

I. Introduction
[I]t must be admitted that there is much we still do not understand about the pharmaceutical innovation process. As always, more work remains to be done.Footnote 1
Commentators often embrace theories when attempting to explain behaviour and justify the law. As we accumulate more knowledge, we refine these theories, making them more nuanced and filling in gaps. This paper concerns improvements to our understanding of the incentives and resources necessary for pharmaceutical innovation (i.e. “drug R&D”).
An often unstated assumption of the innovation theory underpinning pharmaceutical law is that companies are necessary to develop new medical treatments. This assumption consists of three parts. Companies are necessary because they have: (1) expertise in drug R&D; (2) a high appetite for risk; and (3) sufficient capital (or can raise it). Hospitals, universities and research institutes only feature in the broader landscape. They make breakthroughs in fundamental science and sometimes conduct early clinical studies for drugs that receive authorisations.Footnote 2 However, empirical studies show that pharmaceutical companies, as a general rule, conduct the late-stage R&D for drugs that enable regulatory authorisations,Footnote 3 which are necessary before they can be sold.Footnote 4
This paper does not challenge the generalisation that companies obtain almost all regulatory authorisations or that they are necessary for developing new compounds. Rather, it proposes a new, complementary theory of pharmaceutical innovation based on recent evidence that hospitals and universities conduct a similar number of late-stage trials compared to companies for new uses of (already) authorised drugs, particularly once generic versions of the drugs are available.
The process of finding new uses for authorised drugs is commonly known as “repurposing”. Early in a drug’s lifecycle, companies are interested in repurposing and recent data indicate that, on average, they obtain regulatory authorisations for 32 new uses annually.Footnote 5 The authorisations permit the drugs to be marketed for those new uses (as well as the old ones). However, once generic drugs enter the market, companies lose interest in repurposing due to competition. Generic drugs are authorised copies of the compounds that enter the market after key patents expire, and competition from generics leaves hospital and university trials for new uses unauthorised by a regulator, as companies are no longer interested. These trials, in turn, contribute to so-called “off-label” use, in which doctors still prescribe the drugs for the use, despite the lack of regulatory review, a phenomenon described more below, with various negative impacts.
Another assumption of established pharmaceutical innovation theory is that patents are necessary to incentivise drug R&D.Footnote 6 Patents provide 20 years of exclusive rightsFootnote 7 and pharmaceutical innovation theory, which draws heavily on patent justifications, explains that the exclusive rights enable pharmaceutical companies to shore up profitability and investigate all applications of their compounds. No patent, no innovation. Yet, the majority of hospital and university trials described above are almost certainly conducted without the prospect of patents (or other commercial rights), primarily due to challenges enforcing these rights. Thus, established pharmaceutical innovation theory does not fully account for the large numbers of hospital and university trials. A new theory is needed.
What motivates investigators at hospitals and universities (and other similar institutes) to conduct these clinical trials if not patents coupled with market-based incentives?Footnote 8 This paper identifies three groups of developers who respond to other incentives. First, scientists who run trials respond to incentives associated with “open science”. Second, clinicians who run trials respond to a mix of incentives associated with open science and “user innovation” and, third, patients, who often play key roles in trials, respond to incentives associated with user innovation as well. The substance of these incentives is detailed below, but brief explanations of each are helpful now. Incentives from open science explain, for example, why physicists research fundamental laws. In short, scientists need to publish studies to progress their careers and the more original and insightful the studies are, the faster the progression.Footnote 9 In contrast, incentives from user innovation explain why computer programmers write code for open source software when they do not obtain a direct financial benefit. The incentives also extend to surgeons creating new products to assist patient recovery.Footnote 10 The incentives are underpinned by the innovators deriving personal satisfaction and benefitting personally from improved products, amongst other advantages.
Commentators have known for 30 years about hospital and university clinical trials that are not designed to attract the commercial pharmaceutical industry’s attention; however, the topic has been left as a “hidden research system”Footnote 11 that has been “manifoldly present but largely unaccounted for”, especially regarding hospital trials.Footnote 12 One reason these trials have gone unaccounted for is that established pharmaceutical innovation theory assumes that companies are necessary for drug R&D because the resources they control, such as financial capital and manufacturing capabilities, are barriers to innovation for other types of organisations.Footnote 13 This paper does not argue that hospitals and universities have these resources, but it does show that these barriers are lower for hospitals and universities when repurposing, often substantially. For example, data show that the funded costs of hospital and university trials are less than 10 per cent of pharmaceutical companies’ reported costs.
We call the new theory explaining hospital and university drug R&D the “Republic of Translational Medicine”, drawing on pioneering work on incentives in academia that described scientists operating as a decentralised body politic producing new information, a “Republic of Science”.Footnote 14 The Republic of Translational Medicine combines incentives associated with open science and user innovation, distinguishing it from the Republic of Science, which omits incentives from user innovation. The Republic of Translational Medicine has at least two benefits for society. One is that it leads to lower-cost treatments (compared with drugs developed primarily by pharmaceutical companies) and the second is that it will produce treatments that pharmaceutical companies would develop only if they were granted greater financial incentives.
Tacitly aware of the advantages of the Republic of Translational Medicine (but not its details), the EU, US, Australian and UK governments have begun piloting policies, including new legislation, to convert hospital and university trials into authorised treatments.Footnote 15 Commentators propose that the government programmes, coupled with hospital and university trials, represent a new model of drug R&D.Footnote 16 However, these commentators never considered the magnitude of hospital and university trials, nor how the trials are possible, especially regarding incentives and resources. This study does, placing society in a better position to consider the role of the new theory and the new model in society.
Another facet of the Republic of Translational Medicine is that it relies on patients (not patents) to carry parts of the research burden. Patients have played key roles in the pharmaceutical industry for over 20 years, albeit their value is often underestimated or ignored and we are only just beginning to understand their role in repurposing. The insight that patients play roles in R&D is more significant than an honest accounting of contributions: we need to understand their role before we can optimise outcomes from the system.
