


Introduction
Coinfections of multiple pathogens can influence epidemiology and disease severity [Reference Thomas1]. An understanding of ecological drivers of coinfections is important to improve a targeted public health response. Human infections by zoonotic orthohantaviruses and Leptospira spp. are (re-)emerging zoonoses that are almost indistinguishable in their clinical presentation [Reference Sunil-Chandra2] and can often be mistaken for each other.
Leptospira spp. are gram-negative bacteria of the class Spirochaetes, order Leptospirales, family Leptospiraceae and are 6–20 μm in size and 0.1 μm in diameter [Reference Levett3]. They can be divided into saprophytic, intermediate and pathogenic groups (including L. kirschneri, L. borgpetersenii and L. interrogans) [Reference Fischer4]. Human infections can occur after contact with infected animals or indirectly through contact with contaminated water or soil. The disease course is in most cases asymptomatic or mild, but can progress in some cases after a febrile phase to multiple organ dysfunction [Reference Haake and Levett5]. Human incidences vary globally, with amplifying factors (tropical climate, standing water and low sanitation level) being notably absent at higher latitudes [Reference Pappas6]. Rodents and shrews are considered as reservoir hosts for zoonotic Leptospira spp. with prevalences reaching 50% depending on species and season [Reference Fischer4].
Hantaviruses, order Bunyavirales, family Hantaviridae, are enveloped viruses with a three segmented RNA genome of negative polarity [7]. Depending on the species, orthohantaviruses can cause haemorrhagic fever with renal syndrome (HFRS) or hantavirus cardiopulmonary syndrome. There is an estimated 150 000 cases of HFRS each year, with more than half occurring in China [Reference Jonsson, Figueiredo and Vapalahti8]. In Central Europe, Puumala orthohantavirus (PUUV) is the most important hantavirus as reflected by the large number of human cases, in particular during outbreak years. In Germany, the mean incidence is 0.87 per 100 000 inhabitants [Reference Faber9], but it reached 60 per 100 000 inhabitants in the outbreak year 2012 in the districts Göppingen and Heidenheim in Baden-Wuerttemberg [10]. Although the reservoir of PUUV, the bank vole (Clethrionomys glareolus), is distributed throughout Germany, PUUV is present only in the southern and western parts of the country [Reference Drewes11]. The occurrence of Dobrava-Belgrade orthohantavirus (DOBV), genotype Kurkino, in Germany follows the geographical distribution of its reservoir, the striped field mouse (Apodemus agrarius) and is limited to north-eastern and eastern Germany [Reference Faber9, Reference Schlegel12]. Finally, Tula orthohantavirus (TULV) is a broadly distributed orthohantavirus with the common vole (Microtus arvalis) as reservoir, but was also detected in other closely related species such as the field vole (Microtus agrestis), East European vole (Microtus levis) and water vole (Arvicola amphibius) [Reference Schmidt13]. TULV is generally considered to be of no (or low) pathogenicity, with only sporadic evidence of human infections [Reference Schmidt13, Reference Hofmann14].
Coinfection with both pathogens has been confirmed in humans and rodents [Reference Clement15, Reference Kurucz16] and in this study, we screened rodents and shrews from central Germany over the course of a year for pathogenic Leptospira spp., TULV, DOBV and PUUV and evaluated the frequency of dual hantavirus–Leptospira infections.
