


People with psychosis have a life expectancy which is 15 years shorter than that of people without psychosis, with two-thirds of premature deaths being preventable. 1 Psychosis is an illness of young people, with a peak age at onset between 15 and 30 years. Reference Jones2 Early death is mainly due to physical illness, especially cardiometabolic diseases, for which obesity is an important precursor. Obesity increases the risk of heart disease, stroke and death by 1.5–3 times and that of diabetes by 12 times. Reference Kivimaki, Strandberg, Pentti, Nyberg, Frank and Jokela3 Obesity is three times more common in people with psychosis than those without and develops early in the illness. Antipsychotics are prescribed to nearly everyone who develops psychosis but are well-known to be a cause of weight gain leading to obesity. Reference Bak, Fransen, Janssen, van Os and Drukker4 Reducing weight gain is a research priority in psychosis, as highlighted by the James Lind Alliance of patients and clinicians. 5 Currently, obesity and its cardiometabolic consequences in psychosis are addressed with a one-size-fits-all approach, suggesting the same generic interventions to most patients, if any are offered at all. This is not working. We need to better tailor the timing and appropriateness of obesity interventions in psychosis.
Precision medicine has the potential to improve outcomes by targeting the type and timing of interventions according to individual patient characteristics. Reference Lenert, Matheny and Walsh6 Clinical prediction models are widely championed as a way to deliver a precision medicine approach. Reference Studerus, Vaquerizo-Serrano, Irving, Catalan and Oliver7 However, current prediction models are not fully actionable for individuals. At best, they stratify patients on the basis of group averages to recommend interventions selected according to their average treatment effect (ATE) in a population and tell us nothing with certainty about the individual. Further, as current models are based on associations not causes, we do not know whether acting on a predictor variable will actually change the outcome for an individual. For example, yellow tobacco-stained fingers are associated with future lung cancer but clearly do not cause it. Intervening to clean an individual’s fingers does not reduce their risk of future lung cancer. Instead, both finger-staining and lung cancer are confounded by a common cause, smoking, which should be acted on to change the outcome. Existing models such as QRISK and PsyMetRiC are associative rather than causal. Reference Hippisley-Cox, Coupland and Brindle8,Reference Perry, Osimo, Upthegrove, Mallikarjun, Yorke and Stochl9 Like finger-staining in the above example, they identify predictors correlated with outcomes but cannot determine whether intervening on a specific predictor will change the outcome. It is for this reason that we are advised not to causally interpret or act on changes in all individual coefficient values in a multivariable prediction model (this is akin to the ‘table 2 fallacy’ Reference Westreich and Greenland10 ); rather, we should only act on a correctly causally specified variable. Our causal models correctly specify modifiable causes, where intervention will alter outcomes. Nevertheless, the uncertainty of existing association-based models, particularly when not tied to any specific intervention (for instance, PsyMetRiC), may allow more space for shared decision-making processes. Reference Perry, Osimo, Upthegrove, Mallikarjun, Yorke and Stochl9
To be fully actionable for individuals, prediction models need to be based around intervenable causes and show how acting on these causes will materially change the outcome for the individual. To achieve this, we need to model the individualised treatment effect (ITE) of the interventions and predict counterfactuals, which are the different potential outcomes forecast for a specific individual with their unique characteristics following the intervention. Our proposed solution is to develop causal actionable prediction models. These are based on causation, predict counterfactuals and model the ITE by incorporating the proposed intervention into the model, and update predictions over time. Reference Krishnadas, Leighton and Jones11 Correctly causally specifying the actionable predictor variable in our causal actionable prediction models lets us recommend an intervention and/or treatment that we know will actually alter the outcome. Studies exploring these methods exist and have been reviewed recently, but they are underused and not yet applied clinically. Reference Lin, Sperrin, Jenkins, Martin and Peek12,Reference Bica, Alaa, Lambert and van der Schaar13
We hypothesise that the early weight change during the first few weeks of antipsychotic treatment is critical for the development of later obesity and its cardiometabolic consequences in psychosis. Specifically, we aimed to test whether antipsychotic-induced weight gain in individuals with psychosis at 1 year is causally mediated by early weight change in the first 12 weeks of treatment, rather than in the next 12 to 52 weeks. On the basis of this hypothesis, we sought to demonstrate the feasibility of using a causal actionable prediction pathway to prevent antipsychotic-induced obesity at 1 year. We developed two serial causal actionable prediction models: the first was used at baseline to predict clinically relevant antipsychotic-induced weight gain at 12 weeks; and the second was used at 12 weeks to predict obesity at 1 year. We then used counterfactual prediction to demonstrate possible causal changes in the actionable intervention variables that would switch the predicted outcome class to the desired one.