This paper is organised as follows. After this introduction, Section II summarises established pharmaceutical innovation theory and describes the data on clinical trials that established pharmaceutical theory cannot explain. Section III then articulates how incentives associated with open science and user innovation help explain the relatively high number of hospital and university trials and how the barriers to conducting trials (expertise, risk and capital) are lower than commonly thought. This study, in effect, helps to see the whole of a “hidden” research system. The last substantive section, Section IV, combines the new theory with commentary on the new model and provides reflections for the future, particularly regarding the new government programmes. A short conclusion follows.
II. A Collision of Theory and Evidence
A. Background Theories
This section begins by describing patent law justifications that underpin established pharmaceutical innovation theory. The insights provided by these justifications help contextualise the data on hospital and university clinical trials and, therefore, help to understand the differences from the Republic. Two patent justifications are central to pharmaceutical innovation: incentive theory and commercialisation theory.
Incentive theory is perhaps the most influential justification. The theory sees patent law as a legal fix to an economic problem known as “free riding”. Patents are granted for new, inventive and practical applications of “information”,Footnote 17 but information has problematic characteristics under free market conditions. Information is “non-rival”, which means that use by one person does not diminish the opportunities for use by others and it is “non-excludable”, which means that, without legal protection, information producers cannot prohibit others from using it. Absent legal intervention, these characteristics mean that once information is publicly available, third parties can “free ride” on it because they do not have to pay what it costs to produce.Footnote 18
In the pharmaceutical industry, a free rider can enter the market with lower production costs and, therefore, undercut the company that first produced the information. The public would likely benefit from this free riding in terms of lower prices. However, free riders evoke significant concern in drug R&D due to the high risks and upfront costs for originating companies seeking to get a drug to market. Studies estimate the chance of getting a new drug authorised at 10 per cent from the start of human trialsFootnote 19 and the mean out-of-pocket expenses to develop and authorise a new drug at US$322–1,540 million (£240–1,204 million; €288–1,445 million).Footnote 20 Why would any company risk investing in drug R&D if its competitors will prevent it from recouping the costs? Thus, commentators favour patents for drug R&D because patents provide robust, property-based protection for a return on investment on inventions.Footnote 21
It is also important to remember that R&D expenditure does not end the day an inventor discovers something new and applies for a patent. Many inventions require substantial post-invention development. Some people describe efforts to translate new inventions into marketable products as “post-invention” activity and commercialisation theory specifically supports post-invention activity.
The pharmaceutical industry has extensive post-invention development costs. When drugs are first invented, there is no guarantee that they are safe or effective and companies, understandably, proceed with caution. Studies show that the average time from patenting a compound until reaching the market is around 12 yearsFootnote 22 and clinical trials are the best-known aspects of this post-invention activity. In addition, though, companies need to invest in: (1) new manufacturing capacity; (2) distribution channels; (3) marketing strategies; and (4) health technology assessments (which evaluate the value of a drug for healthcare systems). These processes involve various experts, including financiers, chemical engineers, health economists, pharmacists, clinical trial managers, regulatory affairs specialists and lawyers.Footnote 23
According to commercialisation theory, the early privatisation of new inventive information through patents (e.g. 12 years before authorisation) is a feature, not a bug, of the system for several reasons.Footnote 24 First, it provides a secure legal framework for the property owner to cultivate their new resource.Footnote 25 Although (absent legal intervention) information is a public good without scarcity, there is scarcity in the real-world resources required to use information and the costs of hiring scientists and engineers, as well as laboratory space and equipment, soon add up.Footnote 26 Second, commercialisation theory explains that patent law efficiently allocates rights to people (or their employees) with expertise in the technology, who then, operating as market participants, will choose an efficient path to bring products to market.Footnote 27 Third, the early patenting of information helps to reduce the duplication of investment in new technology among competing parties. It is not uncommon for multiple inventors to consider working on the same problem.Footnote 28 Patent applications are often not published until 18 months after they are filed;Footnote 29 but, otherwise, the patent register provides inventors with insight into solutions to their problems or, perhaps, the possibility that no one has solved their problem. If an inventor’s problem has been patented (and published), they must either branch off in a new direction, design around the patent or cooperate with the patentee.
Commercialisation theory offers much insight into the patenting and innovation process, and prospect theory, a type of commercialisation theory, has specific implications for repurposing.Footnote 30 Prospect theory emphasises patentees maximising the value of patents because it is in their interest to find every commercially viable product.Footnote 31 The more uses of an invention discovered during the duration of a patent, the more potential returns on investment. Plus, the innovation process is often cyclical, with new inventions frequently bubbling out of the later stages of earlier ideas.Footnote 32 Repurposing is an example of inventors maximising the value of their patents by finding new uses for an invention originally developed to solve a different problem.
B. The Application of Patent Justifications to Drug R&D in General and Repurposing Specifically
Pharmaceutical innovation theory draws heavily on patent incentive theory and commercialisation theory. The central role of patents in pharmaceutical innovation theory is relatively uncontroversial. Indeed, it is common to hear practitioners and commentators describe the pharmaceutical industry as the “poster child” of the patent system. For instance, Burk and Lemley argue that drug R&D would likely “drop substantially” without patent protection.Footnote 33 They make this argument because pharmaceutical companies that produce new drugs (commonly known as “originators”) expect that generics will rapidly enter the market after the expiration of their patents. If patents were not available, generics would likely enter earlier.