Material and methods
Trapping and dissection
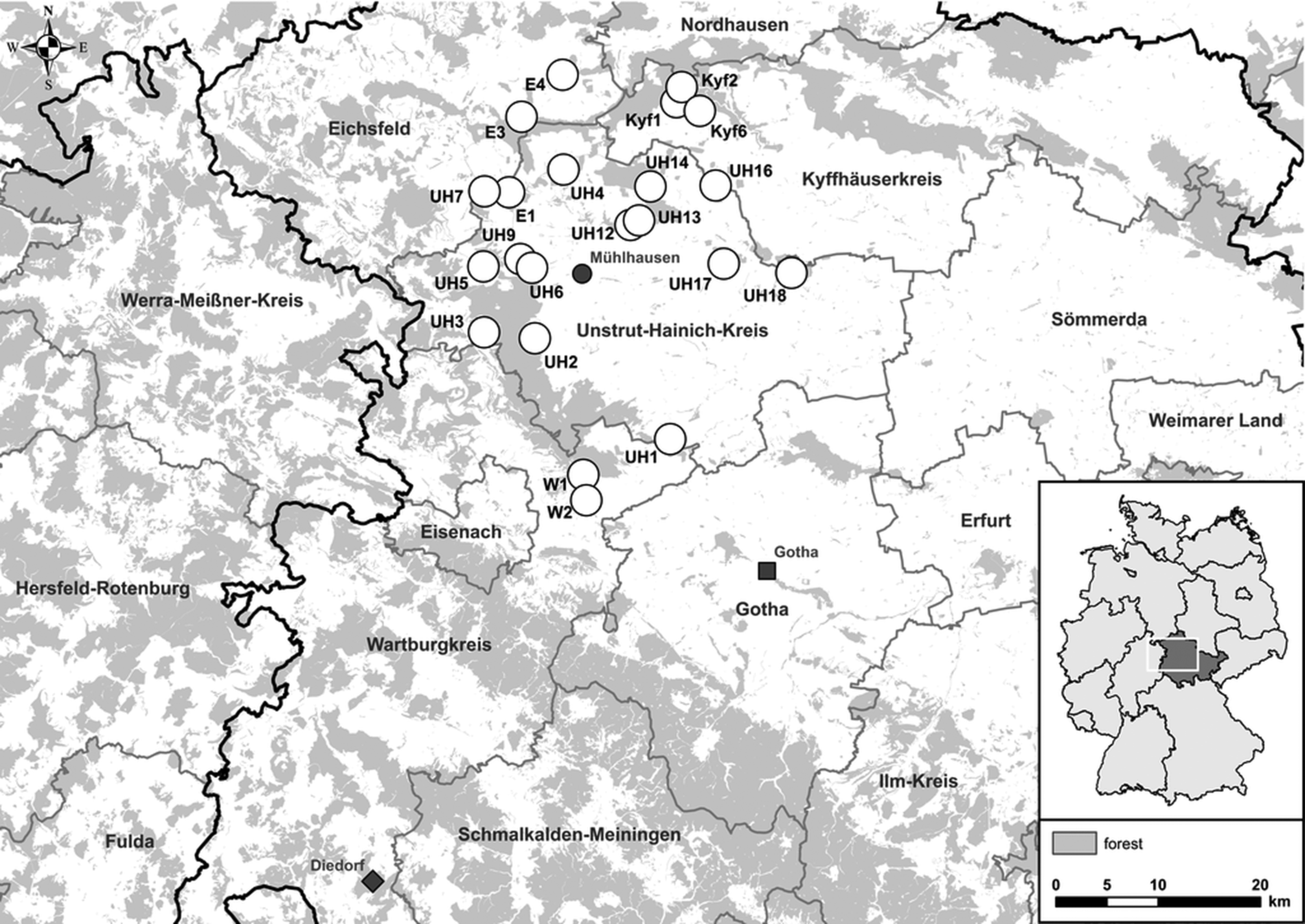
Small mammals were trapped in spring, summer and fall 2017 at 22 sites in Thuringia, central Germany (Fig. 1). In central Germany, the distributional ranges of all abovementioned pathogens and their hosts probably overlap [Reference Fischer4, Reference Schmidt13, Reference Görner17]. Each site consisted of perennial grassland as well as the adjacent grassland-forest ecotone. In each of these habitats small mammals were trapped with 36 snap traps (metal snap traps, Deufa, Neuburg, Germany) set in four rows with 10 m trap spacing. In the ecotone, two rows were set in the grassland section and two rows in the transition to the prevailing forest habitat. The trapping at site UH6 was discontinued after spring season due to logistic reasons. All procedures involving animals were conducted according to relevant legislation and by permission of the Thuringia state office of Consumer Protection (permit 22-2684-04-15-105/16). Small mammal carcasses were frozen at −20 °C until dissection. During dissection, small mammals were measured, weighed and sex was determined. To avoid contamination, sterile instruments for each individual were used. Lung and kidney tissue were collected and stored at −20 °C. If necessary, species and sex were determined by corresponding polymerase chain reaction (PCR) assays using kidney tissue-derived DNA as previously described [Reference Fischer4, Reference Mayer-Scholl18].
Map of 22 trap sites around Mühlhausen (black circle) in Thuringia, Germany (see small overview map). Additionally, the sites Diedorf (diamond) and Gotha (square) are shown where previously Puumala orthohantavirus (PUUV) and Tula orthohantavirus (TULV) were detected, respectively.
Leptospira spp. DNA screening
A pin-head-sized piece of kidney tissue was used for DNA extraction by Tissue DNA Kit according to the manual of the manufacturer (Roboklon, Berlin, Germany). DNA concentration was determined with Nanodrop ND-1000 (peqlab Biotechnologie GmbH, Erlangen, Germany). DNA samples were tested in the conventional lipL32 PCR for the presence of pathogenic leptospires [Reference Fischer4, Reference Mayer-Scholl18]. Genomospecies identification of positive samples was done by secY PCR, dideoxy chain termination sequencing of PCR products with BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems™, Waltham, MA, USA) and sequence comparison to GenBank entries by nucleotide Basic Local Alignment Search Tool (BLASTn) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) [Reference Fischer4].
Hantavirus screening by RT-PCR
RNA was extracted from a lentil-sized piece of lung tissue with QIAZOL reagent (QIAGEN, Hilden, Germany) and eluted in 100 μl DNase/RNase free water (Thermo Fisher Scientific, Schwerte, Germany) [Reference Schmidt13]. RNA concentration was measured with Nanodrop ND-1000. Reverse transcription-PCR (RT-PCR) was performed using SuperScript™ III One-Step RT-PCR with Platinum Taq-Kit (Invitrogen, Darmstadt, Germany). TULV/PUUV S segment RT-PCR screening used the primer pair PUUV342F and PUUV1102R [Reference Essbauer19]. DOBV RNA screening was based on RT-PCR using the S segment primer pair D113M and D955CM [Reference Klempa20]. RT-PCR products of the expected size were directly sequenced using BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems™).