Method
Overview
This was a post hoc analysis of a clinical trial of olanzapine versus haloperidol which recruited 263 participants with first-episode psychosis. We conducted two distinct analyses: (a) causal mediation and (b) prediction modelling, within which there were two sequential models (a baseline model to predict 12-week outcome and a 12-week model to predict 1-year outcome), followed by counterfactual prediction. See Fig. 1 for a schematic overview.
Schematic overview of our methodology.

We adhered to the TRIPOD + AI (Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis + Artificial Intelligence) and AGReMA (A Guideline for Reporting Mediation Analyses) statements. Reference Collins, Moons, Dhiman, Riley, Beam and Van Calster14,Reference Lee, Cashin, Lamb, Hopewell, Vansteelandt and VanderWeele15 All statistical analyses were performed using R, Comprehensive R Archive Network version 4.4.2, for Windows (R Core Team, R Foundation for Statistical Computing, Vienna, Austria; https://cran.r-project.org/bin/windows/base/) and code is provided in the Supplementary Material available at https://doi.org/10.1192/bjp.2026.10561. The Supplementary Material also includes a glossary of causal terms and concepts.
Data and participants
We used data from the Lilly F1D-MC-HGDH trial. The F1D-MC-HGDH trial was a double-blind, multicentre, randomised controlled trial of olanzapine versus haloperidol treatment in 263 participants meeting diagnostic criteria for first-episode psychosis (including schizophrenia, schizophreniform disorder and schizoaffective disorder as defined by the DSM-IV). Inclusion and exclusion criteria were as outlined previously Reference Lieberman, Tollefson, Charles, Zipursky, Sharma and Kahn16 (Supplementary Material). This study was not preregistered and a protocol was not published. Lilly has not contributed to or approved and is not in any way responsible for the contents of this publication.
Data preparation
There were no data cleaning or pre-processing steps other than dummy coding categorical variables.
Outcomes
The outcome time point of 1 year was chosen to provide a clinically meaningful prediction window. We used a continuous outcome for the causal mediation analysis: weight in kilograms at 1 year. We chose to test weight change from weeks 0 to 12 as a mediator for pragmatic clinical reasons, because the majority of people starting antipsychotic medication for the first time will experience remission of symptoms within 3 months, Reference Barnes, Drake, Paton, Cooper, Deakin and Ferrier17 and it is usually inappropriate to offer weight loss interventions when someone is acutely psychotic. In our mediation analysis, we sought to identify whether this period of early weight gain induced by the antipsychotic caused the later weight and obesity outcomes at 1 year. It is only by identifying the factors which cause the outcome that we can know whether changing these causal factors will also change the outcome.
The outcome measure for the baseline prediction model was clinically significant antipsychotic-related weight gain at 12 weeks. This was defined as ≥7% weight gain as per McIntyre et al. Reference McIntyre, Kwan, Rosenblat, Teopiz and Mansur18 The outcome measure for the 12-week prediction model was obesity at 1 year. Obesity was defined as having a body mass index (BMI) ≥ 30 kg/m2. We excluded participants with missing outcomes, as outcome imputation in prediction studies is controversial. Reference Steyerberg19
Sample size
The development sample for the baseline model included 172 individuals followed up to 12 weeks, with 98 individuals with clinically significant weight gain at 12 weeks. The development sample for the 12-week model included 97 individuals followed up for 1 year, with 35 individuals with obesity at 1 year. There was no external validation. The causal mediation analysis described below was performed on the development sample for the 12-week model. This was a feasibility study to illustrate the promise of causal actionable prediction using counterfactual explanations; the models are not intended to be used clinically. No power calculation for the prediction models was performed. Hernán suggests that power calculations should not be a barrier to permitting causal analysis in existing data-sets, and that causal effects are not binary signals which are either statistically significant or not. He further suggests that many studies should be performed and reported, regardless of sample size, and then ultimately meta-analysed to arrive at the true effect estimate. Reference Hernan20
Missing data
For the baseline model, after exclusion of participants who did not complete follow up to 12 weeks (85 of 263 (32.3%) participants), there were 6 of 178 (3.37%) participants with missing height data. Given that the percentage missing was less than 5%, we expected a low risk of bias, and complete case analysis was used, leaving 78 of the 132 (59.1%) original participants in the haloperidol arm versus 94 of the 131 (71.8%) participants in the olanzapine arm (χ 2(1) = 4.1, P = 0.04). We conducted a sensitivity analysis multiply imputing the missing height values and compared the internal validation performance metrics for the baseline model with and without inclusion of participants with missing data. There was no evidence of any difference. This is reported in the Supplementary Material. For the 12-week model, after exclusion of participants with who did not complete follow-up to 1 year (166 of 263 (63.1%) participants; 89 of 132 (67.4%) in the haloperidol arm versus 77 of 131 (58.8%) in the olanzapine arm; χ 2(1) = 1.8, P = 0.2), there were no missing data.