Other legal rights support drug R&D, particularly market and data exclusivity (often called market and data protection) that originators obtain when regulators authorise their compounds for public use. In Europe and the UK, data exclusivity stops companies from obtaining authorisations for generic drugs. Applications for generic drugs rely, in large part, on the data submitted by originators as part of their initial authorisation applications and data exclusivity prevents generic applications from relying on the data for eight years. Similarly, market exclusivity stops generic companies from selling their products for a further two years, even after the generic has been authorised.Footnote 34
As market and data exclusivity only arise after a product is authorised, these legal protections provide limited utility during the R&D process. For example, a university would struggle to attract a licensee if it advertised “potential market exclusivity” for a new compound without a patent. Patents stop all other competitors from using the invention for commercial purposes; thus, one advantage of obtaining a patent is that the licensee can pursue numerous options without competition. If they do not have a patent, any organisation can use the invention unless it is a trade secret; and secrets are hard to keep in the drug industry due to the need to involve many parties, the need for clinical trials and the requirements for regulatory disclosure. Another reason a university would struggle to find a licensee is that market and data protections do not stop competitors from producing and receiving an authorisation for the same treatment. These authorisations would not be as generics; data exclusivity prevents that. Instead, the authorisation application would follow a procedure akin to that for new compounds.Footnote 35
The discussion so far has primarily concerned patent incentives for new drug R&D, not repurposing as such. That said, the theory for repurposing is similar. Theorists and commentators have imported patent justifications for new drugs into the innovation theory for repurposing. Repurposing is generally cheaper and has a higher chance of authorisation compared with developing new drugs. That said, the cost and risks are still significant. Estimates suggest that repurposing has a 20 per cent chance of reaching the market from initial trials in humans and costs hundreds of millions of US dollars.Footnote 36 Thus, commentators state that patents are necessary to encourage companies to repurpose drugs.Footnote 37
Patent considerations, however, pose problems when repurposing. Commentators emphasise two aspects of the law that render patents less valuable, undermining the incentives to repurpose drugs. These aspects have been described for around 20 years in multiple jurisdictions.Footnote 38
The first aspect concerns patent validity and consists of three elements. First, patents are only valid if they describe novel inventions in the sense that the inventions have not been disclosed to the public before the application was made.Footnote 39 Novelty for repurposing means that an inventor’s new use must not be described in the scientific literature (or anywhere else publicly); otherwise, the patent will not be granted (or will be revocable). Second, patented inventions must involve an inventive step.Footnote 40 This requirement demands that the new use is not obvious based on prior knowledge available to the public. This requirement can be challenging to meet for repurposing because determining whether a new use is obvious involves considering what scientists regard as obvious, including expected results from routine experiments.Footnote 41 A third challenge concerns the description of the invention in the patent document, including supporting data. The European Patent Office and various Contracting States to the European Patent Convention 1973 demand that new-use patents provide evidence that the drug plausibly treats a new condition.Footnote 42 Such evidence may include data from clinical trials or animal experiments and there is a risk that it may not meet the threshold, or while developing the data, someone may describe the use publicly, undermining novelty or inventive step.Footnote 43
Traditionally, the validity issues have received significant attention, probably because courts have invalidated patents on them.Footnote 44 Yet, a recent study of patent applications and grants casts doubt on the significance of the validity issues. The study found that applications and granted patents for new uses have increased over the past decade and that,Footnote 45 since 2014, 62 per cent of applications for new uses have been granted, which is higher than the average for all technology at 49 per cent.Footnote 46 Thus, perhaps securing patent protection for repurposing is not such a problem in practice. That said, some companies may avoid developing new uses if they know that, for example, a use has been described publicly before (compromising novelty), even though no competing organisation is researching in the area.
The second, and perhaps more important, aspect of the law that renders patents less valuable for repurposing concerns enforcement.Footnote 47 The problem only applies to drugs for which generics are available. If an originator obtains an authorisation for a new use and has a patent on that use, it can use the patent to stop generic manufacturers from putting the new use on their labels (and other informational inserts). As a result, generic manufacturers are compelled to refer in their packaging to the previously known, now-unpatented uses only – a practice known as “skinny labelling”.Footnote 48 However, skinny labelling often does not stop generics from being used for patented indications due to the way clinicians and pharmacists operate. Clinicians (and other medical professionals) often prescribe drugs by scientific names (e.g. ibuprofen), not brand names (e.g. Nurofen) and without any reference to a patient’s condition,Footnote 49 and then pharmacists frequently dispense generic drugs for patented uses, subject to dosage differences, because they are unaware of the patient’s condition. Indeed, many countries, including the UK, have created systems that financially encourage pharmacists to dispense generics whenever possible.Footnote 50 When generics are dispensed for patented uses listed on other versions, it is called “cross-label use” because the generics are effectively dispensed with reference to the other labels, even if doctors and pharmacists are unaware that the generic does not include the use on its label.
Pharmaceutical companies have litigated cross-label use worldwide because it deprives them of sales. The law is complex and whole books are dedicated to the topic.Footnote 51 However, a common refrain that two commentators have neatly captured is that “if a generic version of a drug is available, developers have little or no opportunity to recoup their investment in the development of the drug for a new [use]”.Footnote 52
One final point needs to be highlighted before moving on. Established innovation theory explains that for-profit companies are needed to repurpose drugs because of the scientific expertise, risk and capital involved. This section has not dwelt on these aspects because they were discussed in the introduction and they often go without saying. Indeed, commentators frequently assume companies are needed and omit to examine whether other organisations could perform the research.Footnote 53 Other times, commentators specifically engage with the idea and conclude, for example, that “the government either does not or cannot offer adequate financial support for this research”.Footnote 54
C. Theory and Evidence
A 2022 study sought to evaluate the claim that “if a generic version of a drug is available, developers have little or no opportunity to recoup their investment” due to cross-label use.Footnote 55 The study interpreted this comment to mean that clinical trials on new uses should significantly slow or stop as generics approach the market.Footnote 56
The study evaluated whether clinical trials slow or stop by tracking the number of clinical trials for new uses over the lifetime of all drugs for which generic authorisation was first granted in the US in 2014 (15 drugs). The study only counted mid- and late-stage clinical trials, respectively known as phase II or III trials.Footnote 57 These are the types that commentators typically think are beyond hospitals’ and universities’ resources and expertise.Footnote 58 Early-stage phase I trials are smaller and less expensive.