Phylogenetic analysis
ClustalW multiple alignments of obtained nucleotide (nt)-sequences were constructed using BioEdit v7.2.5 [Reference Hall21]. The best fitting substitution model was determined by jModelTest v2.1.6 [Reference Posada22]. Phylogenetic trees were reconstructed according to maximum likelihood and Bayesian algorithms via FasttreeMP v2.1.10 and MrBayes v3.2.6 on CIPRES Science Gateway [Reference Miller, Pfeiffer and Schwartz23–Reference Ronquist25]. Subsequently, a consensus tree was established as bootstrap values ≥75 of the maximum likelihood tree were transferred to the Bayesian tree only if branches of both trees were consistent. Probabilities of node support of the Bayesian tree are given when the value was ≥95%.
Statistical analysis
To estimate key drivers of coinfections, a generalised linear mixed model was generated for Microtus spp. in grassland, where the individual coinfection status (binomial variable; TULV RNA positive and Leptospira spp. DNA positive) was the dependent variable. Individual demographic variables (sex, weight as a proxy for age) [Reference Morris26] as well as population level variables (TULV prevalence, Leptospira spp. prevalence, abundance (trap success as individuals per 100 trap nights), abundance in the previous season and season itself) were fixed factors. Site was incorporated as a random factor. The most appropriate model was determined using a multimodel inference approach. Using the dredge function from the MuMIn-package all possible combinations of fixed factors were ranked by their conditional Akaike information criterion (AIC). The best fitting models were defined as being within a ΔAIC of <2 of the best model (lowest AIC). Model coefficients were averaged using the model.avg function. We present the relative importance for each factor as the sum of Akaike weights in the best fitting models where the respective factor occurs as well as the 95% confidence interval (CI) for each factor. Here, a factor can be considered significant if the CIs do not include zero.
As trap success of Microtus spp. in the grassland/forest ecotone precluded a full model, a chi-square test was used to compare the overall prevalence in both habitats. CIs for prevalences were calculated using the exactci-function from the PropCIs-package. All analyses were performed using R [Reference R27].
Results
Small mammal trapping
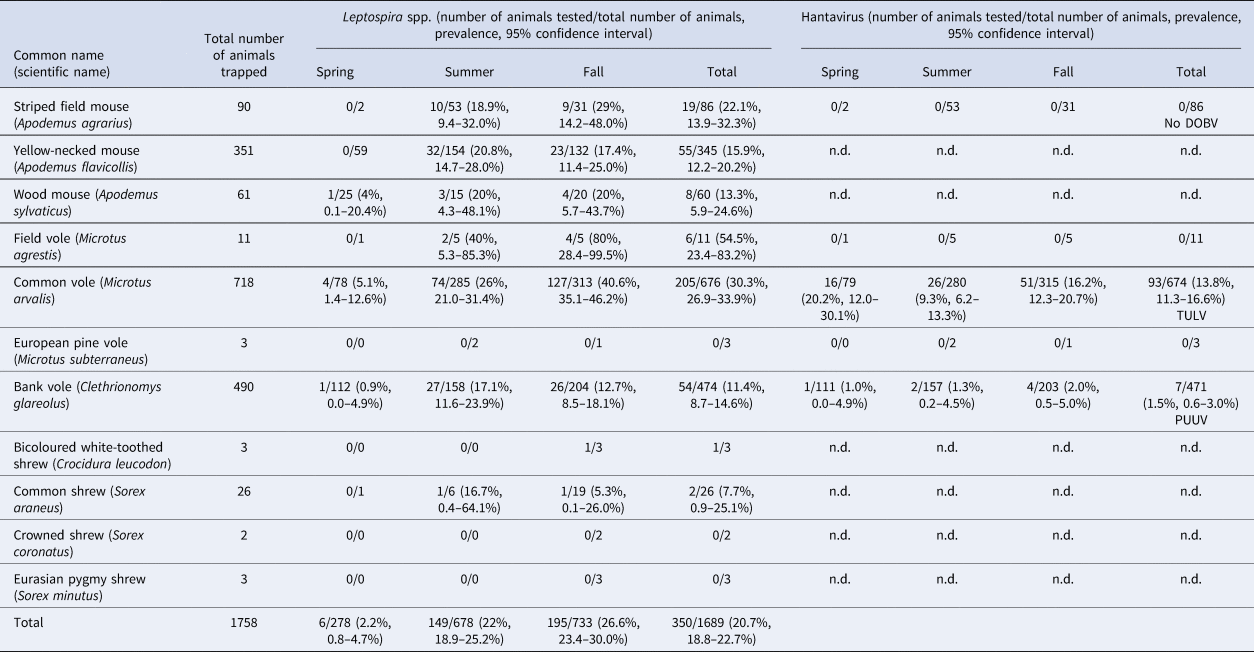
During 2017, 1758 small mammals were trapped, including 90 striped field mice, 351 yellow-necked mice (A. flavicollis), 61 wood mice (A. sylvaticus), 11 field voles, 718 common voles, three European pine voles (M. subterraneus), 490 bank voles and 34 shrews including three bicoloured white-toothed shrews (Crocidura leucodon), 26 common shrews (Sorex araneus), two crowned shrews (S. coronatus) and three Eurasian pygmy shrews (S. minutus) (Table 1).
Small mammals trapped in Thuringia, Germany, and results of Leptospira spp. PCR and hantavirus RT-PCR analyses for Dobrava-Belgrade orthohantavirus (DOBV), Tula orthohantavirus (TULV) and Puumala orthohantavirus (PUUV).
n.d., not done.
