

Introduction
Since its emergence at the end of 2019, coronavirus disease 2019 (COVID-19) has spread worldwide with clinical presentations ranging from asymptomatic to life-threatening [1]. The rise of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the economic and social impact caused a surge of research and the rapid development of several effective vaccines as well as novel therapeutic agents. Camostat mesilate/Foipan is a serine protease inhibitor that has been licensed and extensively used in patients with pancreatitis and reflux esophagitis for more than 20 years in Japan[Reference Talukdar, Saikia, Singal and Tandon2]. After its original approval for the relief of abdominal pain associated with chronic pancreatitis [Reference Talukdar, Saikia, Singal and Tandon2], camostat was found in vitro to inhibit transmembrane protease serine type 2 (TMPRSS2), which plays a critical role in the virus life cycle of SARS-CoV-2 [Reference Hoffmann, Hofmann-Winkler and Smith3]. TMPRSS2 is expressed in the human respiratory tract and contributes to SARS-CoV-2 infection and spread by cleaving and priming the viral spike (S) protein that subsequently binds the ACE2 receptor and allows viral entry into target cells [Reference Hoffmann, Kleine-Weber and Schroeder4]. The potential importance of TMPRSS2 was shown by enhanced SARS-CoV-2 infection in an engineered TMPRSS2 overexpressing cell line (VeroE6/TMPRSS2) and by viral spike (S) protein priming specifically in TMPRSS2 expressing cells (Caco-2) [Reference Hoffmann, Kleine-Weber and Schroeder4, Reference Matsuyama, Nao and Shirato5]. Additionally, camostat was previously found to be effective in protecting mice against death due to a lethal infection by SARS-CoV, with a survival rate of approximately 60% [Reference Zhou, Vedantham and Lu6]. These studies suggested a potential mechanism of action for camostat in treating SARS-CoV-2 infection, and interest in the drug increased globally.