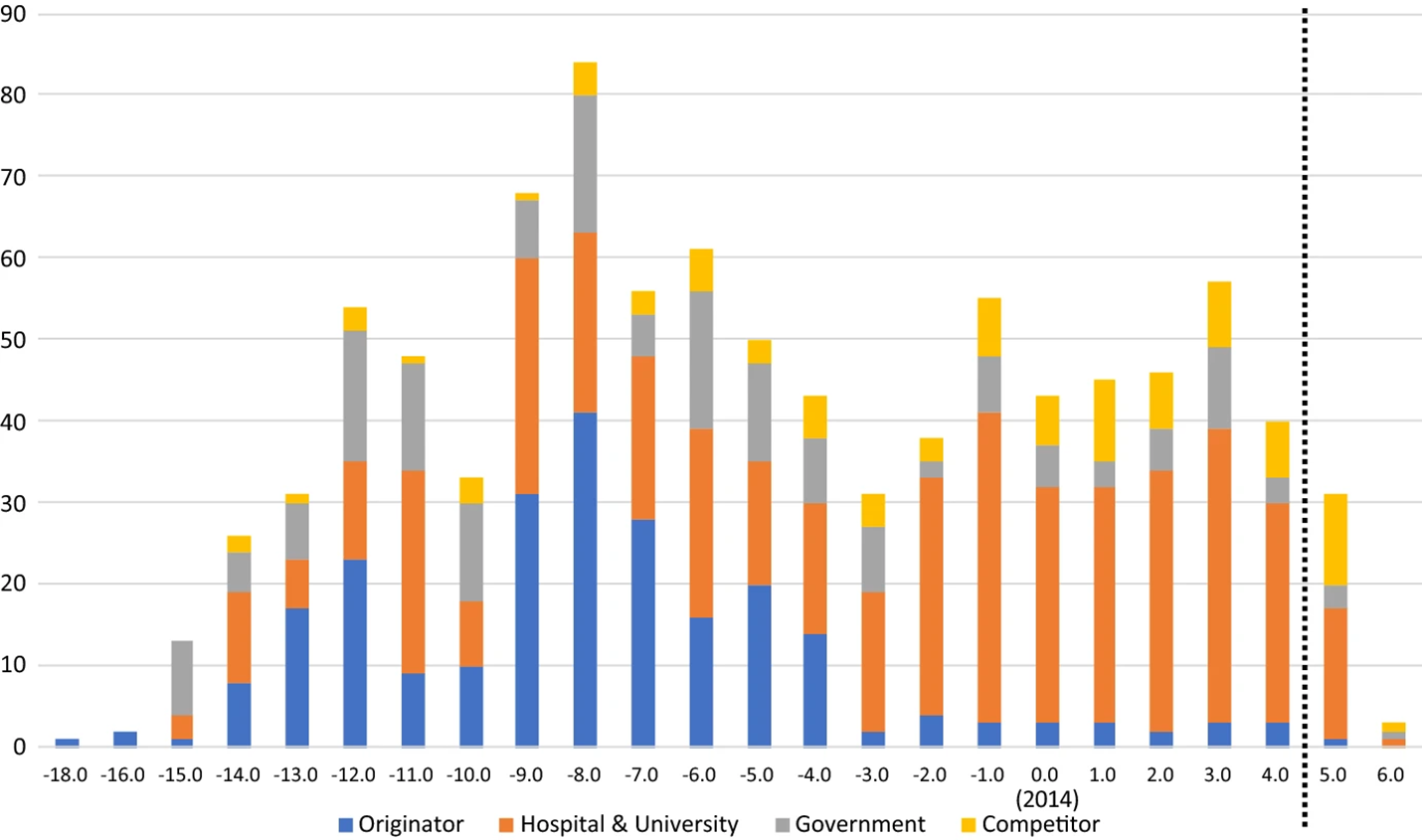
The study’s results align with one aspect of the established theory:Footnote 59 before generics entered the market, originators conducted many clinical trials looking for new uses. Figure 1 shows that eight years before the generics were authorised, the total number of trials for all drugs was 84 trials (on average, 5.6 trials per drug). This finding is consistent with prospect theory; the idea that patentees endeavour to maximise the value of their patents seeking all commercial applications of their inventions.
shows all clinical trials over time grouped by sponsor type. The generic entry date of all the drugs was aligned at day 0 in 2014 and, therefore, the years on the y-axis correspond to the years before and after generic entry. Years after the dotted vertical line are underestimates due to lack of available data.

Although the results align with one part of the established theory, the results show an unpredicted outcome on whether trials slowed or stopped. The study indicates that originators’ trials peak eight years before generic authorisation and then decline as generics approach.Footnote 60 That was to be expected. However, the study also found that the overall number of trials did not slow or stop. What the study found was that hospital and university trials increased once generics were authorised. Indeed, the study shows that hospitals and universities conducted more trials (237) in the five years after generic authorisation compared with the originators’ (223) before generic authorisation.Footnote 61 Moreover, the hospitals and universities generally did so without the patent incentive, due to cross-label use. Established pharmaceutical innovation theory does not predict these trials. Nor does any other theory.
III. The Characterisation of a New Theory of Pharmaceutical Innovation
How can hospitals and universities conduct a relatively large number of trials without the resources of pharmaceutical companies and the promise of enforceable patent rights? The theory proposed in this paper will be described in two steps. First, by analysing the resources required for drug repurposing R&D and, second, by analysing incentives in the absence of patents.
A. Scientific Expertise, Risk and Capital
It is helpful to view the resources needed for drug R&D as three barriers to innovation: expertise, risk and capital. These barriers are high for organisations that develop new compounds. However, the starting point for the Republic of Translational Medicine theory is that all the barriers are lower when repurposing generic drugs.
The first barrier concerns expertise. Originators require extensive expertise in various scientific domains when developing new drugs. However, generic drugs can only enter the market once patents on the compounds expire, which typically means the patent for the drug was first applied for at least 20 years previously. Another way to think about generic drugs is in terms of standards: originators set a standard and generic companies emulate the standard with their versions. High levels of scientific expertise are often not required with all aspects of standards.