Leptospira spp. screening
For 1689 of the 1758 trapped small mammals kidney tissue was available. Overall, 350 of 1689 (20.7%) small mammals tested positive in the lipL32 PCR (Table 1). In rodents, the overall prevalence varied between species: field voles (54.5%; 6/11, CI 23.4–83.3%), common voles (30.3%; 205/676, CI 26.9–33.9%), striped field mice (22.1%; 19/86, CI 13.9–32.3%), yellow-necked mice (15.9%; 55/345, CI 12.2–20.2%), wood mice (13.3%; 8/60, CI 5.9–24.6%) and bank voles (11.4%; 54/474, CI 8.7–14.6%). Two of 26 common shrews (7.7%; CI 0.9–25.1%) were tested positive and one of three bicoloured white-toothed shrews was also positive. None of the European pine voles, crowned shrews and Eurasian pygmy shrews tested positive.
The overall prevalence increased from spring (2.2%, 6/278, CI 0.8–4.7%) to summer (22%, 149/678, CI 18.9–25.2%) and fall (26.6%, 195/733, CI 23.4–30.0%). Leptospira spp. were detected at 21 of 22 sites with an average site-specific prevalence ranging from 2.4% (2/84, CI 0.3–8.4%) at site UH3 to up to 41.5% (22/53, CI 28.1–55.9%) at site W1. The highest prevalence was measured at site E4 with 76.5% (13/17, CI 50–93.2%) in fall just for common voles. The most abundant genomospecies was L. kirschneri (n = 108; 93.1%); L. borgpetersenii was found only in a few individuals (n = 8, 6.9%); no other genomospecies was identified. Common voles only harboured L. kirschneri (n = 92; 100%). Similarly, in striped-field mice (n = 2), wood mice (n = 2), field voles (n = 1) and common shrews (n = 1) also exclusively L. kirschneri was identified. Yellow-necked mice carried L. kirschneri (62.5%; 6/9) or L. borgpetersenii (37.5%, 3/9), and bank voles also carried L. kirschneri (45%, 4/9) or L. borgpetersenii (55.5%, 5/9). L. kirschneri and L. borgpetersenii circulated in the same bank vole population at one site (KYF1) during the same trapping season. Otherwise only a single Leptospira genomospecies was detected per site depending on trapping location and species.
Hantavirus screening
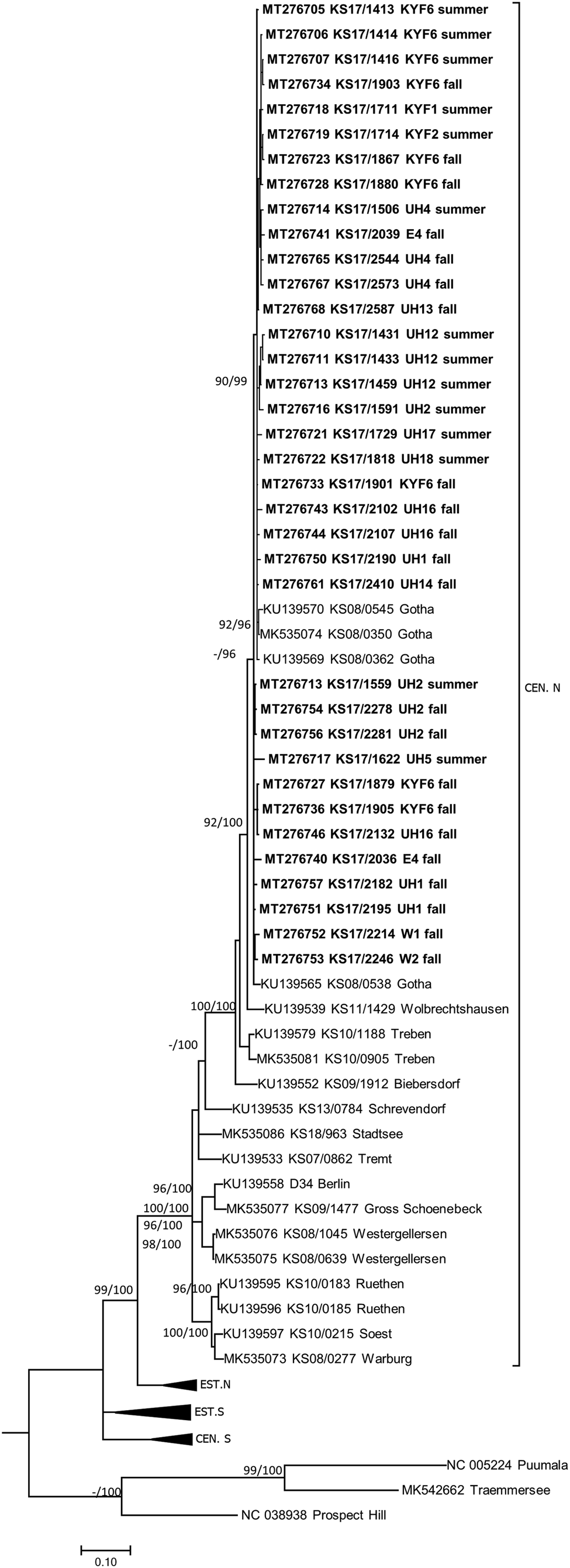
TULV-RNA was detected in 13.8% (93/674, CI 11.3–16.6%) of common voles, in none of the 11 field voles and none of the three European pine voles (Table 1). Overall prevalence in common voles was highest in spring with 20.2% (16/79, CI 12.0–30.1%), followed by fall with 16.2% (51/315, CI 12.3–20.7%) and summer with 9.3% (26/280, CI 6.2–13.3%). No TULV-RNA was found at three sites (E3, UH3, UH9; combined 0/24, CI 0.0–14.2%), while prevalences of up to 33.8% (KYF6; 23/68, CI 17.8–37.4%) were detected among sites where at least 10 common voles were tested. The highest prevalence from sites with 10 or more tested common voles was measured in spring at site UH17 with 58.3% (7/12, CI 27.7–84.8%). TULV RNA positive voles originated from 18 of 21 sites where common voles were trapped. TULV was present at only four sites in spring, despite common vole presence at 15 sites. The overall prevalence at these four sites was 50% (16/32, CI 38.9–68.1%). In summer, TULV was present at 14 sites and at 15 sites in fall. The four sites with high prevalences in spring did not differ significantly from the rest in summer (χ 2 = 0.031, P = 1) or in autumn (χ 2 = 0.474, P = 0.57). Phylogenetic analysis showed that the sequences clustered with TULV sequences from geographically close Gotha, Thuringia, Germany (Fig. 1, square), in the TULV Central North (CEN. N) clade (Fig. 2).
