The scientist must concern himself with what one will say about him in a century, rather than with current insults or compliments.
In 1937, the Spanish neuroscientist and artist Ramón y Cajal commented on an apparent trial and error period of chaotic dendritic and axonic growth, with most of the resulting connections destined to disappear. But over subsequent decades the prevailing concept among neurobiologists and behavioral biologists was that synaptic connections increase with learning. Peter then rigorously showed in 1979 that after birth and an initial burst of growth in synapse numbers in human infants, there is a regression. He and his collaborators went on to show that these connections disappear at different times, with different kinetics, in distinct regions of the brain. The physical housekeeping task of this process is not small – selecting and eliminating billions of inactive synapses in the developing brain. In some regions of the brain, such as the visual system, the clearance mechanism seems to be relatively rapid over weeks or months. In other regions of the brain, for example in areas of higher cognitive function like the frontal cortex, this process of elimination is apparently more prolonged. What process or system has the intrinsic ability to identify and clear unused synapses? What has the ability to do this in a refined way, by targeting the correct contact sites, and without leaving dead cells and debris behind?
Phagocytes are a type of white blood cell that are in essence the vacuum cleaners of the body – sometimes known as the “professional eaters.” Phagocytes eat cellular debris, dead cells and pathogens by engulfing their targets and degrading the debris in specialized intracellular compartments known as the lysosomes. Neutrophils and macrophages are the classic phagocytes, the primary cells of the innate immune system. Neutrophils are fast cells, specialized to remove foreign microbes, but after clearing microbes neutrophils can leave some damage in their midst. This is in contrast tomacrophages, roaming cells that play a surveillance role in tissues. Macrophages move through tissues slowly and work in a more refined manner by targeting debris or microbes using long thin projections, not unlike axons in morphology. Indeed, macrophages clear away neutrophils after neutrophils kill microbes, and limit tissue damage that neutrophils can leave behind. Together, these phagocytes collaborate to clean up and ward off disease. It is interesting that, in the brain, there are generally no neutrophils unless there is infection or abnormal inflammatory insult, as seen in neuroinflammatory disorders like multiple sclerosis. In the brain, there is a specialized type of macrophage, the microglial cell, that functions to clean up tissue damage and debris.
In the late 1800s, phagocytes were discovered by Elie Metchnikoff, the “father of immunology.” Ramón y Cajal viewed neurons in preserved human brains, but Metchnikoff peered through microscopes at simple transparent sea organisms that were alive. In response to rose thorns or microbes, Metchnikoff watched in delight as he witnessed the “cellular act of eating.” He named the cells “phagocytes,” for cells (cytes) that eat (derived from the Greek word “phagein”). It is a dramatic process when phagocytes extend cell projections to engulf and devour microbes, foreign objects or cellular debris. Metchnikoff was a co-recipient of the 1908 Nobel Prize in Physiology or Medicine for this fundamental work.
At a phagocyte meeting in Sicily, where I first saw Professor Beth Stevens show the 1979 Peter Huttenlocher synaptic pruning slide, she was telling a beautiful story of her laboratory’s work, uncovering how synapses in the developing brain are pruned. As her group worked on the visual circuits of mice, she discovered the unexpected. The brain phagocytes, the microglial cells, target and engulf synapses that are being eliminated. It makes sense that during a period of rapid regression in synapses, a phagocyte would be the engineer that makes it happen. During her postdoctoral work she had previously shown that the classic complement cascade mediates synapse elimination in the visual system of mice [Reference Stevens, Allen and Vazquez1]. Complement proteins function in a cascade to activate the innate immune response to pathogens. They call for host defenses. In general, this response is proinflammatory and can contribute to tissue damage. Complement-mediated processes were generally known to be involved in pathologic responses to damage or infection, not normal developmental processes. Stevens’ subsequent work showed that complement components tag synapses that are less active and ready for pruning. These tagged synapses are recognized by the complement receptor CR3 on microglial cells [Reference Schafer, Lehrman and Kautzman2]. Their work demonstrated, for the first time, that pathogen-fighting complement proteins also mediate microglial synaptic pruning activity during normal development. Blocking these complement proteins impaired pruning in mouse models and altered the normal function of the visual circuits during development. This elegant work demonstrates that the complement system is critical for the normal developmental rewiring of brain circuitry that underlies the plasticity of the developing brain.