Primary investigators who run clinical trials to repurpose generic drugs need scientific expertise and they accrue this knowledge through various channels. A common theme from scientists’ presentations at repurposing conferences, which generally focus on the science of repurposing but also include social and legal dimensions, is that the investigators specialise in the area of medicine in which they run trials. Moreover, they often run trials after three events. First, the drugs have been given to patients off-label (as a reminder, an unauthorised use not listed on the drug’s label or any other version of the drug) with promising results. Off-label use is common in some areas of medicine, for example, constituting up to 71 per cent of prescriptions in areas of oncology.Footnote 62 An EU Commission report found that off-label use is generally lawful, too, if certain safeguards are met, such as patient consent.Footnote 63 Second, published articles indicate that the drugs could treat the new disease. The types of articles vary. Some papers might propose theories or describe biochemical studies of how drugs operate in human bodies, suggesting the drug might treat the new disease. Other, more advanced papers might collect data on off-label use, perhaps across multiple hospitals and even compare outcomes to different treatments. And third, the investigators are awarded funding to run the trial, including obtaining ethics approval. In short, the investigators’ experience, scientific studies, off-label use and approvals from grant and ethics bodies mean that investigators have sufficient expertise.
Repurposing may involve changes to the generic. Put otherwise, the standard changes. For example, a greater or lesser dose may be required, the drug might need to be administered differently (e.g. via injection instead of orally), or it might need to be taken in combination with another drug. Often, these changes are still within the primary investigator’s (or the wider research team’s) capacity. For instance, it is common for clinicians to prescribe dosages different to those on labels (e.g. for children)Footnote 64 and drugs are frequently given in combination with other treatments (again, after 20 years, often plenty of data will be available on drug combinations).Footnote 65
Some changes may exceed the team’s skill set, such as changing the route of administration from a tablet to a patch. However, whether this change exceeds the skill set depends on the team. Many teams have access to formulation scientists or pharmacists within their institutes. Two examples illustrate this point. First, two of the authors were recently invited to a university trial of a vitamin D patch that had been reformulated at the university. Second, numerous hospital and university trials show that bevacizumab, which is authorised for intravenous infusion (e.g. via a catheter in the arm) for various cancers,Footnote 66 can be used to treat age-related macular degeneration (“AMD”) at a much lower dose via intravitreal injections (i.e. injection into the eye).Footnote 67 Bevacizumab is not authorised for AMD in the UK. Yet, the National Institute of Clinical Evidence, the government body that reviews the cost-effectiveness of drugs, recommended it be used off-label for AMD despite the availability of authorised alternatives.Footnote 68 The issue of off-label uses going unauthorised is a topic that will be returned to below. Suffice for now, though, investigators (and their teams) can have the skills to tweak the composition and delivery of generic drugs.
When considering expertise, it is also important to acknowledge areas of expertise that hospitals and universities do not need. Two areas are manufacturing and distribution. While companies invest heavily in these capabilities, hospitals and universities generally do not need to replicate them, since originators and generic companies already supply the drugs (even if tweaking is needed). If hospitals and universities were required to manufacture at scale, their trials would likely not occur. As it stands, their role is more oriented to providing intellectual contributions.
The second barrier concerns risk. From the established theory’s perspective, the issue is that a high appetite for risk is required because one organisation is responsible (or perhaps a few under contract) for developing a drug, and hospitals and universities cannot bear this risk. As a reminder, pharmaceutical companies risk large sums of money (hundreds of millions of US dollars) on a relatively small chance of regulatory authorisation (20 per cent from the start of trials).
In the Republic, governments, hospitals and universities assume the risk for drug R&D. Governments are mentioned because they are the primary organisations funding the clinical trials. These three organisations, however, have different risk–reward structures and, in general, the rewards are likely to be realised.
Governments generally have no interest in directly selling drugs. Their concern lies in improving health outcomes and expanding the number of available treatments while keeping costs manageable for healthcare systems. Thus, they typically award grants when the social value of the research exceeds its market value.Footnote 69 Indeed, this foundational idea for grants helps explain why so many trials are funded for repurposing: commercial science has little interest in repurposing generic drugs due to patent-related challenges, whereas patients and health systems will benefit via off-label use and new government programmes (discussed below). Moreover, whereas companies view results showing a new treatment is ineffective as a failure, governments (via health systems) often see them as valuable: off-label use arises when patients have no better options, and such “negative” results redirect clinicians and patients towards other treatments. Thus, governments have clear and likely rewards when awarding grants for repurposing.
Hospital and university risk–reward structures are also starkly different from company structures. Hospitals’ and universities’ interests primarily revolve around treating patients and academic outputs. Investigators can generally publish papers if the trials are well designed, even if the results are “negative” because they inform clinical practice.Footnote 70 Patient treatments (or impact from universities’ perspectives) will also likely follow, especially via off-label use (including the possibility that “negative” results will redirect treatments). Thus, hospitals and universities have clear and likely rewards, too.
Primary investigators also manage risk and their roles should not be ignored. The investigators already have “a win” when they obtain a grant. Their projects are usually self-contained and conclude when the trial ends, regardless of the trial’s outcome. The value of the paper to their careers depends on the results, but this is a risk scientists eagerly take and they do not face the risk that poor trial results would undermine the aim of selling a drug. Perhaps they will want to conduct a follow-up trial (e.g. a more extensive phase II or phase III trial), but no money rides on that decision either (and their institutes typically have little expectation they will). All these perspectives demonstrate that the high appetite for risk under the established theory is immaterial in the context of the Republic.
There are criticisms of government funding of clinical trials. One is that grants are often difficult to obtain, with success rates of 20 per cent or less; these low success rates increase the risk that positive results in phase I or II might not be followed up in later trials because investigators cannot secure additional funding. However, this argument omits another feature of the Republic: risk decentralisation. In the Republic, the development of compounds is divided into numerous organisations, akin to the accretion of scientific knowledge in open science, in which promising results attract broad interest. The development of bevacizumab for AMD demonstrates this decentralisation. A non-exhaustive review of the clinical studies shows over 25 institutes involved across several continents.Footnote 71
The third barrier is capital. The most cited study on the cost of clinical trials found that the average out-of-pocket cost for phase III trials is US$255 million (£191 million; €228 million).Footnote 72 Commentators have criticised these values because the underpinning data are not publicly available.Footnote 73 However, even if the values were assumed to overestimate the cost fourfold, the “real” cost of US$64 million (£48 million; €57 million) would still exceed what hospitals and universities could routinely afford. Or, put another way, one does not hear of enough grants worth this much being awarded to hospitals and universities to explain the large numbers of trials in Figure 1.