Analytical methods
Causal mediation analysis
There was no correlation between weight change from 0 to 12 weeks and that from 12 to 52 weeks (r(95) = −0.0180, P = 0.9). Therefore, we conducted a parallel causal mediation analysis using ordinary least squares linear regression per the directed acyclic graph (DAG) in Fig. 2. The exposure of interest (the intervention) was antipsychotic choice at baseline (olanzapine versus haloperidol). The first mediator of interest was weight change from 0 to 12 weeks (12-week weight − baseline weight). The second mediator of interest was weight change from 12 to 52 weeks (52-week weight − 12-week weight). The outcome was weight at 1 year. We adjusted for mediator–outcome confounders measured at baseline (age, ethnicity (White, Black or Asian/other), gender, current smoking status, weekly alcohol intake (number of drinks) and BMI). We assumed that the exposure was unconfounded as it was randomised. Parallel causal mediation was performed using the PROCESS macro in R. Reference Hayes and Little21 We obtained bootstrapped confidence intervals (n = 10 000) for the (pure) natural direct effects, the (total) natural indirect effects and the total effects. This also tested for moderation of the mediators’ effects on the outcome by the exposure (i.e. exposure-by-mediator interactions).
Directed acyclic graph outlining our causal assumptions for the parallel causal mediation analysis and the causal actionable prediction models. Green paths represent causal paths from antipsychotic choice at baseline to weight at 1 year. Confounders of the weight change mediators and outcome included age, ethnicity (White, Black or Asian/other), gender, current smoking status, weekly alcohol intake (number of drinks) and baseline body mass index. As the exposure (antipsychotic choice) was randomised and therefore unconfounded, the minimal adjustment set for estimating the causal mediation effects consisted of these mediator–outcome confounders only. For the baseline prediction model (predicting 12-week weight change dichotomised at ≥7%, i.e. clinically significant antipsychotic-related weight gain), the causal actionable predictor of interest was antipsychotic choice. This was randomised so that the estimated causal treatment effect would not be subject to bias. For the 12-week prediction model (weight at 1 year dichotomised at BMI ≥ 30 kg/m2, i.e. obesity), the causal actionable predictor of interest was weight change from 0 to 12 weeks. Time flows from left to right. The diagram was created in DAGitty, a popular browser-based environment for creating, editing and analysing causal diagrams (directed acyclic graphs).

We denote the potential outcome by Y(t,M(m)), which depends on treatment status t ∈ {0, 1} and mediator M set to status m ∈ {0, 1}. Then, the (pure) natural direct effect (NDE) is obtained by holding the potential mediator at the level under the control condition while changing the treatment from the control to the experimental condition
 $$ NDE= \mathbb{E}\,\left[Y\left(t=1, M\left(m=0\right)\right)-Y\left(t=0, M\left(m=0\right)\right)\right], $$
$$ NDE= \mathbb{E}\,\left[Y\left(t=1, M\left(m=0\right)\right)-Y\left(t=0, M\left(m=0\right)\right)\right], $$
where NDE is the natural direct effect, and
 $\mathbb{E}$
denotes mathematical expectation. The (total) natural indirect effect is the difference between the total treatment effect and the (pure) natural direct effect. This represents how much the outcome would change if the treatment condition was controlled at the experimental level, while the mediator was changed from the level under the control condition to the level under the experimental condition:
$\mathbb{E}$
denotes mathematical expectation. The (total) natural indirect effect is the difference between the total treatment effect and the (pure) natural direct effect. This represents how much the outcome would change if the treatment condition was controlled at the experimental level, while the mediator was changed from the level under the control condition to the level under the experimental condition:
 $$ {\rm NIE}= \mathbb{E}\left[Y\left(t=1, M\left(m=1\right)\right)-Y\left(t=1, M\left(m= 0\right)\right)\right], $$
$$ {\rm NIE}= \mathbb{E}\left[Y\left(t=1, M\left(m=1\right)\right)-Y\left(t=1, M\left(m= 0\right)\right)\right], $$
where NIE is the natural indirect effect. The (pure) natural direct effect and the (total) natural indirect effect add up to the total treatment effect:
 $$ \textrm{TE}={\rm NDE}+{\rm NIE}, $$
$$ \textrm{TE}={\rm NDE}+{\rm NIE}, $$
with TE denoting the treatment effect.