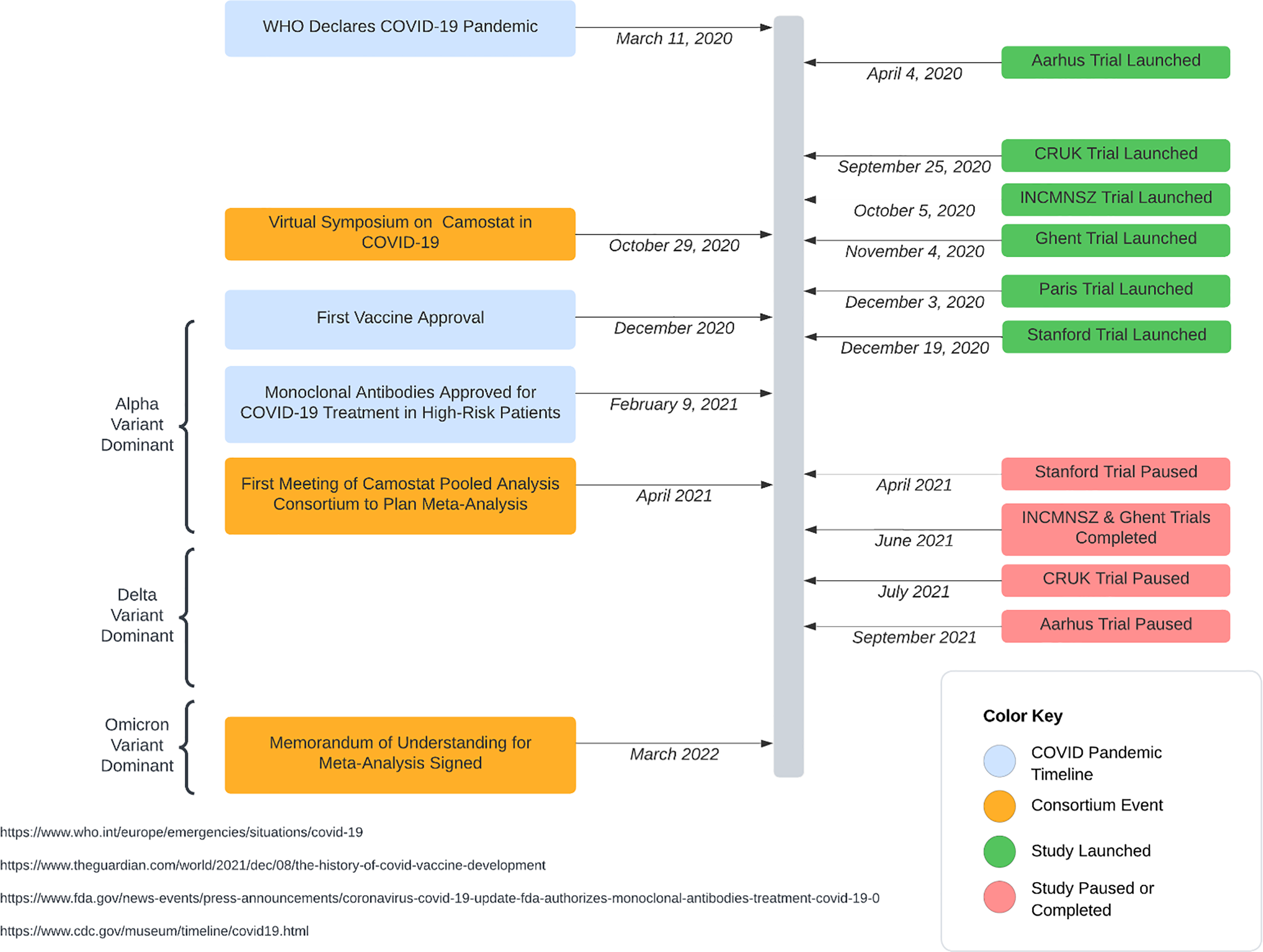
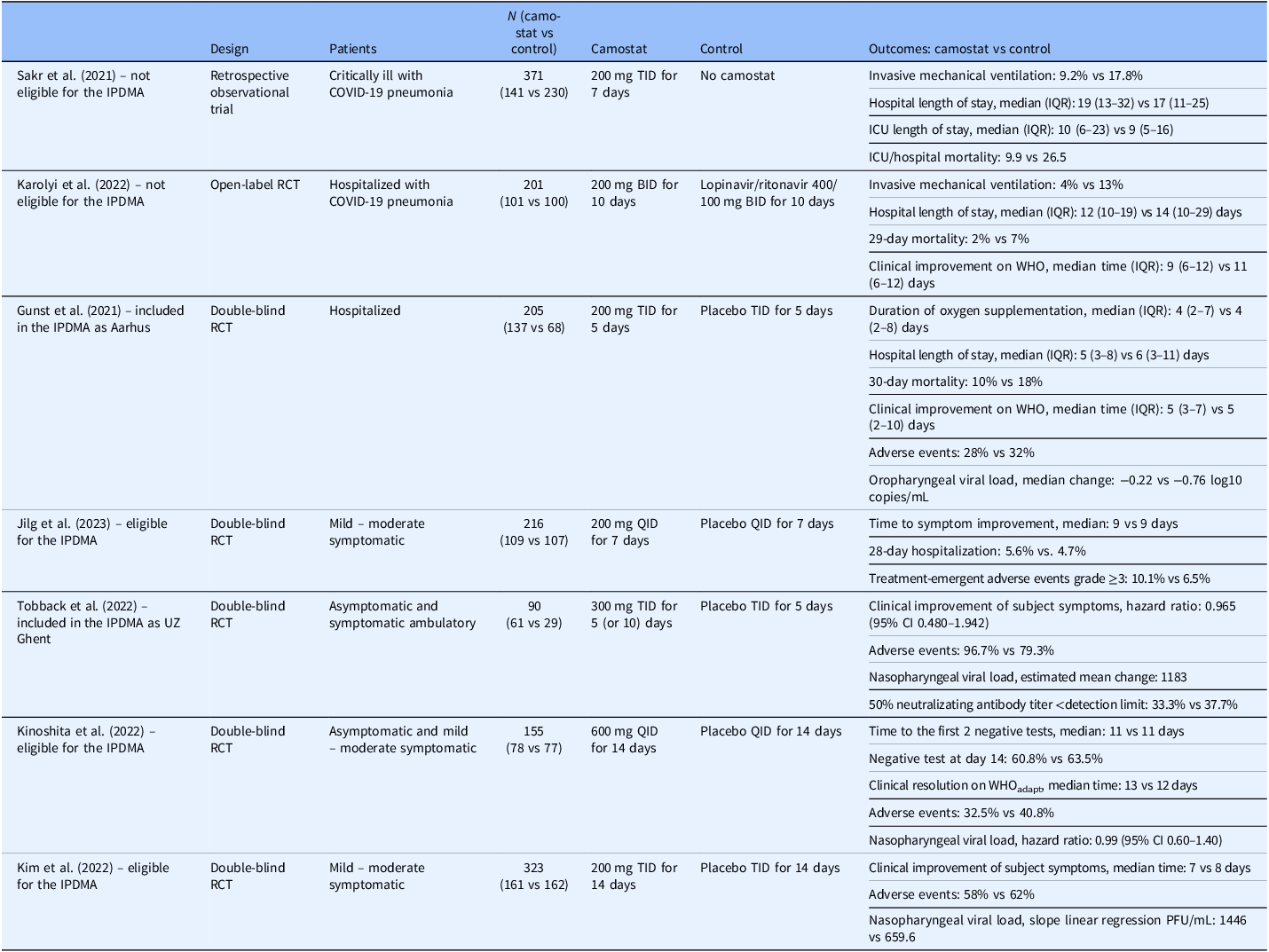
By the autumn of 2020, studies evaluating camostat as a potential treatment for COVID-19 had been launched [Reference Shah and Bryce7]. The landscape for treatment and prevention of SARS-CoV-2, however, rapidly changed. Remdesivir was approved for the treatment of hospitalized patients in October 2020 and monoclonal antibodies were available by the end of 2020 for outpatients; the first COVID-19 vaccine was administered on 8 December 2020. Investigators were concerned that the emerging treatments altered equipoise for placebo-controlled trials; the onset of vaccines also reduced the frequency of serious illness, the endpoint of most of the trials. Consequently, many of the trials investigating camostat stalled and only a few published their findings (Table 1, Figure 1) [Reference Jilg, Chew and Giganti8–Reference Kim, Jeon and Kim14]. Those that have been published demonstrated inconsistent results, each focusing on different target subgroups with limited ability to draw informative conclusions.
Timeline of studies in the camostat pooled analysis consortium in context of the COVID-19 pandemic.

Summary of published peer-reviewed trials evaluating camostat in COVID-19

BID = two times daily; CI = confidence interval; ICU = intensive care unit; IPDMA = individual patient-data meta-analysis; IQR = interquartile range; PFU = plaque forming unit; QID = four times daily; RCT = randomized controlled trial; TID = three times daily; WHO = World Health Organization’s 7-category scale; WHOadapt = WHO scale adapted to 8 categories.