Related work on microglial cells was also going on in the laboratory of Cornelius Gross, but from a different perspective. With clear evidence that activity across synapses determines which ones are preserved or removed, Gross was also interested in what determines the structural remodeling of synaptic connections during development. His group observed the presence of synaptic material inside microglial cells, providing direct evidence that these cells can engulf synapses [Reference Paolicelli, Bolasco and Pagani3]. Gross focused on another small molecule,fractalkine, a chemokine that regulates phagocyte recruitment to damaged tissues. His group found that this small chemokine is expressed in synapses that are less active and, if you deplete it in mouse models, the circuitry of neurons in the brain is altered. His work provides further mechanisms for how microglial cells regulate synapse elimination.
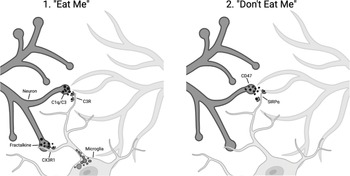
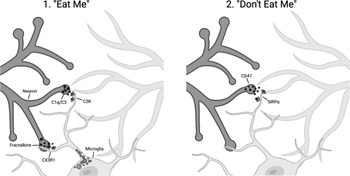
Part of understanding a biological process is to identify the molecular players. The work of the Stevens and Gross laboratories places the complement system and fractalkine into the targeting piece of the puzzle. But in addition to having an on switch you need an off switch – the pathways that limit this irreversible change to brain circuitry. The Stevens laboratory identified a known pathway that protects from macrophage engulfment, a cell surface “cluster of differentiation” protein, CD47, that binds to the signal regulatory protein alpha (SIRPα) on macrophages and is known as a “don’t eat me” signal. CD47 protects synapses from elimination. In mice deficient in CD47, the Stevens group found that there was increased microglial engulfment of synapses, and increased pruning [Reference Lehrman, Wilton and Litvina4]. Figure 17.1 summarizes some of the known pathways that positively and negatively regulate microglia-mediated synaptic pruning [Reference Rivest5].
Schematic of microglial-cell-mediated synaptic pruning, showing the signals that mediate synapse removal by microglial cells (“eat me”) including complement components C1q and C3 recognition by complement receptors on microglial cells and fractalkine–CX3R1 interactions. Microglial-cell-mediated pruning is inhibited by “don’t eat me” signals mediated by CD47 and SIRPα.
Despite this progress and glimpse of understanding, many challenges remain. In an interview with Beth Stevens in January 2022, she noted that “pruning is a permanent removal of synapses.” The brain is adept at plasticity – the ability to change – to reform connections and edit out ones that are not needed. There are not just pruned synapses, but some connections are “weakened,” like bad memories, and can erupt at later times. These types of “silent synapses” or weakened connections can be tapped and “called back into action.” Another important concept is that some of the connections are inhibitory rather than excitatory, and a shift in the balance of excitatory and inhibitory connections can affect output. During development there are simply too many connections, and permanent changes to brain circuitry are needed to control this “cacophony” of signals. But, at later stages, these modifications in synapses may be more subtle, and not structural. The synapse may only become quiescent rather than eliminated – and be available for future use when needed. It is likely that there is a continuum of modification, from elimination of synapses to their modification and, in essence, “turning off or on.”
However, there is evidence that synaptic pruning, just like pruning a bush, occurs throughout life – and its aberrant activity can contribute to neurodegenerative diseases including Alzheimer’s disease. Remarkably, work from Stevens and colleagues now has shown a genetic connection between complement protein expression and risk for Alzheimer’s disease – with too much pruning and permanent removal of brain circuits later in life. In the case of Alzheimer’s disease, distinct complement components have been implicated, including complement proteins C1q and C3, and this has implications for the treatment of Alzheimer’s disease [Reference Hong, Beja-Glasser and Nfonoyim6].
Stevens also commented on Feinberg’s work and the hypothesis related to pruning and schizophrenia. “He leaned on Peter’s data to back up this hypothesis and was inspired by him.” Feinberg made the connection between sleep, adolescence and pruning. As Stevens noted:
When you sleep, what happens during sleep – active cognitive processes go offline – may be to make sure noncognitive things happen like lymphatic flux. “Sleep on it” – consolidating memories – strengthening them – the idea of “use it or lose it” – may occur in part at night. Maybe it is pruning of the connections that are not needed that occurs during sleep. Sleep and pruning.