We conducted sensitivity analysis with respect to unobserved residual confounding by injecting synthetic mediator–outcome confounders of increasing strength into the causal mediation analysis. We also explored two plausible alternative potential mediators of 0- to 12-week change for the effect of antipsychotic choice on the weight outcome at 1 year available in the trial data-set (0- to 12-week change in total serum cholesterol as the only available measure of lipids, and 0- to 12-week change in random serum glucose as the best available marker of insulin resistance, adjusting for baseline values).
Causal actionable prediction model development and internal validation
We developed two serial models because a model using only predictors at baseline may not be applicable at later time points. For both the baseline and 12-week causal actionable prediction models, we used binary logistic regression fitted by maximum likelihood estimation. We chose to develop binary logistic regression prediction models as these mirror clinical decision boundaries, which are mostly based on predefined thresholds (e.g. offering a weight intervention only to people who have a BMI ≥30 kg/m2 and are therefore classified as obese).
Internal validation adjusting performance metrics for optimism was conducted by bootstrap (n = 500) as described by Harrell. Reference Harrell22 Discrimination was quantified by the concordance statistic (c-statistic). For binary outcomes, the c-statistic is equal to the area under the receiver operating characteristic curve, which plots the sensitivity versus 1 − specificity for consecutive probability thresholds of the predicted risk. The c-statistic can be interpreted as the probability that a randomly selected participant with the outcome will be ranked higher than a randomly selected participant without the outcome. Reference Steyerberg and Vergouwe23 Calibration was assessed with the logistic calibration framework first proposed by Cox in 1958. Reference Cox24 Herein, calibration was assessed by regressing the observed binary outcomes (Y) on the log odds of the predictions (the linear predictor (LP)) with a logistic model: logit(Y) = a + b LP × LP. The coefficient b LP is known as the calibration slope and is ideally 1 when a model is well calibrated. If b LP < 1, the prediction model overfits and its risk estimates are too extreme (high risks are overestimated and low risks are underestimated). If b LP > 1, the prediction model tends to underfit the data and the opposite pattern is observed (i.e. its predictions are too modest). We did not assess the calibration-in-the-large, i.e. the intercept a when the slope is fixed at unity (a|b LP = 1). This was because if model fitting is obtained by standard statistical estimation methods such as maximum likelihood, the calibration-in-the-large at internal validation is guaranteed to be ideal. Reference van Calster, McLernon, van Smeden, Wynants and Steyerberg25,Reference van Calster, Nieboer, Vergouwe, De Cock, Pencina and Steyerberg26 Calibration, an important and often overlooked aspect of model performance, measures how reliable a model’s predictions are. It is the agreement between the observed and predicted risks. Specifically, a model is well calibrated if the event rate is X% among patients with a predicted risk of X%. For example, if we predict a 10% risk that a patient will relapse from a disease, the observed proportion should be 10 relapses per 100 patients with such a prediction. Reference Steyerberg and Vergouwe23,Reference van Calster, Nieboer, Vergouwe, De Cock, Pencina and Steyerberg26 We also provide flexible internal validation calibration curves (which plot the predicted probabilities on the x-axis against the actual observed proportions on the y-axis using a smoothing technique). Reference van Calster, Nieboer, Vergouwe, De Cock, Pencina and Steyerberg26 We assessed the clinical usefulness of using a treatment strategy based on the prediction model compared with treating all or treating none. Hereto, decision curve analysis was performed. Reference Vickers and Elkin27
The final models were recalibrated by multiplying the coefficients by the internal validation calibration slope (b LP), then re-estimating the intercept. This procedure is known as logistic recalibration or uniform shrinkage. Shrinkage reduces regression coefficients towards zero, such that less extreme predictions are made. Steyerberg suggests that such shrinkage improves predictions from models, especially in small data-sets. Reference Steyerberg and Vergouwe23 Shrinkage is recommended by the PROBAST (Prediction model Risk Of Bias ASsessment Tool) best practice guidelines, Reference Moons, Wolff, Riley, Whiting, Westwood and Collins28 although more recent research cautions against ‘reflexive recalibration’, i.e. mathematically adjusting the model in response to evidence of miscalibration without consideration of underlying causes. Reference Swaminathan, Srivastava, Tu, Lopez, Shah and Vickers29