Consensus phylogenetic tree of the partial S segment sequences of Tula orthohantavirus (TULV) (alignment length 549 nucleotides (nt), positions 406–951, counting according to TULV S segment, accession number NC_005227). TULV is sorted in the clades Central North (CEN.N), Central South (CEN.S), Eastern North (EST.N) and Eastern South (EST.S). The consensus tree is based on Bayesian analyses with 107 generations and a burn-in phase of 25%, and maximum-likelihood analyses, with 1000 bootstraps and 50% cut-off using the general time reversible (GTR) substitution model with invariant sites and a gamma distributed shape parameter for both algorithms. Posterior probabilities exceeding 95% from Bayesian analyses are given behind and bootstrap values are given before the slash for major nodes if exceeding 75%. The tree reconstructions were done via CIPRES [Reference Miller, Pfeiffer and Schwartz23]. Alignments were constructed with Bioedit V7.2.3. [Reference Hall21] using the Clustal W Multiple Alignment algorithm implemented in the program. Names in bold indicate newly generated sequences (see Supplementary Table S1). Triangles indicate compressed branches (see Supplementary Table S2 for used sequences). Clade designation followed previous publications for TULV [Reference Saxenhofer28].
In 1.5% (7/471, CI 0.6–3.0%) of tested bank voles PUUV-RNA was detected. Positive voles were trapped at neighbouring sites UH2, UH3, UH9 and UH6 (Fig. 1). Phylogenetic analysis revealed that the novel PUUV strains belong to the PUUV Central European (CE) clade. The novel sequences clustered closest to sequences from western and northwestern parts of Germany such as Gilserberg (Hesse), Goettingen and Sennickerode (both in Lower Saxony) (Fig. 3). Interestingly, PUUV sequences from Diedorf (Thuringia, Fig. 1, diamond), a site only 50 km away from the trapping locations in this study (Fig. 1), clustered differentially, i.e. with sequences from southern Germany, like Swabian Jura and Bavarian forest.
Consensus phylogenetic tree of partial S segment sequences for Puumala orthohantavirus (PUUV) (alignment length 711 nt, positions 355–1065, counting according to PUUV S segment, accession number NC_005224). PUUV is sorted in the clades Alpe-Adrian (ALAD), Central European (CE) clade including Belgium (BE), France (FR), Germany (DE), Slovakia (SK), Danish (DAN), Finnish (FIN), Latvian (LAT), Northern-Scandinavian (N-SCA), Russian (RUS), Southern-Scandinavian (S-SCA) as well as the PUUV strains Hokkaido, Muju and Fusong. The consensus tree is based on Bayesian analyses with 1.5 × 107 generations and a burn-in phase of 25%, and maximum-likelihood analyses, with 1000 bootstraps and 50% cut-off using the general time reversible (GTR) substitution model with invariant sites and a gamma distributed shape parameter for both algorithms. Posterior probabilities exceeding 95% from Bayesian analyses are given behind and bootstrap values are given before the slash for major nodes if exceeding 75%. The tree reconstructions were done via CIPRES [Reference Miller, Pfeiffer and Schwartz23]. Alignments were constructed with Bioedit V7.2.3. [Reference Hall21] using the Clustal W Multiple Alignment algorithm implemented in the program. Names in bold indicate newly generated sequences (see Supplementary Table S1). Triangles indicate compressed branches (see Supplementary Table S2 for used sequences). Clade designation followed previous publications for PUUV [Reference Drewes11, Reference Castel29].

DOBV infection was not detected in any of the 86 tested striped field mice (Table 1).
Coinfections
In 6.6% (44/671, CI 4.8–8.7%) of common voles, we detected a coinfection of Leptospira spp. with TULV. There was no statistical difference between coinfection prevalence detected in forest ecotone (7.7%; 3/39, CI 1.6–20.9%) and in grassland (6.5%; 41/632, CI 4.7–8.7%) (χ 2 = 0.0114, P = 0.91). Seasonal differences became apparent. While the prevalence of common voles infected with both pathogens differed significantly (χ 2 = 6.563, P = 0.01) between summer 4.3% (CI 2.2–7.4%, 12/280) and fall 10.2% (CI 7.1–14.1%, 32/313), no coinfections were detected in spring (0/78).
