


Introduction
Dilated cardiomyopathy is a major cause of paediatric heart failure, transplantation, and sudden death despite its rarity. Reference Robinson, Zaklyazminskaya and Povolotskaya1,Reference Falik-Zaccai, Barsheshet and Mandel2 While typically linked to diverse genetic defects, some cases occur within syndromic contexts such as cardio-cutaneous syndromes. iASPP, encoded by PPP1R13L, interacts with transcription factors such as NF-κB and p53 to inhibit their transcriptional activity; beyond its role in desmosomal integrity, iASPP modulates apoptosis through the p53 pathway, thereby linking cell survival and inflammatory signalling in cardiomyocytes. Reference Falik-Zaccai, Barsheshet and Mandel2 Recently, biallelic PPP1R13L loss-of-function variants, affecting iASPP, have been identified as a novel cause of autosomal recessive arrhythmogenic cardiomyopathy with variable ectodermal abnormalities. Reference Tulbah, Alruwaili and Alhashem3,Reference Kalayinia, Mahdavi, Houshmand, Hesami, Pourirahim and Maleki4 We report a childhood-onset dilated cardiomyopathy case due to a novel homozygous PPP1R13L frameshift variant.
Molecular methods
In this study, isolated genomic DNA from peripheral blood sample was fragmented using the KAPA HyperCap Heredity Panel Kit (Roche, Ref No: 09462511001). Adapter ligation was performed with the MGI Easy DNA Adapter-96 Plate Kit (MGI, Shenzhen, China). Library purification was carried out using KAPA HyperPure Beads (Roche, Basel, Switzerland), followed by amplification with MGI Primer Mix. Library concentrations were measured using the Qubit 4 Fluorometer (Thermo Fisher Scientific, USA), and qualified libraries were pooled for hybridisation.
Target enrichment was performed using the KAPA HyperCapture Bead Kit and KAPA HyperCapture Reagent Kit (Roche, Basel, Switzerland) together with a custom-designed Nanodigmbio probe set. Single-stranded DNA molecules were generated using the MGIEasy Circularisation Module (MGI, Shenzhen, China), and sequencing-ready libraries were prepared.
Sequencing was carried out on the MGI DNBSEQ-G400 platform (MGI, Shenzhen, China) using DNBSEQ-G400RS Sequencing Flow Cell and DNBSEQ-G400RS High-throughput Sequencing Kit (FCL PE150), generating paired-end 150 bp reads.
This study aimed to analyse 3324 genes associated with common inherited disorders, covering over 99% of the coding regions annotated in CCDS, RefSeq, and GENCODE databases. The target sequencing was designed to achieve an average coverage depth of 20x–50x.
Raw sequencing data were aligned to the GRCh37 (hg19) human reference genome and processed using the Genomize-SEQ platform (Genomize, Istanbul, Turkey). Low-quality variants were filtered out, and the remaining variants were annotated and evaluated using publicly available databases including ClinVar, HGMD Public, gnomAD, ExAC, and dbSNP. Variants with potential clinical relevance were classified and interpreted according to the 2015 guidelines of the American College of Medical Genetics and Genomics.
Case presentation
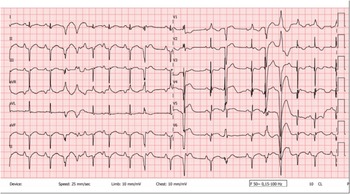
A previously healthy 4-year-old boy presented with complaints of fatigue and abdominal pain starting one week prior to admission, and swelling of the eyelids and rapid breathing that developed two days prior to presentation. His past medical history was unremarkable. His parents were first-degree cousins, and the family’s other two children were healthy. The patient’s father had five siblings who died during the infantile period, with the underlying cause remaining unknown. On physical examination, the patient’s body weight was 13.9 kg (−1.42 SDS), height was 98 cm (−1.26 SDS), and head circumference was 50 cm (−0.68 SDS). He appeared in poor general condition, was tachypnoeic and tachycardic, with bilateral periorbital oedema and rales in both lungs. Chest X-ray revealed findings consistent with bilateral pleural effusion, and cardiomegaly was observed on telecardiography. The patient was admitted to the ICU and received respiratory and circulatory support with a diagnosis of decompensated heart failure. Echocardiographic evaluation revealed biventricular dysfunction with a severely reduced left ventricular ejection fraction of 20% and a left ventricular end-diastolic diameter of 52 mm (Z-score: +4.62). Inotropic infusions, diuretics, ACE inhibitors, and antiplatelet therapy were initiated due to heart failure, and intravenous antibiotic therapy was started due to elevated acute phase reactants. At initial evaluation, respiratory viral PCR testing was positive for rhinovirus. Laboratory analysis revealed an elevated troponin level of 291 ng/L and an NT-proBNP level of 7373 ng/L (Supplementary Figure S1). The patient had elevated cardiac enzymes, and cardiac MRI demonstrated findings consistent with myocarditis, leading to administration of intravenous immunoglobulin. Despite intravenous immunoglobulin therapy, troponin levels did not normalise. Due to persistent troponin elevation, corticosteroid therapy was initiated. However, recurrent troponin peaks were observed during steroid treatment. Intermittent intravenous immunoglobulin administrations were given, yet troponin levels remained persistently elevated. Follow-up cardiac MRI revealed advanced global biventricular hypokinesia and dilatation, with left ventricular ejection fraction of 15% and right ventricular ejection fraction of 16% (Figure 1, Supplementary Video S1). CT angiography did not reveal any coronary artery anomalies. On electrocardiography, ventricular extrasystoles were observed, and 24-hour Holter monitoring detected 18% multiform ventricular extrasystoles, coupled ventricular extrasystoles, non-sustained and sustained ventricular tachycardia episodes, leading to initiation of beta-blocker therapy (Figure 2). Metabolic disease screening performed as part of the aetiological evaluation was unremarkable. Due to the presence of sparse, dry hair, a high anterior hairline, a broad and flat nasal bridge, and pointed teeth, genetic evaluation was planned (Figure 3). Next-generation sequencing identified a homozygous PPP1R13L(NM_006663.4):c.2368_2375dup p.(Pro793GlyfsTer32) frameshift variant, classified as pathogenic according to American College of Medical Genetics and Genomics criteria. The variant is located in exon 12 out of 13 of the gene and is predicted to undergo nonsense-mediated decay, as it is not situated in the last exon or within the last 50 base pairs of the penultimate exon. Segregation analysis in family members could not be performed. However, assuming that the parents are related and considering the family history, it is highly likely that both parents are heterozygous carriers. This would be consistent with the autosomal recessive inheritance pattern of the disease. Ophthalmologic and audiologic evaluations were within normal limits. Histopathological examination of the skin biopsy specimen demonstrated numerous hair follicles with telogen morphology, varying in diameter, and no trichodysplastic changes were identified. Heart transplantation was planned; however, the patient unfortunately died due to cardiac arrest following ventricular fibrillation during follow-up.
Cardiac MRI (cine steady-state free precession sequence) at the mid-ventricular level in short-axis view. The image demonstrates significant left ventricular dilatation with an end-diastolic diameter of approximately 52 mm.