Choice of predictors, causal motivations and generation of counterfactual explanations
For the baseline causal actionable prediction model, we specified antipsychotic treatment at baseline as the causal actionable predictor for intervention. Olanzapine is the antipsychotic agent most likely to be associated with an increase in BMI according to a recent meta-analysis, Reference Pillinger, McCutcheon, Vano, Mizuno, Arumuham and Hindley30 but it is recommended for use as a first-line medication in psychosis as it is well tolerated (low extrapyramidal side-effect profile) and the most effective choice (other than clozapine, which is reserved for cases of treatment resistance owing to potentially life-threatening side-effects). Reference Taylor, Barnes and Young31 Haloperidol is the antipsychotic least likely to be associated with BMI increase. Reference Pillinger, McCutcheon, Vano, Mizuno, Arumuham and Hindley30 Given that the antipsychotic at baseline was randomised, the effect was expected to be unbiased. Other baseline predictor variables were included and represented competing exposures of the outcome, increasing the precision of the estimate of the treatment effect of the antipsychotic and the performance of the prediction model. For the 12-week prediction model, given our hypothesis that antipsychotic-induced obesity at 1 year would be causally mediated by 0- to 12-week weight change, we specified this as the causal actionable predictor variable for intervention. It was therefore necessary to adjust for the other baseline variables, which were exposure–outcome confounders of the actionable predictor (12-week weight change), otherwise the causal effect would have been subject to bias. This also improved the precision of the effect estimate and performance of the prediction model. Covariates were measured with precision at specific times according to protocol, reducing bias from measurement error compared with routine clinical practice.
We then used counterfactual prediction to demonstrate possible causal changes in these actionable intervention variables that would switch the predicted outcome class to the desired one. Choosing an alternative type of antipsychotic medication at baseline or trying to lose weight gained 12 weeks after starting such a medication may be possible and therefore actionable, whereas altering age or gender is not. Hereto, we used Dandl’s multi-objective counterfactuals method, implemented via the ‘counterfactuals’ R package, to predict counterfactual explanations that would change the predicted outcome class from positive (e.g. probability of obesity ≥50%) to negative (e.g. probability of obesity <50%) (Supplementary Material). Reference Dandl, Molnar, Binder and Bischl32
Patient and public involvement
Three people with lived experience of psychosis reviewed the proposed analysis plan and contributed to the decision to develop causal actionable prediction models that target physical health problems in psychosis.
Results
The baseline demographics and clinical characteristics of the baseline and 12-week model development samples are reported in Table 1. There were no significant differences between the two development samples with respect to any of the baseline characteristics. In the baseline sample, 98 of 172 (57.0%) participants met criteria for clinically significant antipsychotic-related weight gain at 12 weeks. In the 12-week model sample, 35 of 97 (36.1%) participants met criteria for obesity at 1 year.
Patient characteristics (recorded at baseline except weight change from 0 to 12 weeks, which was recorded at 12 weeks, and weight change from 12 to 52 weeks, which was recorded at 52 weeks)

a. t values were from Welch’s two sample t-test; χ 2 values were from Pearson’s chi-squared test with Yates’s continuity correction.
b. Weight change from 0 to 12 weeks was not used in the baseline prediction model but was used in the 12-week prediction model and was considered as a parallel mediator in the causal mediation analysis per the directed acyclic graph in Fig. 2.
c. Weight change from 12 and 52 weeks was not used in the development of either prediction model but was considered as the other parallel mediator in the causal mediation analysis per the directed acyclic graph in Fig. 2.