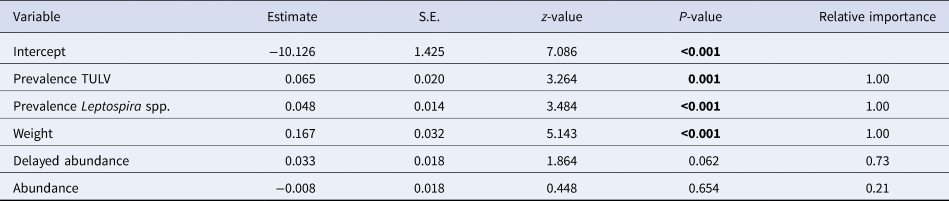
The initial global generalised linear mixed model had a R 2marginal of 0.52 and no overdispersion, but the factor season was associated with increased multicollinearity (variance inflation factor >4) and was subsequently omitted from the model. Table 2 shows the comparison of candidate models as well as their respective AIC and model weights. The first three models were included in the AIC cut-off value of Δ2 and subject to model averaging. Averaged parameter estimates and respective relative importance are presented in Table 3. Individual coinfection probability with TULV and Leptospira spp. was driven by both, individual and population-level factors. Individual age and population-level TULV and Leptospira spp. prevalences are significant factors, in determining coinfections. Both abundance measures (delayed and direct) were selected in the averaging process, but only the abundance in the previous season seemed to influence subsequent coinfection dynamics (delayed density dependence). Parameter effect sizes (mean and 95% CI) are shown in Figure 4a. Individual weight had the most dominant effect, while the CIs of the delayed abundance marginally incorporated zero. Model predictions for each factor are shown in Figure 4 (b, c), where for each factor all other factors were kept constant at their respective mean value. Predictions show that older individuals have a higher probability of being coinfected and that a higher abundance of common voles in the previous season increased the probability of subsequent individual coinfections. For both pathogens an increasing prevalence (while keeping the other pathogen constant) increased the probability of coinfections. As both pathogens differ in their range of detected prevalences, this effect is more prominent in Leptospira spp. compared to TULV. However, the relationship between the increase in prevalences of single pathogens and coinfections is significantly better explained by an exponential increase (Leptospira spp.: R 2 = 0.99; TULV: R 2 = 0.99) compared to a linear one (Leptospira spp.: R 2 = 0.86; TULV: R 2 = 0.90) (comparison Leptospira spp.: F = 334.88; P < 0.001, TULV: F = 451.06; P < 0.001). This indicates that prevalences near the upper end of the potential range result in disproportionally more coinfections compared to lower prevalences. Two of 469 bank voles (0.4%) tested positive for PUUV and Leptospira spp. These originated both from site UH2 in fall.
Graphical representation of the model averaging following multimodel inference. (a) Averaged factor mean estimates and their 95% confidence interval. (b–d) Prediction for each factor in the average model. For each predicted factor all other factors were kept constant at their respective mean value. (b) Relationship between individual weight and prevalence of coinfections. (c) Density dependence (direct and delayed) of coinfections. (d) Relationship between single pathogen infections and the prevalence of coinfections.

Binomial generalised linear models explaining the probability of the occurrence of coinfections between Leptospira spp. and TULV. Estimates of continuous variables and presence of categorical (indicated by+) population-level and individual variables are presented. Models with Δ AIC >2 were excluded. DF = degrees of freedom, logLik = log-likelihood value
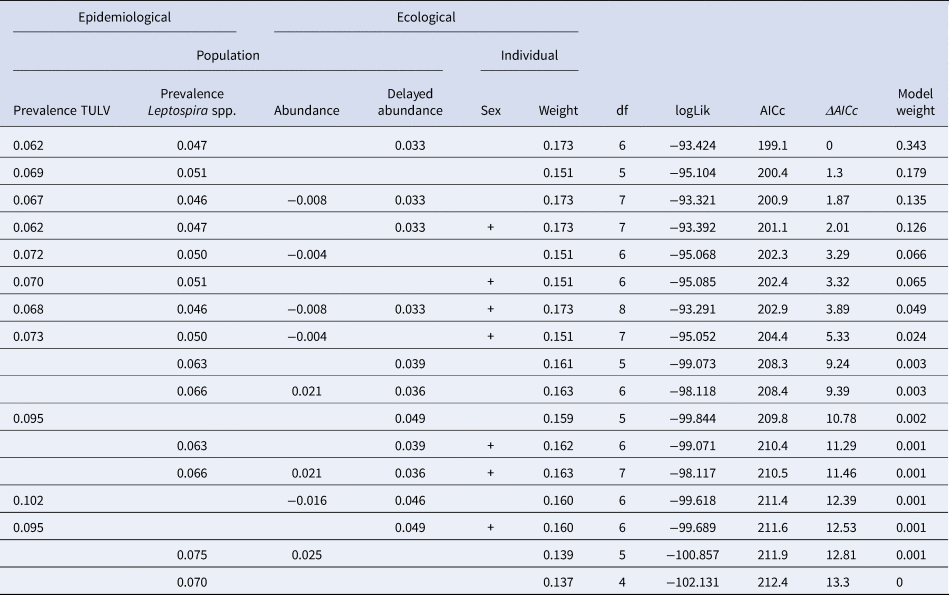
Model averaged estimates for the probability of the occurrence of coinfections between Leptospira and TULV. Relative importance as the sum of Akaike weights of all best fitting model where the specific variable is included. Significant factors are highlighted in bold. S.E. = Standard Error
Discussion
We detected Leptospira spp. in several small mammal species in central Germany. Compared to a previous study in the same region (Fig. 1, square) [Reference Fischer4], overall prevalence was higher in this study. However, the tendency that Microtus spp. had, on average, higher prevalence compared to most other species is mirrored here. In a European context, studies that screened at least 10 individuals of one species, generally reported similar prevalence for striped field mice (12.0–19.6%) [Reference Fischer4, Reference Kurucz16, Reference Mayer-Scholl31], common voles (14.0–30.0%) [Reference Fischer4, Reference Kurucz16, Reference Mayer-Scholl31–Reference Blagojevic33] and field voles (12.0–30.1%) [Reference Fischer4, Reference Mayer-Scholl31]. For the yellow-necked mouse only a study in Serbia detected a higher average prevalence with 34.3% [Reference Blagojevic33], for the wood mouse studies detected similar average prevalence with 15.4% and 18.0% [Reference Mayer-Scholl31, Reference Vitale34] and for bank voles our study is in line with a previously published prevalence [Reference Fischer4, Reference Mayer-Scholl31].