Because camostat remains a plausible and likely safe treatment, and because some previously useful treatments – such as monoclonal antibodies – are no longer effective, we asked investigators with ongoing, terminated, or paused outpatient randomized controlled trials (RCTs) of camostat to participate in an individual patient data meta-analysis (IPDMA). Our aim was to synthesize evidence to make a collectively stronger conclusion than could be achieved with any individual trial [Reference Gunst, Staerke and Pahus11, Reference Tobback, Degroote and Buysse12, Reference Terada, Fujita and Kawahara15, Reference Gurevitch, Koricheva, Nakagawa and Stewart16]. This pooled study, which was facilitated by novel analytic methods and data sharing efforts during the global health emergency [Reference Petkova, Antman and Troxel17–Reference Bunning, Hedlin and Purington21], assessed the efficacy of camostat in patients with mild COVID-19 in reducing viral shedding (viral endpoints) and symptom duration (clinical endpoints).
Methods
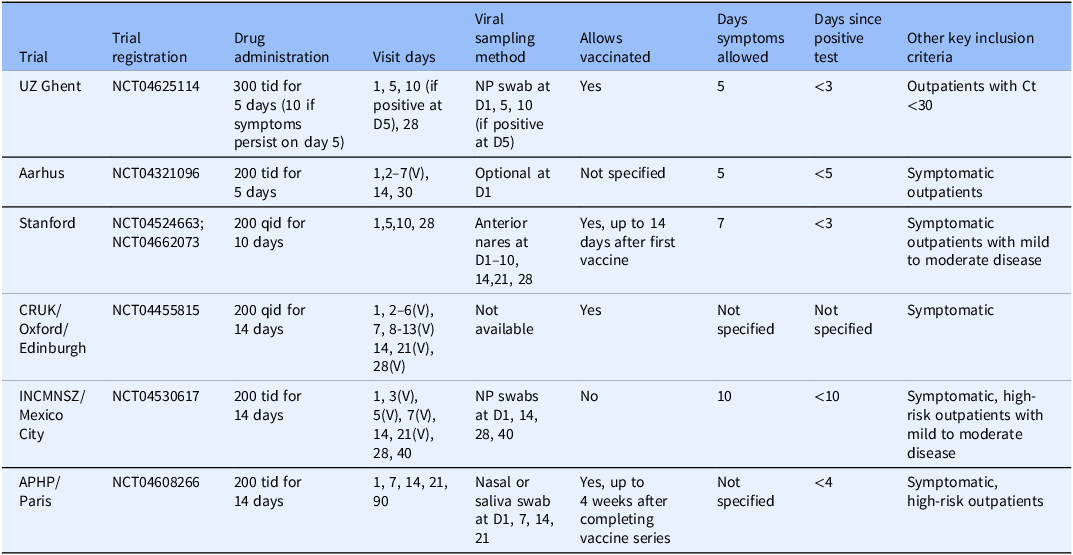
Eligible studies of camostat were identified through www.clinicaltrials.gov. To be included, studies needed to be a RCT evaluating the efficacy of camostat in adults with acute SARS-CoV-2 as compared to the standard of care. The studies also needed to collect data on either viral or clinical endpoints to allow harmonization across studies in a meta-analysis. Ten studies were identified, and all were invited to participate. Out of these, six provided individual patient data for the meta-analysis: Stanford University (Stanford), Cancer Research UK (CRUK), Ghent University Hospital (UZ Ghent), National Institute of Health Sciences and Nutrition Salvador Zubiran (INCMNSZ), Aarhus University Hospital (Aarhus), and Assistance Publique–Hôpitaux de Paris (APHP). Table 2 describes each participating trial, including key inclusion/exclusion criteria, dose, and schedule of camostat administration, measurements collected, and sample size. Four eligible studies did not have individual patient data available for inclusion in this analysis. The individual patient data for one study became available only after the endpoint harmonization process described below [Reference Jilg, Chew and Giganti8]. The corresponding author from the other three studies were contacted and, while the authors initially responded, they ultimately did not share individual patient data with no reason provided [Reference Kinoshita, Shinoda and Nishizaki13, Reference Kim, Jeon and Kim14, Reference Chupp, Spichler-Moffarah and Søgaard22]. Results from all four studies have been reported. Due to the inconsistent endpoints collected across early studies of COVID-19, most endpoints reported in the publications cannot be combined with our endpoints in a traditional meta-analysis. However, one endpoint in Jilg. et al. aligned with our study endpoints and was incorporated in our analysis [Reference Jilg, Chew and Giganti8].
Summary of trials included in individual patient data meta-analysis
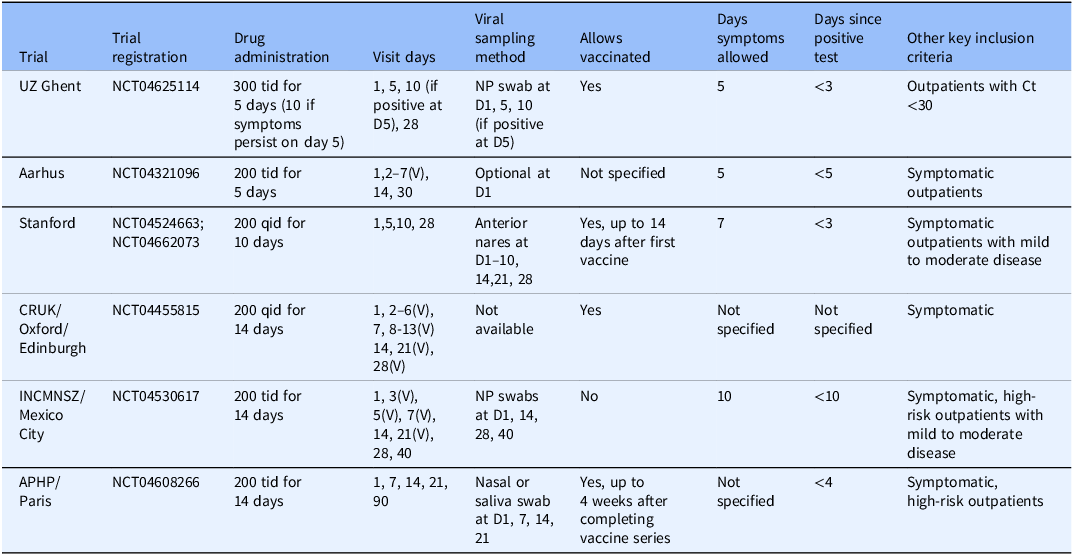
D = Day; NP = nasopharyngeal; V = Virtual visit.