And if sleep is disturbed, as it is in schizophrenia, this may contribute to abnormal pruning during adolescence and risk for schizophrenia.
Stevens went on to say that “immune system pruning is still a new concept,” and much remains to be done to understand it. “Your dad inspired what we are doing. We do not know if schizophrenia and pruning is true, but now we have the tools to study it.” The problem with this early work, and more recent studies, is that “it is a snapshot of a small region of the brain.” It has been hard to repeat his work because of the difficulty getting fresh tissue and, because of the intrinsic variability of synapse number in different areas of brain tissue, there is a lot of “noise.” To increase confidence in a finding you need to increase numbers of samples studied. There are plentiful data to support synaptic pruning in the sensory regions of the brain like the visual system – but there is a gap in understanding the frontal cortex, areas that control higher cognitive functions. Some of this gap can be addressed using animal models like mice or marmosets. Just as with Peter’s work, “we have to understand the normal first in the frontal cortex of mammalian models – once you have the normal then you can study disease.”
The idea that phagocytes and complement proteins have been repurposed in the brain for normal neurodevelopment is intriguing. The complement system, for example, is best known for its roles during infection and inflammation, but infection and inflammation can also affect the brain. “Neuroinflammation” arises when the immune systems of the brain – microglial cells, the complement system and other mediators – go into overdrive. This overdrive can contribute to the abnormal synaptic pruning and changes in brain circuitry recently noted in neuroinflammation. For example, in patients with the autoimmune disease systemic lupus erythematosis (lupus), neuroinflammation has recently been thought to be associated with abnormal pruning that can lead to neurological deficits. In addition, in the demyelinating autoimmune disease multiple sclerosis (MS), studies in both humans and mouse models have shown increased synapse loss in the visual system. A recent study reported increased C3 complement proteins at synapses in MS, and treatment with a complement inhibitor improved visual function in mouse models of MS [Reference Werneburg, Jung and Kunjamma7]. In another example, Matt Johnson, a scientist at the Broad Institute at the Massachusetts Institute of Technology, studying synaptic pruning in Alzheimer’s disease, said that the association of flu infection in pregnancy and the subsequent onset of schizophrenia may be related to neuroinflammation and pruning. In addition, the development of neuroinflammation around the time of birth is thought to disrupt synaptic pruning, potentially leading to neurodevelopmental disorders.
It is important to consider that microglial cells are likely not the whole story. Other cell types in the brain have been implicated in synaptic pruning, including the star-shaped astrocyte, the most abundant cell type in the human brain. Similar to microglial cells, astrocytes can engulf synapses in an activity-dependent manner. Indeed, mice that lack a key receptor for astrocyte engulfment, the multiple EGF-like-domains 10 (MEGF10), have impaired synaptic pruning in the visual system [Reference Chung, Clarke and Wang8]. The astrocyte also releases factors that regulate microglial-cell-mediated pruning of synapses, suggesting that microglial cells and astrocytes likely collaborate to mediate the refined synaptic pruning necessary for normal development.
What is clear is that the next steps to understanding synaptic pruning in humans will require improved imaging. For example, imaging at the cellular level to determine if microglial cells directly eat synapses. The challenge remains that synapses are tiny and beyond the resolution of most light microscopes. Super-resolution imaging and three-dimensional reconstruction are needed to provide convincing evidence that synapses are inside microglial cells or in the lysosomal compartments of these cells that degrade debris. The possibility also exists that microglial cells act with more restraint, nibbling at synapses and altering their function. Could that contribute to a population of quiescent synapses that later are re-energized by activity of these quiescent connections? In addition to improved imaging of the cellular events, in humans, improved imaging of brain connectivity over time is needed. The advent of PET imaging to look at synapses in live human brains is a first step in this direction. But, in addition to imaging, analysis of human samples over time – like the levels of complement proteins in the cerebrospinal fluid that bathes the brain, may provide biomarkers to determine the extent of damage in disease states like schizophrenia or Alzheimer’s disease. The ultimate goal for this work is to target specific pathways, such as particular complement constituents, to treat schizophrenia and Alzheimer’s disease before the disease has irrevocably progressed. Some of these kinds of studies are in the works.