One might wonder: how can clinicians and scientists afford to run the clinical trials in Figure 1? The answer is that non-commercial trials are much cheaper, as demonstrated in two studies on the cost of non-commercial trials. Unfortunately, it is challenging to identify precisely how these trials can be so much cheaper because the values are calculated differently. Nevertheless, the studies are good enough to show marked differences in price and explain the numbers in Figure 1.
The first study on trial costs aimed to develop a cost-prediction tool for non-commercial trials. One of the study’s underpinning ideas was to ensure that trials are not underfunded.Footnote 74 The tool was in its fourth version, and the article details how the authors identified the costs and how investigators can use the tool to estimate costs. The study did not calculate the costs of proposed trials; however, the article notes that 12 multicentre, interventional randomised trials have been funded with the tool, with a median cost of “nearly €2,000,000” (£1,710,000; US$2,150,000).Footnote 75 The article did not state the phase of the trials, but the trials were interventional and multi-site, indicating they were probably phase III.Footnote 76 Relative to the average cost for phase III commercial trials (i.e. US$255 million), the funding for these non-commercial trials is 0.8 per cent the price.Footnote 77
The second study obtained cost estimates for clinical trials from five academic clinical trial units in the UK. The units are specialist research entities that design and coordinate clinical trials. Each unit provided an estimate for the same study: a 5.5-year, surgical (non-pharmaceutical) trial conducted across 20 hospitals, recruiting ∼830 patients.Footnote 78 In terms of numbers and duration, this is equivalent to a drug-based phase III trial. The average funded cost for these trials was £770,000 (US$970,000; €919,000) with a range from £660,000 to £1,380,000 (∼US$879,000–1,838,000; €788,000–1,647,000).Footnote 79
The costs for the surgical intervention included 24 months of recruitment and 24 months of follow-up with patients, but, as the trials were surgical, they excluded the costs of pharmaceuticals.Footnote 80 Thus, it is necessary to account for drug costs if the study is to be useful for current purposes. Although the first study on the cost-prediction tool omits details of the funded studies, it includes an example of costs and describes the main cost drivers. The cost example lists the purchase of drugs at €150,000 (7 per cent of the total grant) and the authors describe this cost as a “low impact” driver relative to other high-impact drivers (e.g. patient monitoring and project management).Footnote 81 That said, the authors acknowledge that the costs of drugs will vary; thus, if the cost of drugs is to be factored into the costs in this second study, a safe estimate is to double the cost, changing the impact from 7 to 14 per cent.Footnote 82 For purposes of estimating the costs of clinical trials, it is better to overestimate drug costs than to underestimate them.
If 14 per cent is added to the second study’s costs, the numbers are within the range of what grant bodies routinely award. The mean cost becomes £878,000 (US$1,106,000; €1,048,000) with a range from £752,000 to £1,573,000 (US$1,002,000–2,095,000; €898,000–1,878,000).Footnote 83 Another way to look at these values is as a percentage of the commercial costs for phase III trials mentioned above. In this case, the average funded cost is 0.5 per cent of the cost of commercial phase III trials, with a range of 0.4 to 0.8 per cent. Thus, the two studies on non-commercial clinical trials provide relatively concordant results, with the first study’s average funded cost coming to 0.8 per cent the cost of commercial trials.
Before moving on, it should be noted that data on the cost of commercial trials can vary significantly and that lower estimates alter comparisons with non-commercial trials. For instance, another study found the average cost of phase III commercial trials is US$33,000,000 (£24,773,000; €29,571,000).Footnote 84 However, even with this lower value, the two estimates for non-commercial trials remain less than one-tenth of the price.
B. If Patents Are Not Incentivising Innovation, What Is?
Before searching for new incentives, we need to reconsider whether patents are playing a role. One could argue that most hospital and university clinical trials after generic entry are all responding to patent incentives. Support for this argument can be found in the study that shows obtaining patents (i.e. validity) is not a significant issue. However, the argument is almost certainly wrong for three reasons. First, there is a consensus in the literature from the last 20 years that enforcing patents for repurposing new uses is challenging, primarily due to cross-label use and no evidence or body of literature contradicts this point.Footnote 85 Second, the authors frequently hear these issues repeated from industry executives, university technology transfer experts, clinicians and scientists. And third, a recent interview with experts at government repurposing programmes suggests most investigators are aiming for off-label use, which does not require patent protection.Footnote 86 Thus, while patents might incentivise some trials with generic drugs, most are likely driven by something else.
If patents are not incentivising the hospital and university trials, what is? Open science provides the first part of the answer. The term “open science” can be used in several related contexts. Recently, the term has been used to emphasise a “set of principles and practices that aim to make scientific research from all fields accessible to everyone for the benefits of scientists and society as a whole”.Footnote 87 However, the term can also be used as a synonym for academic science, with openness referring to the practice of being open to all people, as well as open publishing. In this sense, the term also extends to describing the culture, norms and institutions that exist within it.Footnote 88 This study uses the term in this wider sense.