When the investigators of the individual studies began collaborating, the first step was to compare protocols to assess feasibility of harmonizing key variables to pool individual patient data collected from the individual. After endpoint overlap across studies was assessed, we defined the objectives, endpoints, and methods appropriate to achieve our goals in a prespecified SAP (see Supplemental Materials) [Reference Berman and Parker23]. We modeled our design after the methods of Troxel et al. in following a Bayesian monitoring approach to avoid conflicts with the type I error control in the individual study designs, as several of the included studies had paused enrollment and could possibly continue to completion [Reference Troxel, Petkova and Goldfeld18].
Enrolled patients had been randomized into two arms: (1) the control arm, comprised of standard supportive care in addition to placebo in all trials except CRUK where the comparator was standard supportive care alone and (2) the treatment arm, comprised of camostat in addition to standard supportive care (see Table 2 for dosage). Randomization was performed in a 1:1 ratio camostat:control in all trials except UZ Ghent, which randomized in a 2:1 ratio. Randomization at Stanford was stratified by age and sex.
The individual studies were approved by the ethical review boards at their respective institutions. We used the Cochrane risk of bias tool for randomized trials to evaluate the risk of bias for each of the individual studies.
Data sharing/MoU
A Memorandum of Understanding (MoU) was put in place between all participating institutions sharing data and Ono Pharmaceutical Co., Ltd., the manufacturer and supplier of camostat. The MoU included (1) the institution serving as the data coordinating center to store, manage, and analyze the data, (2) the outline of the statistical analysis to be performed, and (3) the description of the technical and organizational measures to ensure the security of data transfer and storage.
Harmonizing viral endpoints for IPDMA
Patient-level data for one or more viral endpoints were available from four studies: Stanford, APHP, UZ Ghent and INCMNSZ. For this IPDMA, the harmonized primary viral endpoint was rate of change in viral load as measured by cycle threshold (Ct) obtained from nasal swabs at Day 1 and 5 (UZ Ghent) and anterior nares swabs at Days 1 through 5 (Stanford). We chose to model the rate of change in viral load to be consistent with other studies of infectious disease reporting viral load endpoints. A secondary viral endpoint was an indicator for testing positive at Day 7 and 14 (anterior nares swabs from Stanford and APHP with nasopharyngeal samples from INCMNSZ).
Harmonizing clinical endpoints
All studies collected clinical endpoints of some type but at varying time points. The following symptoms collected by Stanford, CRUK, UZ Ghent, and Aarhus at consistent time points were included in the symptom resolution endpoint: shortness of breath, fatigue (excluding mild fatigue), nausea, sore throat, nasal congestion, myalgia, cough (excluding mild cough), and diarrhea (not available in patients contributed by CRUK). All questionnaires used an ordinal scale to collect the severity of each symptom. The Stanford trial assessed symptoms daily through Day 28 by sending participants a link to the COVID-19 Outpatient Symptom Survey [Reference Bunning, Hedlin and Purington21]. The CRUK trial assessed symptoms via daily phone or video calls using the FluiiQ Influenza Intensity and Impact Questionnaire [Reference Osborne, Norquist and Elsworth24]. A questionnaire was emailed to patients enrolled in the UZ Ghent trial through Day 14 (or to Day 28 if symptoms persisted at Day 14) [Reference Tobback, Degroote and Buysse12]. Participants in Aarhus were emailed a questionnaire daily until symptoms resolved or Day 30, whichever occurred first (see Supplemental Materials for symptom surveys).
The harmonized primary clinical endpoint for the meta-analysis was time to initial symptom resolution within 14 days, defined as time from randomization until the first of two consecutive days when no symptoms reported. Patients who did not meet the symptom endpoint on their last completed survey were censored.
The secondary clinical endpoint was a composite endpoint of time to hospitalization, initiation of supplemental oxygen use, and death within 28 days from time of randomization. Patients who were never hospitalized, never received supplemental oxygen, and were alive at 28 days were censored.
Safety
Additional secondary outcomes included adverse events (AEs) and a composite of hospitalizations, supplemental oxygen use, and death during the study.
Statistical analyses
All analyses followed the intent-to-treat (ITT) principle and, as such, we grouped patients according to their randomized treatment and included all patients randomized by the time data were shared for analysis. All analyses were adjusted for age and sex, i.e. the randomization stratification variables used at Stanford.
All models were fit using a Bayesian framework, unless otherwise noted. As noted above, we followed a Bayesian monitoring approach to avoid conflicts with the type I error control in the individual study designs for the primary viral and primary clinical endpoints. The distributions and parameters in the model were refined during simulations prior to unblinding. After fitting the models, we evaluated our findings’ robustness to the choice of prior distributions, e.g. we centered the distribution of the beta coefficient at a nonzero value. For example, our primary analyses relied on uninformative priors, where the prior distribution for our parameter of interest is centered at 0. In sensitivity analyses, the center of the prior distribution for our parameter of interest would be nonzero.
To evaluate the harmonized viral endpoint, we derived the rate of change in the viral load over Days 1–5 by fitting a linear regression to each patient’s Day 1–5 data. Rate of change was compared between the two arms using a linear mixed effects model with a random intercept and random slope for trial to account for correlation between observations from the same trial and heterogeneity of effects across trials.
