestock that graze on plants such as Oxytropis lambertii (Gardner et al. Reference Gardner, Molyneux and Ralphs2001). Endophytic fungi such as Undifilum oxytropis (Pleosporales) have also been reported to produce swainsonine (Pryor et al. Reference Pryor, Creamer, Shoemaker, McLain-Romero and Hambleton2009). To our knowledge, Pseudogymnoascus spp. have not been found to be endophytic, and they are mostly found in soil, marine sponges or plant/leaf litter. However, the putative cluster in P. griseus (strain AKSP4) shows a high possibility of producing a compound similar to swainsonine, as most of the genes are shared with good AA similarity. Given that this possibility exists, extra care should be taken to prevent P. griseus from being introduced to environments outside of Antarctica. However, in a clinical context, swainsonine is also a potential cancer therapy drug, as in vivo and in vitro experiments with the compound were able to induce apoptosis and inhibit the growth of cancerous human oesophageal squamous cells (Li et al. Reference Li, Huang, Dong, Li, Ding and Yu2012). The combinatorial usage of swainsonine also helped to increase the sensitivity of Ehrlich ascites carcinoma cells to cisplatin in vivo (Santos et al. Reference Santos, Latorre, Hueza, Sanches, Lippi, Gardner and Spinosa2011). This renders investigating the final structure of the swainsonine derivative from P. griseus (strain AKSP4) valuable, as it may show similar, if not more potent, therapeutic effects to other swainsonines.
As regions 96.1 and 40.1 both have the required core gene (brasilane synthase, braA) to modify farnesyl pyrophosphate (FPP) into trichobrasilenol (Feng et al. Reference Feng, Surup, Hauser, Miller, Wennrich and Stadler2020) and other additional genes that are predicted to encode for unknown putative enzymes (Figs 9c & 10c), they are likely to produce trichobrasilenol derivatives, as their shared genes show moderate AA similarity (Tables S6 & S7). However, the effects and significance of trichobrasilenol in the context of polar Pseudogymnoascus remain unclear. Lastly, while regions 110.1 and 40.1 (Figs 9d & 10b) share a high proportion of genes, the AA similarity percentages for each matched gene are only moderate (Tables S6 & S7), suggesting that these regions may only be able to produce a geodin derivative. A previous study has shown that geodin derivatives are able to exhibit significant antibacterial and insecticidal effects (Chao et al. Reference Chao, Said, Zhang, Qi, Hu and Zheng2022). The introduction of a halogenated benzyl, particularly fluorobenzyl, and the substitution of a 4-OH may be the driving factors underlying the increased effects observed. Potentially, P. griseus (strain AKSP4) and P. australis (strain HNDR4) might be able to produce geodin derivatives with similar or more potent effects, giving them great industrial potential and significance.
Additionally, antiSMASH has also shown several BGCs deposited in MIBiG as having 100% of their genes shared with the putative regions found in P. griseus (strain AKSP4) and P. australis (strain HNDR4; Tables S4 & S5). However, these putative BGCs may not necessarily be able to produce derivatives of the linked compounds. This is because they are poor matches due to their very low AA sequence similarities, even though the BGCs have high proportions of shared genes. Additionally, many of the putative BGCs are not linked to any compounds, representing cryptic BGCs. These cryptic BGCs may yet be involved in the production of reported SMs from Pseudogymnoascus. However, due to the lack of experimental work investigating the genes responsible for these reported SMs, they would not be present in the MIBiG database, and hence the antiSMASH results would not show these compounds originating from Pseudogymnoascus. Cryptic BGCs often require specific triggers such as microbial competition through co-culturing of fungi with bacteria or induced environmental stress through changes in temperature, pH or nutritional availability (Fischer et al. Reference Fischer, Schroeckh, Brakhage, Schmoll and Dattenböck2016, Begani et al. Reference Begani, Lakhani and Harwani2018). However, it is difficult to replicate such triggers in the laboratory so as to produce consistent results, possibly due to the effects of multi-environmental parameters and their variations in culture conditions. Hence, attempts to optimize BGC activation and production are complex and challenging. Other strategies also include the refactoring of BGCs into other microbial hosts for heterologous expression (Harvey et al. Reference Harvey, Tang, Schlecht, Horecka, Fischer and Lin2018) or involving gene manipulation by using CRISPR-Cas9 to activate minimally transcribed BGCs (Roux et al. Reference Roux, Woodcraft, Hu, Wolters, Gilchrist and Chooi2020).