Central to this study, economists have examined the motivations of scientists in open science, articulating two connected incentives. The first is a basic wage, typically tied to teaching,Footnote 89 and the second is variable and tied to research. In open science, the quality of individuals’ research is evaluated by peers who value rigour and insight. The second incentive is captured by the description “collegiate reputational reward system”.Footnote 90 The higher the quality of an individual’s research, the faster their career will develop, including promotions and increases to their basic wage. In addition, they will earn prizes, honours, prestige, paid speaking and consulting opportunities and positions in venerable institutions (e.g. scientific societies), among other possibilities.Footnote 91
Researchers in academic science occasionally respond to market incentives, for example, when they spin out a company. However, these spinouts usually stop operating in the open science framework at this stage. Indeed, this is one of the key differences between open science, which the Republic relies upon, and commercial science, which is part of the established theory of pharmaceutical innovation. Open science responds less to market incentives (except insofar as it concerns basic wages and teaching), whereas commercial science is based on them.Footnote 92
A second incentive in play is the same as that found in the phenomenon of “user innovation”. User innovation focuses on the relationship between developers and their inventions. The term “user” is apposite because users develop products, at least partially, for themselves.Footnote 93 Whereas individuals in open science typically develop new ideas that do not immediately benefit themselves (e.g. theories of gravitation), individuals in user innovation do. In addition, user innovation distinguishes between an individual who makes a product to sell and one who makes a product to use. Selling falls within the remit of market incentives, whereas producing for one’s own use does not (directly) respond to market mechanisms. Instead, researchers in user innovation develop their products or ideas as improvements to their everyday work or interests.Footnote 94 Moreover, because users are not seeking to commercialise their innovations, they typically do not pursue intellectual property rights.Footnote 95 One of the classic examples of user innovation is a computer programmer writing code for open source software.Footnote 96
User innovation has another aspect that some researchers find surprising: users often reveal their innovations without seeking financial gain.Footnote 97 A review of user innovation across various fields found that innovation was coupled with gratis disclosure in industries as diverse as semiconductors, library information systems, sporting equipment and medical devices.Footnote 98 The review explains that users reveal their innovations gratis because it is often in their best interests, providing them with benefits such as peer recognition, satisfaction and reciprocated ideas for improvement. Indeed, the benefits of improvement connect to the heart of user innovation, as the innovations are for users who develop them.Footnote 99 The review also suggests that one of the reasons user innovation has gained prominence in recent decades is that improved technology has: (1) eased the designing and making processes (e.g. computer-aided design and small-scale manufacturing) and (2) enhanced communication between user communities.Footnote 100
Open science has long played a role in drug R&D. Scientists often develop new drug candidates, perhaps through early-stage phase I trials, that they subsequently transfer to pharmaceutical companies to obtain the necessary authorisations. It is true that universities or hospitals occasionally run phase II and phase III trials that companies then obtain authorisations for;Footnote 101 however, these instances are generally outliers. A review of the most clinically impactful drugs over a 25-year period found that pharmaceutical companies ran the bulk of late-stage development that led to authorisations.Footnote 102
The lack of hospital-and university-authorised drugs is probably one reason why commentators have avoided using open science to explain large numbers of phase II and III trials. Another three reasons to avoid thinking open science could run the trials are the barriers discussed above (i.e. expertise, risk and capital). Yet, when open science is combined with user innovation and the lower barriers to conducting repurposing trials, it is easier to see how it contributes to the number of trials in Figure 1.
User innovation, by contrast, is infrequently associated with drug R&D but is likely playing a significant role. For example, the authors have spoken with medical practitioners who run trials and publish case studies of off-label use, yet place less value on publishing than university scientists. Instead, they want to do their job well, solve problems, treat their patients, help others through publishing and, hopefully, continue to improve their activities through feedback. These rewards are the same as those associated with user innovation.
It is also possible that a mix of incentives from open science and user innovation motivates clinicians.Footnote 103 For example, they derive satisfaction from treating their patients and accelerate their careers through publications. Indeed, many clinicians’ contracts include practice and research. A mix of incentive systems likely encourages at least some clinicians to research new uses.
Clinicians, however, are not the only people responding to user-innovation incentives. Patients, caregivers and patient organisations, which for simplicity will be referred to as “patients”, play a role that might be overlooked if it were not touted in academic circles and corners of commercial science. It is easy to mistake patients as recipients of treatments only, rather than as recipients and developers. Yet, patients often facilitate repurposing research, as demonstrated at leading repurposing conferences, which all include patient sessions, featuring the roles they play. The reason patients get involved in repurposing is straightforward and consonant with user innovation: patients want better treatments (or sometimes, a treatment).
Academics and regulators have championed patients’ roles in commercial science for at least 20 yearsFootnote 104 and some companies, or at least individuals within companies, have engaged with patients for at least as long,Footnote 105 although uptake has been uneven.Footnote 106 A primary reason pharmaceutical companies have sought to integrate patients into their R&D processes is that some of the industry’s scientific feats have been commercial failures due to limited patient utility.Footnote 107 Even so, far less attention has been paid to patients’ roles in the Republic. The vast majority of studies on patients as innovators focus on their interactions with pharmaceutical companies, not hospitals and universities. Thus, viewing patients as part of the Republic brings new life to a well-known but often overlooked topic.
Patients are also likely to play more prominent roles in the Republic compared with commercial science. The literature on patients’ roles in commercial science describes patients taking on tasks such as helping with recruitment, publicising results and providing feedback on trial design, including scientific aspects (e.g. how a disease is measured) and practical elements (e.g. how easy the drug is to use).Footnote 108 Perhaps patients perform these roles equally prominently in the Republic, but it is reasonable that their roles are more prominent and diverse in the Republic due to fewer resources relative to pharmaceutical companies.
In drawing this part of the paper to a close, it is important to acknowledge that considerable content has been traversed. Consequently, the next section summarises the new theory’s defining characteristics before explaining how the Republic can be seen as part of a new model of drug R&D aimed at authorising treatments, connecting this study with other research on new government programmes.
C. Connecting the Dots: The Republic in Summary and R&D Models
An underpinning aspect of the new theory is that developers do not respond to market incentives in the form of exclusive rights to sell medicines. Instead, three groups of developers respond to different mixes of incentives. Scientists who run trials respond to incentives associated with open science, especially reputation building through publications; clinicians who run trials respond to a mix of incentives associated with open science and user innovation, such as improved job performance and job satisfaction (e.g. treating sick patients); and patients primarily respond to incentives associated with user innovation, too, namely, better treatments.
Established pharmaceutical innovation theory assumes that companies are necessary for drug R&D because of three barriers to innovation: scientific expertise, risk and capital. Yet, in each case, these barriers are lower for repurposing than for new drug development. The expertise required when repurposing is lower because the drug is well studied and manufactured (i.e. readily available), investigators select projects based on their expertise, and the trials are approved by grant and ethics bodies. Risk is lower because no organisation’s financial viability depends on the authorisation and sales of the drug, and the capital is lower because the funded cost of hospital and university phase III trials is around one-tenth or less of the cost of commercial trials.