Odds of testing positive on Day 7 and Day 14 were evaluated by treatment arm using a generalized linear mixed-effects model with a random effect for trial, fit using frequentist methods. A separate model was fit to estimate odds ratios comparing camostat to placebo for Day 7 and for Day 14 because different studies had data available for each time point. Jilg et al. reported the number of participants testing positive on both Day 7 and Day 14 by arm in their Figure 2. We included these participants in a post hoc sensitivity analysis [Reference Jilg, Chew and Giganti8].
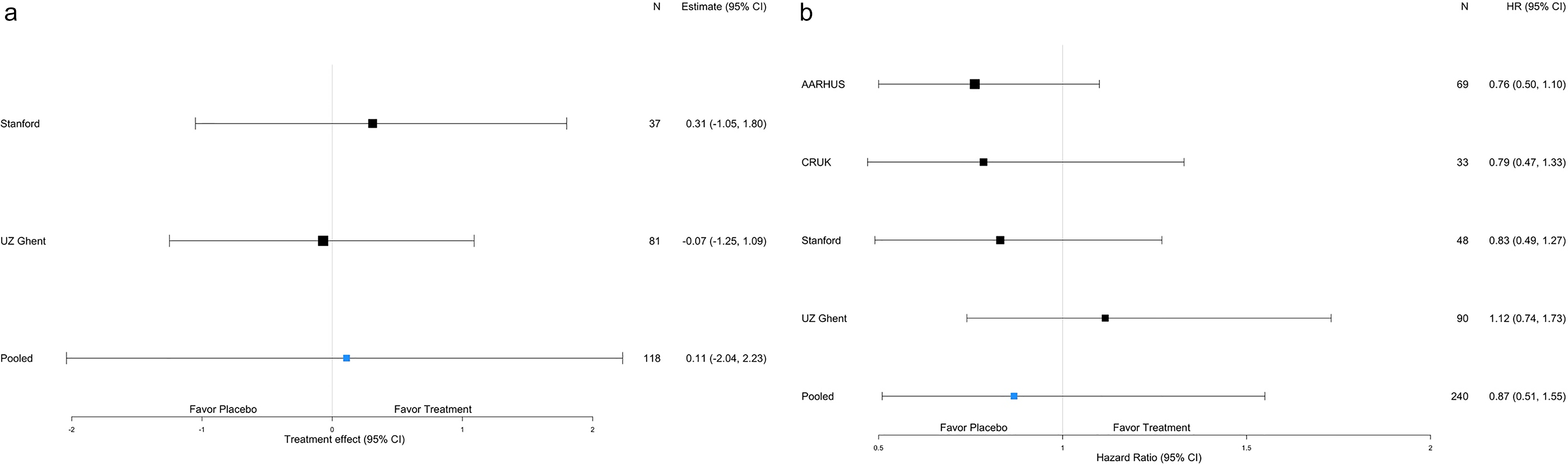
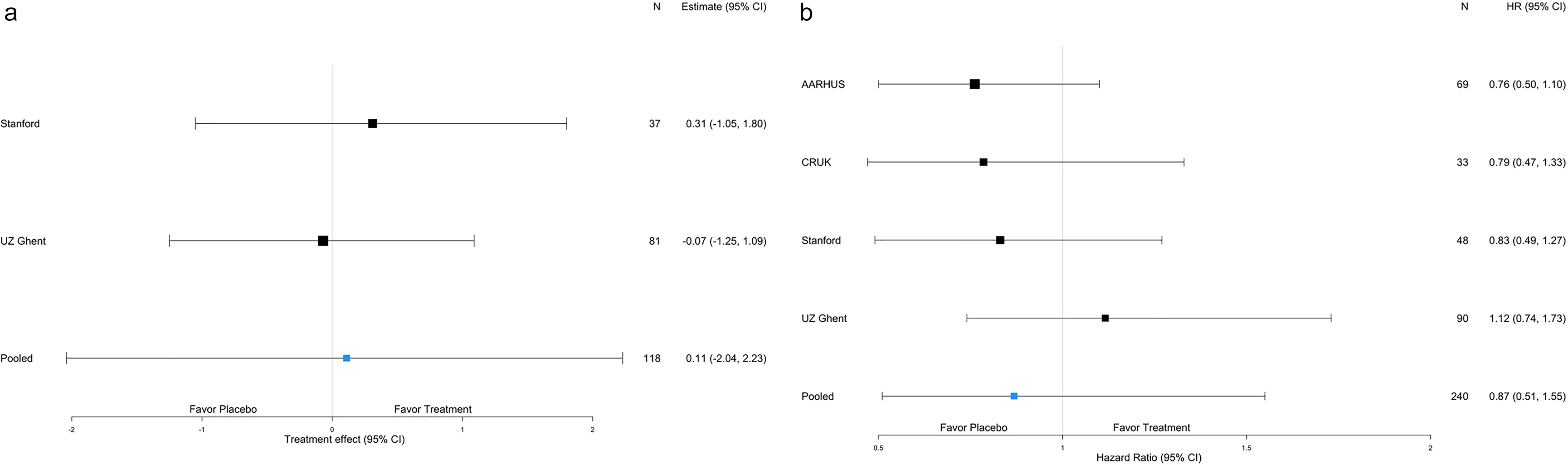
Forest plot displaying estimates and credible intervals by trial for the harmonized viral endpoint (Panel a) and the harmonized clinical endpoint of time to symptom resolution (Panel b).
To evaluate the harmonized clinical endpoints, time until symptom resolution was compared between treatment arms using a two-parameter frailty proportional hazards model to account for within-trial correlation. The hazard ratio for time to clinical improvement was estimated from a Weibull model with Gamma frailties.