In general, prevalence increased from spring to fall, likely reflecting more favourable conditions for survival outside the host at high temperature and in moist soil [Reference Levett3]. Interestingly, the strong variance of Leptospira spp. prevalence was not only dependent on season but also on site. In fall, the season with the highest overall prevalence, there was high spatial variability in Leptospira spp. prevalence. While there was no Leptospira spp. at some sites, three sites exhibited >65% prevalence for the common vole in fall (E4, KYF6, W2). A comparable Leptospira spp. prevalence is often reported only in Norway rats (Rattus norvegicus) collected in sewage systems [Reference Krojgaard35].
High prevalence of Leptospira spp. in certain sites arise from local environmental conditions such as soil composition (e.g. mineral and salt composition), soil humidity [Reference Thibeaux36] and the presence of water bodies. Irrigation can be a significant factor for Leptospira prevalence in rodents [Reference Morand37] and human outdoor activity, mainly watersports, is related to localised outbreaks of leptospirosis in humans [Reference Jansen38, Reference Lau39]. The effect of livestock on human or even rodent infection risk is still unclear [Reference Mwachui40] and requires further investigation. On a larger scale, weather effects like intense rainfall with subsequent flooding have been shown to cause more widespread outbreaks of leptospirosis [Reference Lau39]. Further studies should incorporate these risk factors to estimate the spatial persistence of Leptospira in their natural reservoirs.
In grassland, prevalence was especially high in common and field voles, which were exclusively infected with L. kirschneri [Reference Fischer4, Reference Mayer-Scholl31, Reference Obiegala41]. Forest rodents were found to carry either L. kirschneri or L. borgpetersenii; L. interrogans was not detected here. We detected L. kirschneri in wood mice and either L. kirschneri or L. borgpetersenii in bank voles and yellow-necked mice. Other studies reported lower prevalence for L. borgpetersenii but high prevalence for L. interrogans in forest rodents [Reference Fischer4, Reference Mayer-Scholl31, Reference Obiegala41]. All these studies are consistent with our finding that L. kirschneri is the most frequently found Leptospira genomospecies in small mammals in Germany [Reference Fischer4, Reference Mayer-Scholl31, Reference Obiegala41].
The detection of TULV-RNA at 18 of 21 sites in this study where common voles were trapped is in line with the German-wide distribution of this pathogen [Reference Schmidt13]. The overall prevalence of 13.9% in common voles is comparable to previously published values of 6.2–23.4% in Europe including Austria, Czech Republic, France, Germany and Hungary [Reference Schmidt13, Reference Kurucz16, Reference Saxenhofer28, Reference Schmidt32]. Field voles and European pine voles were not infected with TULV, even though TULV-positive common voles were present in the sites. This finding confirms the common vole to be the main reservoir for TULV and other Microtus spp. to be rather accidental hosts [Reference Schmidt13] even though it is based on a small number of individuals from these two species that were available for analyses. As expected, the sequences clustered in the CEN.N clade of TULV together with sequences from geographically close origin (see [Reference Saxenhofer28]).
The very low prevalence of PUUV in this study was most likely a result of the study location at the distributional edge of this hantavirus in Germany. High PUUV prevalence was detected earlier in bank voles during the hantavirus outbreak year 2010 in the western part of Thuringia. Those published PUUV sequences (site Diedorf, see Fig. 1) formed a separate clade ‘Rhön Mountains’ [Reference Drewes11, Reference Faber30]. Thuringia is situated at the eastern distribution border of PUUV in Germany [Reference Drewes11] and features zones with previously reported disease clusters in humans and infected bank voles only in the western part of the state [Reference Faber9, Reference Drewes11, Reference Faber30], while the exact extent of the distributional range is largely unknown. The presented phylogeny provides further information on the dynamics of PUUV in bank voles along its distribution border, as sequences from this study did not cluster with sequences from the abovementioned site Diedorf in Thuringia, but instead with sequences from Lower Saxony and Hesse. This observation may suggest two immigration routes of PUUV-infected bank voles into Thuringia over time, which presents an interesting opportunity to study the short- and long-term dynamics of zoonotic pathogens along the edges of their distributional range in the future.