Twelve-lead surface electrocardiogram obtained at 4 years of age, showing multiform ventricular extrasystoles, QRS axis deviation, and repolarisation abnormalities.

Frontal facial photograph of the patient demonstrating syndromic features, including sparse and dry hair, a high anterior hairline, and a broad, flat nasal bridge.

Discussion
The identified novel PPP1R13L variant, c.2368_2375dup p.(Pro793Glyfs*32), represents a single-base duplication that causes a frameshift and introduces a premature termination codon 32 amino acids downstream. This alteration is predicted to result in nonsense-mediated mRNA decay, effectively abolishing normal protein production. As a result, it meets the American College of Medical Genetics and Genomics PVS1 criterion at a very strong level (PVS1: null variant in a gene where loss of function is a well-established mechanism of disease). Additionally, the variant is absent from large population databases such as gnomAD, fulfilling the PM2 criterion at a moderate level (PM2: extremely low allele frequency in population databases, supporting rarity and potential pathogenicity). Initially, the variant was classified as likely pathogenic based on the combination of PVS1 and PM2. However, after a more detailed clinical investigation and careful comparison of the patient’s phenotype with the known features of PPP1R13L-related disease, the diagnosis became clinically definitive. The patient exhibited a highly specific phenotype that matched the gene-associated syndrome, allowing application of the PP4 criterion at a strong level (PP4_Strong: patient’s phenotype is highly specific for a disease with a single genetic aetiology). Incorporating this additional line of evidence led to the reclassification of the variant as pathogenic, underscoring the value of integrating genotype and phenotype data in accordance with American College of Medical Genetics and Genomics/AMP guidelines to ensure accurate diagnosis in rare Mendelian disorders.
Genetic factors play a central role in the aetiology of dilated cardiomyopathy, with more than 40 genes implicated, most commonly inherited in an autosomal dominant manner. Reference Falik-Zaccai, Barsheshet and Mandel2 In a subset of patients, dilated cardiomyopathy occurs within a broader syndromic context involving extra-cardiac organ systems. Cardio-cutaneous syndromes, most notably Naxos and Carvajal syndromes, represent prototypical examples of this association, characterised by distinctive early-onset ectodermal features such as woolly hair and palmoplantar keratoderma, followed by the development of cardiomyopathy due to defects in desmosomal proteins. Reference Falik-Zaccai, Barsheshet and Mandel2
Pathogenic variants in PPP1R13L, encoding the iASPP protein, have recently been implicated in a distinct form of cardiocutaneous syndrome characterised by severe childhood-onset dilated cardiomyopathy with variable ectodermal features. iASPP, a member of the ASPP protein family, plays a pivotal role in stabilising desmosomes and the intermediate filament network through its interaction with desmoplakin and desmin, thereby maintaining cardiomyocyte integrity. This pathomechanism overlaps with that described in Naxos and Carvajal syndromes, in which desmosomal disruption leads to combined features of arrhythmogenic cardiomyopathy and dilated cardiomyopathy. Reference Coudert, Thevenon and Testard5 Experimental models further support this link: Ppp1r13l-deficient mice develop right ventricular dilatation and sudden death with features of ARVC, while human myocardial samples demonstrate impaired desmoplakin–iASPP binding and reduced iASPP expression. Reference Notari, Hu and Sutendra6
Beyond its structural role, iASPP is essential for regulating NF-κB mediated inflammatory responses. Loss of function promotes excessive inflammation, maladaptive cardiac remodelling, and aggressive cardiomyopathy, consistent with observations in animal models. Reference Falik-Zaccai, Barsheshet and Mandel2 Similarly, inflammation has been recognised as a complex and central contributor to the pathogenesis of arrhythmogenic cardiomyopathy. It may act not only as a secondary response to myocyte necrosis but also as an active driver of disease progression, even in the absence of infectious triggers or immune cell infiltration. Experimental models have shown that desmosomal gene pathogenic variants can intrinsically activate inflammatory pathways within cardiomyocytes, leading to structural instability, myocardial remodelling, and arrhythmogenesis. Reference Meraviglia, Alcalde, Campuzano and Bellin7