The whole genomes in this study could be further refined by incorporating long-read sequencing to enhance contiguity and resolve repetitive regions. Future experimental investigations such as transcriptomics and heterologous expression are necessary to confirm the gene expression and function of these in silico predicted BGCs, especially the production of potentially significant natural compounds. Although Pseudogymnoascus SMs have been identified to have antimicrobial properties, other compounds may have specific ecological and adaptive roles (e.g. fungal development, protection from ultraviolet damage and acting as signalling molecules for interspecies interaction; Keller Reference Keller2019) that have yet to be investigated. Studies looking into SMs produced by Pseudogymnoascus under different stressors will offer insights into its adaptations towards environmental changes. Moreover, both P. griseus (strain AKSP4) and P. australis (strain HNDR4) exhibited high numbers of genes annotated as GHs, which are CAZymes involved in the synthesis and degradation of carbohydrates. Many GHs are important ecological chemicals, having roles in nutrient cycling, stress adaptation, microbial competition and development (Lyu et al. Reference Lyu, Shen, Fu, Xie, Jiang, Li and Cheng2015, Van Munster et al. Reference Van Munster, Nitsche, Akeroyd, Dijkhuizen, Van der Maarel and Ram2015, Rafiei et al. Reference Rafiei, Vélëz and Tzelepis2021). Further annotations to elucidate the identities and functions of GHs in polar Pseudogymnoascus would be valuable to understanding their various survival and adaptation mechanics in harsh cold environments.
Conclusion
The whole genomes assembled in this study are the first for polar Pseudogymnoascus fungal strains, generating ~35.60 Mb and 32.00 Mb genomes for P. griseus (strain AKSP4) and P. australis (strain HNDR4), respectively. This study is also the first to report P. australis inhabiting the Arctic region. Totals of 58 and 51 BGC regions were predicted for P. griseus (strain AKSP4) and P. australis (strain HNDR4), respectively, showing their potential to produce compounds similar to choline, swainsonine, trichobrasilenol and geodin. These metabolites have potential applications in the biotechnological industry as antibacterials, anticancer drugs and insecticides. Finally, the high-quality genomes produced in this study can serve as valuable references for future studies involving polar fungi.
Availability of data and materials
All sequence reads for both strains are deposited in the NCBI Sequence Reads Archive (SRA) database: P. griseus (strain AKSP4) at SRA ID: SRR31077044 and P. australis (strain HNDR4) at SRA ID: SRR31077043. The whole-genome assemblies generated for P. griseus (strain AKSP4) is available at JBJRFJ000000000 and that for P. australis (strain HNDR4) is available at JBJRFK000000000. The related BioProject is also deposited under PRJNA1156921, whereas the BioSample for P. griseus (strain AKSP4) can be found at SAMN43503273 and that for P. australis (strain HNDR4) can be found at SAMN43503274.
Supplementary material
To view supplementary material for this article, please visit http://doi.org/10.1017/S0954102026100741.
Acknowledgements
We thank the Data Intensive Computing Centre (DICC), Universiti Malaya, for providing the computational resources to carry out the required analyses. All fungal samples were provided by the National Antarctica Research Centre (NARC), Universiti Malaya.
Author contributions
MRI, SAA and TTC conceptualized the project and acquired project funding; TTC obtained the preliminary whole-genome sequence dataset of fungal samples; SAA and JS aided in the collection of the AKSP4 sample; SAA and MGA aided in the collection of the HNDR4 sample; LSY constructed the methodology; LSY performed the formal analysis and the original draft preparation; LSY, MRI, SSL, SAA, TTC, JS, MGA and ZHL reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Financial support
This project was supported by the Ministry of Higher Education (HICoE IOES-2023E), Yayasan Penyelidikan Antartika Sultan Mizan (GA006-2022), the Flagship GA006-2014FL Grant and the Long-Term Research Grant Scheme (LRGS) LR002-2011A from the Ministry of Higher Education Malaysia and Universiti Malaya Scholarship Scheme (UMSS).
Competing interests
The authors declare none.













 Open access
Open access