In secondary analyses, we used a Cox proportional hazards model, fit using frequentist methods and stratified by trial to compare the time to the first of hospitalization, supplemental oxygen use, and death. The proportional hazards assumption was visually assessed. Patients who did not experience the composite endpoint were right-censored at the end of the patient’s follow-up or Day 28, whichever was earlier. We performed the hospitalization efficacy analysis in (1) all trials collecting data on hospitalization and supplemental oxygen and (2) the subset of patients from the INCMNSZ and APHP trials.
The model parameterizations and additional details are provided in the SAP (Supplemental Materials). Estimates obtained using Bayesian methods are presented with 95% credible intervals. When fitting frequentist models, we performed two-sided tests with alpha =0.05 and present 95% confidence intervals. All analyses were conducted in R version 4.2.1 [25]. We used the “rstanarm” package to fit the Bayesian models in R.
Role of the funding source
The funders had no role in data collection, analysis, or the decision to publish.
Results
431 patients were included in at least one individual patient-level data meta-analysis. The number enrolled in individual trials ranged from 34 to 114. Viral endpoints through Day 5 were derived from 118 patients at UZ Ghent and Stanford; 75 patients from Stanford and APHP contributed viral data at Day 7; 74 patients from Stanford, INCMNSZ, and APHP had viral data available at Day 14; and 240 patients contributed symptom or other clinical data from Stanford, CRUK, UZ Ghent, INCMNSZ, and Aarhus. In a sensitivity analysis, we additionally included 178 patients for whom we were able to extract the secondary viral endpoint of testing positive at Day 7 and 14 from Figure 2a of Jilg et al. [Reference Jilg, Chew and Giganti8].
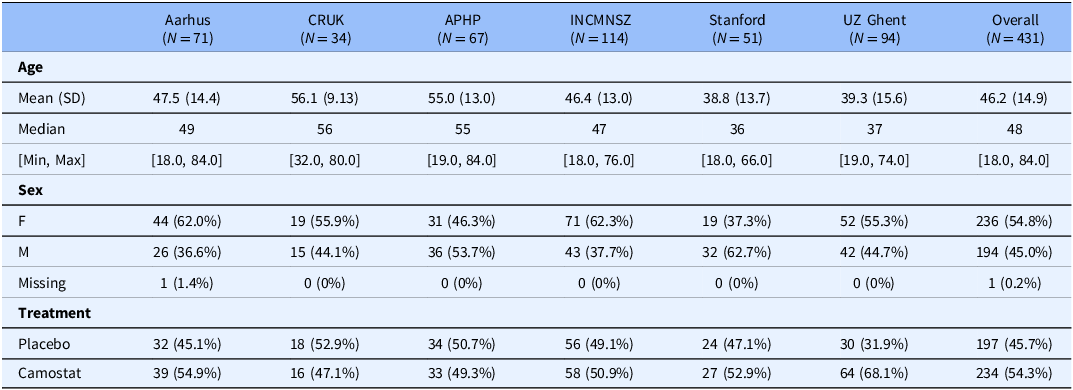
The mean age of participants in the IPDMA studies ranged from 39 to 56 years with a minimum age of 18 and a maximum of 84. Most studies had slightly more females with a range of 37–62% female by study (Table 3). We assessed the risk of bias in the individual studies to be low using the Cochrane risk of bias tool for randomized trials.
Baseline characteristics of all patients overall and by trial
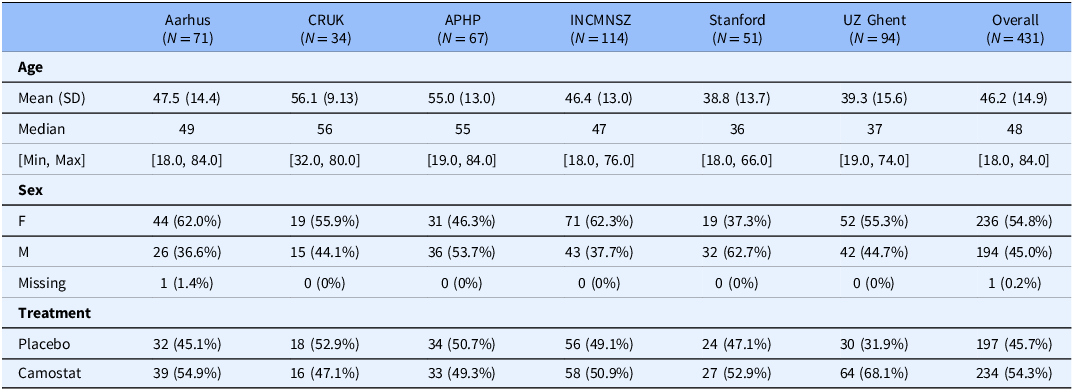
Harmonized viral endpoint IPDMA
Ct values correlate inversely with viral load. The rate of change of Ct values through Day 5 was 0.11 greater in the control arm as compared to the camostat arm (95% credible interval (CrI): 2.04 lower, 2.23 higher), meaning that the amount of detectable virus decreased somewhat more in the placebo than in the control arm. This observed difference, however, was not statistically significant (Figure 2). These findings were unchanged in a sensitivity analysis shifting the prior distribution for the parameter of interest.