So far, this paper has described a new theory of the incentives and resources that underpin trials for new uses of known drugs. Yet, one aspect of drug R&D that has been minimised is regulatory authorisation and the process of getting drugs to patients. Unless treatments are authorised, many clinicians will be reluctant to prescribe off-label because of concerns about efficacy, whether they could be liable for patients’ adverse effects and how the cost of the drug will be covered.Footnote 109
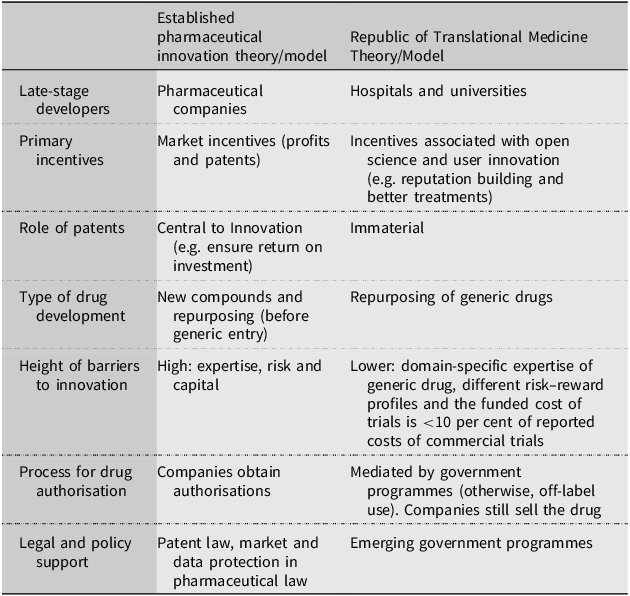
Commentators often describe the whole drug R&D process, from discovery to authorisation and patient delivery, as a model. At the heart of the established drug R&D model is a pharmaceutical company that coordinates late-stage R&D, including obtaining regulatory authorisations.Footnote 110 Scientists have called for new models for repurposing (particularly due to a lack of patent incentives)Footnote 111 and a recent study evaluated whether seven recently established government repurposing programmes in the US, UK and EU constitute a new model. The study suggested that three new government programmes, two from the US and one from the UK, represent a new model because they do not rely on patents, and the programmes rely on hospitals and universities to conduct clinical trials. The programmes search for new uses (e.g. via literature searches and asking clinicians) and if they find sufficient evidence for the authorisation of a new use, two of the programmes negotiate with companies to apply for the authorisations, while the third empowers the regulator to force companies to update their labels.Footnote 112 This means companies are still involved in the new model, but their roles are significantly reduced. Table 1 summarises the differences between the established theory/model and the Republic theory/model.
Summary of differences between the established pharmaceutical innovation theory/model and the Republic of Translational Medicine Theory/Model

Although the study suggested the government programmes represent a new model of R&D, it never considered how the trials were possible, especially regarding incentives and barriers to innovation. This study examined those issues. As a result, we are now able to reflect on necessary and perhaps more difficult questions regarding the future of the Republic and the new model, the “Republic model”. These are addressed in the next section.
IV. Open Questions and the Future of the Republic
The emergence of the Republic model presents society with options. Do we aim to realise the full potential of the new model, cultivating all the areas it might benefit society? Do we decide that the established model has worked well enough so far and, therefore, politely acknowledge the emergence of Republic model as a footnote and primarily focus our efforts on resolving cross-label use? Or, do we consider a mix of the two options?
For at least a decade, commentators have proposed reform to overcome cross-label use. The proposals range from reforming patent law to changes in how drugs are dispensed and from cash incentives to extended data exclusivity periods.Footnote 113 Many of these proposals have been examined elsewhere and they will not be discussed here, except to say that no reforms have been enacted for several reasons, including the idea of making physicians and pharmacists liable for infringement and the possibility that pharmaceutical companies will game the new laws to keep their prices high for longer.Footnote 114 Instead, it is reasonable to infer from the lack of reform that the Republic model offers society a plausible alternative. Indeed, that is effectively the approach adopted by many governments in their new repurposing programmes.
The problem is, though, that the Republic theory raises more questions than it answers. The questions include fundamental issues, such as how hospital and university trials can be so inexpensive compared with companies’ trials and to what extent are patents still incentivising hospital and university repurposing. Another question concerns patients; are patients sufficiently involved in the Republic, or is further integration required to ensure useful treatments?
The questions also include more applied issues, such as how frequently hospital and university trials produce sufficiently high-quality data to support a label change and whether society should shift more resources to the Republic model, with the aim of authorising more treatments. The EU is currently investing more resources, with new legislation in final stages of negotiation that would allow the European Medicines Agency to authorise new uses developed by hospitals and universities.Footnote 115 On the other hand, the UK Government has recently suspended its programme, indicating that off-label use is sufficient to get repurposed drugs to patients, amongst other reasons.Footnote 116 Is the UK or the EU taking the best approach? Or, perhaps both are taking optimal courses in their circumstances?
In short, we have a wealth of questions and relatively few answers. The productivity of the new theory and the future of the Republic model in society will likely depend on these answers, amongst others. This paper has made progress by outlining the contours of the Republic and the new model; however, compared with the research needed to explain the theory and optimise the new model, this study is only a candle when a floodlight is needed.
V. Conclusion
Established pharmaceutical innovation theory holds that organisations rarely repurpose generic drugs in the absence of enforceable patents, and that hospitals and universities typically avoid phase II and III trials because they lack the necessary expertise, appetite for risk and capital. Yet, empirical evidence shows that hospitals and universities do, in fact, conduct substantial numbers of late-stage clinical trials. This apparent paradox is explained by the concept of the Republic of Translational Medicine: clinicians, scientists and patients repurpose generic drugs in response to incentives outside the patent system, previously associated with open science and user innovation. And, with regard to resources, hospitals and universities can mount such efforts with fewer resources than industry, as data show that the funded costs of hospital and university trials are less than 10 per cent of pharmaceutical companies’ reported costs.
One of the central aims of this study has been to illuminate this parallel “hidden” innovation system. In doing so, it takes some of the first steps towards filling a 30-year gap in the literature, explaining the incentives and resources that underpin the Republic and situating it within a potential shift towards a new model of translational medicine.

 Open access
Open access