Causal mediation analysis
There was evidence for a (total) natural indirect effect of antipsychotic choice (exposure) on weight at 1 year (outcome) mediated by weight change from 0 to 12 weeks in our parallel causal mediation analysis (5.70, 95% CI 2.83 to 8.66). We did not find strong evidence for a (total) natural indirect effect mediated by weight change from 12 to 52 weeks (2.52, 95% CI −0.797 to 6.55), or for a (pure) natural direct effect (−1.60, 95% CI −5.51 to 2.30). We found evidence for a total effect of the exposure on the outcome (6.62, 95% CI 1.44 to 12.3). We did not find evidence for moderation of either of the mediators’ effects on the outcome by the exposure (suggesting that there were no exposure-by-mediator interactions). The full output from PROCESS is available in the Supplementary Material. In the sensitivity analysis, evidence of the (total) natural indirect effect of antipsychotic choice on weight at 1 year mediated by weight change from 0 to 12 weeks remained until an average correlation r > 0.6 of the synthetic confounder with the mediator and outcome (strong unobserved residual confounding), whereas evidence for the total effect of the exposure on the outcome remained until r > 0.3 (moderate unobserved residual confounding). Neither plausible alternative potential mediator for 0- to 12-week weight change (0- to 12-week cholesterol change or glucose change) showed evidence for a (total) natural indirect effect. The full results of these sensitivity analyses are available in the Supplementary Material.
Prediction model presentation and internal validation
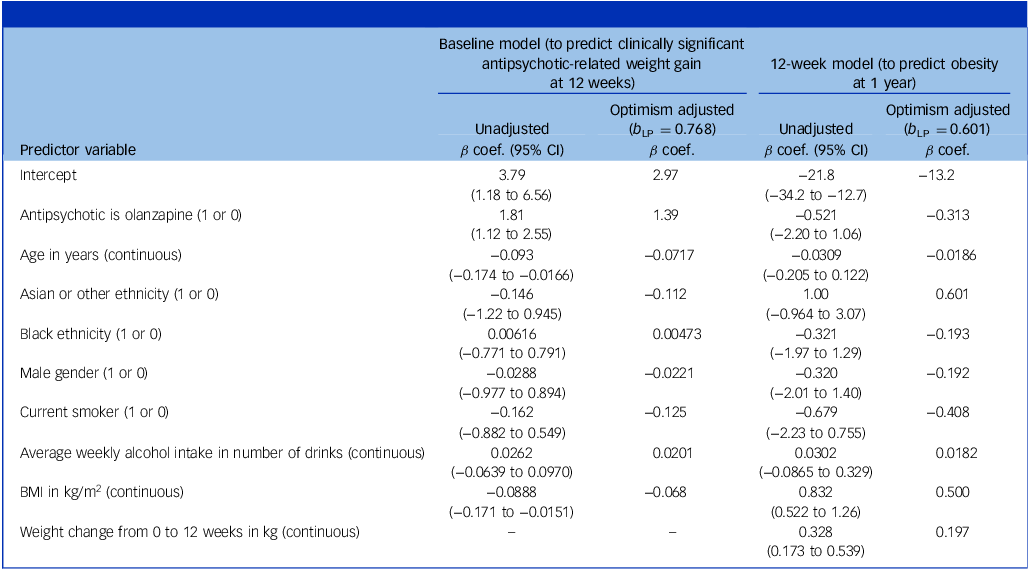
The coefficients for the unadjusted and optimism-adjusted coefficients for the baseline model (predicting clinically significant antipsychotic-related weight gain at 12 weeks) and the 12-week model (predicting obesity at 1 year) are presented in Table 2. At internal validation after correction for optimism by bootstrap, the discrimination c-statistic for the baseline model was 0.728 (95% CI 0.661 to 0.801), and the calibration slope b LP was 0.768 (95% CI 0.436 to 1.21). The internal validation c-statistic for the 12-week model was 0.904 (95% CI 0.820 to 0.961), and the calibration slope b LP was 0.601 (95% CI −0.0633 to 1.21). The original and shrunken linear predictors and optimism-adjusted calibration slopes and decision curves are presented in the Supplementary Material.
All variables were measured at baseline in both models, except weight change from 0 to 12 weeks which was measured at 12 weeks

The baseline prediction model outcome was clinically significant antipsychotic-related weight gain at 12 weeks. The 12-week prediction model outcome was obesity at 1 year. The reference antipsychotic was haloperidol, the reference ethnicity was White, and the reference gender was female. b LP, internal validation calibration slope (the shrinkage factor); β coef., beta coefficient.