Of 75 patients from Stanford and APHP with RT-PCR test results available at Day 7, 69 continued to amplify SARS-CoV2. The odds of a positive result were 2.20 times higher (95% CrI: 0.36 times lower, 13.4 times higher; P = 0.40) in the camostat than in the placebo arm. At Day 14, 35 out of 74 patients from Stanford, APHP, and INCMNSZ continued to yield SARS-CoV2 RNA by RT-PCR and the estimated odds ratio of a positive test was 0.66 times lower (95% CrI: 0.25 times lower, 1.72 times higher; P = 0.40) in the camostat than in the placebo arm. The results were similar when including the viral data published in Jilg et al. (Day 7 OR = 1.24, 95% CrI: 0.76 times lower, 2.02 times higher; P = 0.38 and Day 14 OR = 0.94, 95% CrI: 0.26 times lower, 1.64 times higher; P = 0.84) [Reference Jilg, Chew and Giganti8].
Harmonized clinical endpoints IPDMA
No difference was observed in the time to symptom resolution between treatment arms (Figure 2). The estimated hazard ratio was 0.87 (95% CrI 0.51, 1.55) in favor of placebo. The hazard ratio estimated for each study ranged from 0.76 (95% CrI 0.50, 1.10) to 1.12 (95% CrI 0.74, 1.73) with all credible intervals including a hazard ratio of 1.
Safety IPDMA
Adverse event data were available from Stanford (25 AEs), CRUK (69 AEs), UZ Ghent (257 AEs), INCMNSZ (216 AEs), and Aarhus (19 AEs). A total of 586 AEs were reported in 229 participants across the five studies. The number of AEs reported in each system organ class by treatment arm are provided in the Supplemental Materials.
Hospitalization was rare and no subjects died or received supplemental oxygen prior to hospitalization, so the composite endpoint is equivalent to time to hospitalization. No difference was observed in time to hospitalization between the treatment arms (HR 0.76; 95% confidence interval 0.34, 1.67). The proportional hazards assumption was found to be appropriate, based on a visual assessment of the Schoenfeld residual plot (see Supplemental Materials). There were two deaths in the camostat arm and two deaths in the control arm, all in hospitalized patients.
Discussion
This study is the most comprehensive evaluation of the effectiveness of camostat to treat COVID-19 to date. Treatment with camostat in these studies, primarily in outpatient settings, showed no difference in decline in viral shedding and no improvement in symptoms as compared to standard supportive care in patients with mild COVID-19. While there was some heterogeneity in patient populations and how the drug was administered across the trials, little heterogeneity was observed in results prior to pooling.
At the onset of the COVID-19 pandemic, numerous clinical trials were swiftly initiated, mainly targeting hospitalized patients with severe illness. However, the majority of COVID-19 patients are diagnosed and treated in outpatient settings, where there is a significant demand for treatments that can be administered conveniently to prevent worsening of the condition. The search for effective preventive and therapeutic treatments against COVID-19 in outpatient clinical trials is challenging with regard to constructional and organizational issues as well as within constantly shifting landscapes from changing virus variants to rise and fall in incidence [Reference Grenier, Loniewski and Plazy26]. Recruitment slowed in the present individual studies in parallel with the roll-out of the COVID-19 vaccination campaign along with the introduction of monoclonal antibodies and other therapeutics that perturbed equipoise and made placebo control ethically unacceptable.
At the same time the SAP was being developed, a MOU was being drafted between the institutions conducting the participating studies and Ono, the sponsor and supplier of camostat for some participating studies. The effort required to gain consensus across institutions on the MoU, including the data sharing agreement adhering to the requirements of the GDPR covering the trial data collected in the European Union, was by far the most-challenging and time-consuming step of the process in this IPDMA.
The lack of efficacy of camostat on SARS-CoV-2 viral load in the present meta-analysis is in agreement with the findings in previously published RCTs where swab specimens from the nasopharynx or oropharynx were taken [Reference Jilg, Chew and Giganti8, Reference Gunst, Staerke and Pahus11–Reference Kim, Jeon and Kim14]. Regarding the role of TMPRSS2 in viral entry into the cells, it might be thought that the inhibitory effect of camostat may especially be effective in the early phase of COVID-19. SARS-CoV-2 RNA levels peak around symptom onset and then gradually decline, supporting that administration of camostat might best be started as early as possible in the course of infection [Reference Puhach, Meyer and Eckerle27]. In the present study, patients’ treatment was started within 5–7 days after symptom onset and the optimal time point for therapeutic intervention might have been exceeded. However, data from Kinoshita et al. and UZ Ghent did not show significant reductions in viral load when treatment was started at 3 days median time of symptom onset (interquartile range 0–5 and 1–4, respectively) [Reference Tobback, Degroote and Buysse12, Reference Kinoshita, Shinoda and Nishizaki13]. In conclusion, SARS-CoV-2 viral load reduction by camostat is unlikely to be effective, even when treatment is started as early as possible in clinical practice. These results do not, however,exclude potential effects of this drug if administered as a prophylactic, for instance to close contacts of patients.
There was insufficient evidence to conclude camostat affected the time to subjective symptom resolution or the time to hospitalization in the present meta-analysis, which is in agreement with all previous studies evaluating main COVID-19 symptoms [Reference Jilg, Chew and Giganti8, Reference Tobback, Degroote and Buysse12–Reference Kim, Jeon and Kim14]. In contrast, therapeutic benefits in terms of clinical endpoints have been described by Sakr et al. and Karolyi et al. [Reference Sakr, Bensasi and Taha9, Reference Karolyi, Pawelka and Omid10]. These studies included moderate to severely ill patients hospitalized with COVID-19 pneumonia and concomitant therapies may also have had confounding effects on their outcomes. Taken together, camostat is unlikely to be an effective treatment for symptoms in COVID-19 outpatients.