In this study, we did not detect DOBV infections in 86 striped field mice. DOBV infections have been detected only in striped field mice from more eastern and northern located sites, including the eastern part of Thuringia [Reference Schlegel12, Reference Rasche42]. Likewise, human infections were detected exclusively in eastern and north-eastern Germany [Reference Faber9, Reference Hofmann43].
Coinfection with both, Leptospira and TULV in common voles were observed before in Hungary with a prevalence of 3.7% [Reference Kurucz16]. We identified both, individual and population-level factors associated with coinfection of Leptospira and hantavirus in common voles. Individual-level drivers seemed to be mostly associated with age. For each pathogen this has been previously described [Reference Fischer4, Reference Krojgaard35, Reference Khalil44]. The possibility of infection increases over each individual's lifetime and common voles are probably persistently infected with both pathogens, although we have to acknowledge that weight might be an imperfect proxy for age, especially when chronically infected, coinfected individuals could potentially suffer from malnourishment.
Overall, coinfections of Leptospira spp. and TULV did depend on host density. Rather than coinfections increasing with immediate density, there was a time-lagged response, where individual coinfections were positively correlated to the density 3 months ago. For other pathogens, this time delay has been shown to be an integral part of the transmission process where an increase in density enhances the availability of susceptible hosts that later can become infected [Reference Burthe45]. In coinfections, this aspect might even be amplified, as the transmission process for two pathogens has to be completed. The route of transmission can potentially add to the delayed effect. Rodriguez-Pastor et al. [Reference Rodríguez-Pastor46] detected delayed density dependence in Bartonella rochalimae and attributed it to the flea life-cycle as a potential cause for the delayed response. In our context, Leptospira spp. can survive outside of their host up to 9 weeks in soil [Reference Thibeaux47] and up to 20 months in freshwater [Reference Andre-Fontaine, Aviat and Thorin48, Reference Casanovas-Massana49]. Long periods of environmental survival might preclude any association with immediate host abundance and rather favour delayed responses.
Unsurprisingly, both pathogens are positively associated with increased coinfections, representing the underlying mathematical probability of coinfections to occur when prevalences of both pathogens increase. However, this relationship is best characterised by an exponential regression rather than a linear one (Fig. 4d), indicating that high prevalences are associated with disproportionally more coinfections. This could be interpreted as increased availability of individuals susceptible to coinfections in high prevalence scenarios for both pathogens. Telfer et al. [Reference Telfer50] highlighted the importance of pathogen community interaction in determining the overall individual susceptibility to subsequent infections. This would imply that an infection with one of the two pathogens would compromise immunocompetency of the infected individual facilitating a ‘more efficient’ infection with the other pathogen. Our methodology is, however, not suitable to track individual changes within a population across time and might therefore miss subtle individual effects.
Consequently, frequent coinfections were observed in areas where a particularly high prevalence of Leptospira spp. was detected. We conclude that, at least for TULV in grassland, high levels of coinfections with Leptospira spp. are rather driven by the spatial assemblage of high Leptospira spp. prevalences than by TULV prevalence. Despite the low zoonotic potential of TULV [Reference Reynes51], coinfections are of general concern. At sites with a high prevalence of Leptospira spp. in rodents and an associated increase in human leptospirosis cases, our results suggest that there is also an increased risk of hantavirus coinfections, that might go undetected in humans when coinfections exhibit similar clinical presentations. The spatial assemblage of high Leptospira spp. prevalence is therefore of concern as it might also present hot-spots for coinfections with other pathogens. The environmental and epidemiological drivers associated with the patchy occurrence of those hot-spots should be the topic of future research.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268821000443.
Acknowledgements
We thank Philipp Harpering, Mechthild Budde, Kyra Jacoblinnert, Philipp Koch, Maysaa Dafalla, Ulrike M. Rosenfeld, Diana Below, Marco Rump, Hannah Schmuck and Vivien Lukas Hartmann for support of small mammal trapping. We also thank Dörte Kaufmann, René Ryll, Stefan Fischer, Vanessa Schulze, Robin Brandt, Maria Justiniano Suarez, Marie Luisa Schmidt, Elfi Schlohsarczyk, Maysaa Dafalla, Dewi Murni Alfa, Florian Binder, Lea Tölken, Beate Matzkeit and Donata Hoffmann for dissection of small mammals, and Patrick Wysocki for preparing Figure 1.
Financial support
This study was commissioned and funded within the Environment Research Plan of the German Federal Ministry for the Environment, Nature Conservation, Building and Nuclear Safety (JJ, grant number 3716484310) and the German Federal Ministry of Education and Research through the Research network for zoonotic infectious diseases (RoBoPub consortium, FKZ 01KI1721A, awarded to RGU and 01Kl1721E awarded to JJ; ZooKoInfekt, FKZ 01KI903B awarded to RGU and 01KI903A awarded to MP). GH was supported by the Swiss National Science Foundation (grant 31003A-176209).
Conflict of interest
All authors declare that they have no conflict of interest.
Data availability statement
The data for the study are available from the corresponding author.










 Open access
Open access