Clinical reports increasingly illustrate the pleiotropic spectrum of PPP1R13L-related disease. Tulbah et al. described a consanguineous family in which six children carrying a homozygous frameshift variant presented with dilated cardiomyopathy and woolly hair, several also exhibiting cleft lip/palate, developmental delay, or congenital glaucoma. Reference Tulbah, Alruwaili and Alhashem3 Kalayinia et al. subsequently reported a stop gain variant associated with autosomal recessive arrhythmogenic cardiomyopathy, Reference Kalayinia, Mahdavi, Houshmand, Hesami, Pourirahim and Maleki4 and Coudert et al. expanded the phenotypic spectrum of PPP1R13L-related disease by describing patients with biallelic variants and anorectal malformations, including imperforate anus in one case and an intermediate-type anorectal anomaly in another. Reference Coudert, Thevenon and Testard5 Robinson et al. reported 7 children from 5 unrelated families who had severe dilated cardiomyopathy, with or without ectodermal or neurodevelopmental features, and pathogenic variant in the PPP1R13L gene. One patient experienced sustained ventricular tachycardia. Three of the children died before 5 years of age due to progression of heart failure; 3 underwent cardiac transplantation, and 1 was awaiting transplant. In 4 of the children, hair was described as sparse/thin and wiry, curly, or woolly, and 2 children had dystrophic nails. Cleft lip and palate were present in 2 brothers. Reference Robinson, Zaklyazminskaya and Povolotskaya1 Similarly, Henry et al. reported a three-year-old girl with a homozygous PPP1R13L pathogenic variant who developed inotrope-dependent heart failure during viral infections, eventually requiring left ventricular assist device placement. She exhibited classic ectodermal features including wiry hair, bifid central incisors, and recurrent paronychia, in addition to gastrointestinal anomalies such as constipation and rectal prolapse. Reference Henry, Bernhardt, Hayes and Mitchelson8
Our patient displayed a comparable phenotype, with rapidly progressive dilated cardiomyopathy accompanied by ectodermal features including sparse hair, craniofacial dysmorphism, and abnormal dentition. Importantly, malignant ventricular arrhythmias culminating in sudden cardiac death suggest that, as in other desmosomal cardiomyopathies, the dilated phenotype may represent the terminal stage of an arrhythmogenic process. Persistent elevation of cardiac troponin levels in our patient, despite anti-inflammatory treatment and intravenous immunoglobulin administration, may reflect ongoing myocardial injury and active disease progression. Rather than reflecting only acute myocardial injury, sustained troponin elevation may indicate chronic subclinical inflammation, ongoing myocyte apoptosis, or adverse remodelling, particularly in the context of genetic cardiomyopathies. In PPP1R13L-related disease, where inflammation and desmosomal instability play central roles, persistent troponin elevation may serve as a surrogate for the severity and progression of the cardiomyopathic process.
PPP1R13L pathogenic variants are listed in OMIM as “Arrhythmogenic cardiomyopathy with variable ectodermal abnormalities” underscoring the intrinsic arrhythmogenic substrate irrespective of left ventricular size. Thus, recognition of this entity has major implications for clinical management.
Conclusion
This case further expands the genotypic and phenotypic spectrum of PPP1R13L-related cardiomyopathy. PPP1R13Lpathogenic variants represent an emerging cause of syndromic paediatric dilated cardiomyopathy with a high risk of malignant arrhythmias and sudden death. Our case underscores the need to consider this diagnosis in early-onset cardiomyopathy with syndromic features. Early recognition is essential for appropriate genetic counselling, multidisciplinary care, and timely subspecialty referral, while continued case reporting will further refine genotype–phenotype correlations.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1047951126111883
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Competing interests
No potential conflicts of interest relevant to this article was reported.
Ethical standards
Written informed consent was obtained from the patient’s parents for the publication of this case report and any accompanying images, including identifiable photographs. The consent includes the use of clinical data and diagnostic images for academic and scientific dissemination. The authors affirm that the patient’s identity has been adequately protected, and all ethical standards for case report publication have been met.





 Open access
Open access