This study has numerous strengths, including its sample size, patient level data, and diversity in population as a result of participation by six trials across six different countries. The analysis highlights how a pooled analysis can be used in circumstances where equipoise may be lost and/or other extenuating circumstances can halt the progress of a clinical trial, threatening its ability to yield informative results. Unlike meta-analyses performed on published results, our meta-analysis is unlikely to be subject to the publication bias encountered with traditional meta-analyses, because we did not identify participating studies by searching published studies. Identified studies that were not included in the IPDMA all similarly concluded that camostat is not an effective treatment of COVID-19 in this patient population. Limitations of our study include the heterogeneous study designs resulting in different doses administered, inconsistent measurement schedules across trials, differing ascertainment of outcomes, and varying amounts of data available to contribute to the findings. This heterogeneity likely biases the estimates towards the null. Not all studies made their safety data available, and we observed heterogeneity in reporting safety data for the reporting studies, a common problem in clinical trials [Reference Schroll, Maund and Gøtzsche28]. The heterogeneity of study conduct is particularly apparent in the collection of AEs across trials. As in all meta-analyses, the interpretation of the results is limited by data from eligible trials that are unavailable at the time of the analysis. While the eligible trials that were not included in this individual patient-data meta-analysis were all published, a traditional meta-analysis was not conducted in addition to the IPDMA because the endpoints collected and reported across studies differed widely.
In the future, strong consideration should be given to the idea of multiple centers joining forces to design one multi-center study of the efficacy of a given compound. This is particularly important in a pandemic, where the difficulties of initiating such studies are great, and the landscape is fast-moving and unpredictable. A multi-center study designed in collaboration at the start could very well have resulted in arriving at these findings much earlier. Furthermore, the authors here advocate for other similar efficiencies of trial design including platform or master protocols where multiple drugs can be studied simultaneously. At Stanford University, the camostat agent was indeed being studied as one of the sub-protocols in a platform protocol for this purpose [Reference Bunning, Hedlin and Purington21]. Conducting a clinical trial requires a great deal of resources to establish its infrastructure. We believe it is critical for academic and research institutions to pool resources to study multiple agents simultaneously, and that for any given agent, that it be studied in a multi-center framework. Not only will this yield huge gains in efficiency allowing us to address our questions faster and with fewer resources, but it will also allow us to achieve a higher generalizability of findings.
In summary, our team joined together in a creative manner to salvage much effort that was invested in the launching of our respective trials so that we could draw conclusions with strength. We did so in the middle of a pandemic and applied novel principles to design an overarching study that enabled our respective initial study designs to continue if needed. While harmonization of outcomes was key to the pooled analysis, we did not harmonize on triggering rules. Each trial had the flexibility to develop their own stopping rules to inform next steps. In addition, we considered both virologic and clinical endpoints. Such flexibility was critical for enabling participation by as many trials as possible. Our biggest challenge was gaining consensus in data sharing agreements that worked across our international regulatory bodies. With increased experience in collaborating on global clinical trials, we believe these challenges to inefficiencies can and should be learned and overcome.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/cts.2026.10769.
Data availability statement
The statistical analysis plan is included in the supplement and code is publicly available at https://github.com/haleyhedlin/camostatIPDMA. The individual patient data or study-level data from any specific study included in this analysis can be requested from the corresponding trial investigators.
Acknowledgments
The authors thank the participants, clinical investigators, coordinators, and contributors who have participated in the clinical trials and the individuals who helped with the data sharing and agreements required to allow us to perform this meta-analysis.
Author contributions
Haley Hedlin: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing, Els Tobback: Methodology, Writing – original draft, Writing – review & editing, Justin Lee: Formal analysis, Writing – review & editing, Yiwen Wang: Formal analysis, Writing – review & editing, Ilaria Dragoni: Project administration, Writing – review & editing, Daniel C. Anthony: Conceptualization, Methodology, Supervision, Writing – review & editing, Kevin Dhaliwal: Writing – review & editing, John Norrie: Methodology, Writing – review & editing, Sarah Halford: Project administration, Writing – review & editing, Jose Gotes: Writing – review & editing, Mariana Moctezuma: Writing – review & editing, Antonio Olivas-Martinez: Writing – review & editing, Chaitan Khosla: Writing – review & editing, Upinder Singh: Writing – review & editing, Jesper Damsgaard Gunst: Writing – review & editing, Alonso Valdez: Writing – review & editing, David Kershenobich: Writing – review & editing, David Boutboul: Writing – review & editing, Ole Søgaard: Conceptualization, Writing – review & editing, Marie-Angélique De Scheerder: Conceptualization, Methodology, Supervision, Writing – review & editing, Manisha Desai: Conceptualization, Methodology, Supervision, Writing-original draft, Writing – review & editing, Julie Parsonnet: Conceptualization, Methodology, Supervision, Writing-original draft, Writing – review & editing.
Funding statement
This manuscript is partially supported by the National Institutes of Health, the funding source of Stanford’s Center for Clinical and Translational Education and Research award, under the Biostatistics, Epidemiology and Research Design Shared Resource: UL1TR003142 and Stanford Cancer Institute’s Biostatistics Shared Resource (BSR): P30CA124435 (HH, JL, YW, MD). The trial and work at Stanford was supported in part by anonymous donors to Stanford University and also under a Materials Transfer Agreement with Ono Pharmaceutical Co., Ltd. (HH, JL, YW, CK, US, MD, JP). The trial at CRUK was funded by LifeArc (ID, DCA, KD, JN, SH). The trial in Aarhus was funded by The Lundbeck Foundation (JDG, OSS). The trial at APHP was funded by the Assistance Publique Hôpitaux de Paris (DB). Ono Pharmaceutical Co., Ltd., provided study drug.
Competing interests
US is an advisor to Regeneron and Gilead and her institution received research support from the National Institutes of Health, the Agency for Healthcare Research and Quality, and Pfizer, Inc. All other authors declare no competing interests.



 Open access
Open access