Counterfactual prediction
We generated counterfactual explanations for three example patients, as illustrated in Fig. 3 and detailed in Supplementary Table 1.
Patient A was a 28-year-old Black male non-smoker who drank no alcohol. His baseline body mass index (BMI) was 27.6 kg/m2. He was randomised to olanzapine (X). Our baseline model predicted that he would have clinically significant antipsychotic-related weight gain (CSARWG) at 12 weeks (P( Y | X) = 0.60). His factual outcome matched our factual prediction. To avoid the outcome of CSARWG at 12 weeks, we should substitute olanzapine (X) for haloperidol (X′) (P(Y | X′) = 0.27). Given that he remained on olanzapine, at 12 weeks he had gained 12.5 kg of weight, representing a 3.65 kg/m2 increase in BMI. Our 12-week model predicted that he would be obese at 1 year (P(Y | X) = 0.86), which matched his factual outcome. To avoid obesity, the patient’s 12-week weight gain should be restricted to 3.2 kg, representing a 0.938 kg/m2 increase in BMI (P( Y | X′) = 0.49). The actual counterfactual outcome was unobserved. In other words, if a real-world patient with identical baseline characteristics had taken haloperidol rather than olanzapine, we predict that they would not develop CSARWG. However, it may have been that olanzapine was required (e.g. owing to more tolerable side-effects). So, at 12 weeks, our second causal actionable prediction model could be used to determine how much weight loss (or restriction of weight gain) was required to avoid obesity at 1 year. In this case, the patient would need to lose 9.3 kg, representing a 2.71 kg/m2 reduction in BMI. Patient B was a 19-year-old White female smoker who drank no alcohol. Her baseline BMI was 29.8 kg/m2. She was randomised to olanzapine. Our baseline model predicted she would have CSARWG at 12 weeks (P( Y | X) = 0.69). However, the factual prediction did not match the factual outcome (possibly because the treatment effect was heterogeneous). The counterfactual prediction derived by changing olanzapine to haloperidol resulted in a lower probability of CSARWG at 12 weeks (P( Y | X′) = 0.38). Although the patient did not develop CSARWG on olanzapine, she had gained 3.2 kg of weight by 12 weeks, representing a 1.24 kg/m2 increase in BMI. Using this information, our 12-week model predicted that she would be obese at 1 year (P( Y | X) = 0.77), matching the factual outcome. To avoid obesity at 1 year, she would be required to lose 2.9 kg by 12 weeks (P(Y | X′) = 0.49), representing a 1.15 kg/m2 reduction in BMI; this would translate to a 6.12 kg loss in the real world (i.e. more than the patient had gained in the 12 weeks since starting olanzapine), representing a 2.39 kg/m2 reduction in BMI. Finally, patient C was a 22-year-old Black smoker who drank five alcoholic drinks per week. His baseline BMI was 25.6 kg/m2. He was randomised to haloperidol, and the patient’s factual outcome at 12 weeks did not match the factual prediction (P(Y | X) = 0.40). In this case, the counterfactual explanation would not have been desirable, as changing haloperidol to olanzapine would have increased the probability of this outcome (P( Y | X′) = 0.73). By 12 weeks, the patient had gained 8.2 kg of weight on haloperidol, representing a 2.37 kg/m2 increase in BMI, and our 12-week model predicted that he would be obese at 1 year (P(Y | X) = 0.51), matching the factual outcome. To avoid obesity at 1 year, weight gain should be restricted to 7.8 kg (P(Y | X′) = 0.49), representing a 2.23 kg/m2 increase in BMI; this would translate to a 0.49 kg loss, representing a 0.14 kg/m2 reduction in BMI. This figure is illustrative only and not to scale.

Discussion
Overview
The results of our causal mediation analysis support the hypothesis that early weight change (during the first 12 weeks rather than the next 40 weeks) causally mediates antipsychotic-induced weight gain at 1 year, rather than just being associated with it. The distinction between causation and association is crucial because it shows that intervening during this early period could materially change the outcome. This is in contrast to association models, which cannot distinguish between causal and non-causal relationships. The causal actionable prediction models we developed on the basis of this finding were used for counterfactual prediction to show what would happen to specific individuals under different scenarios (e.g. Patient A’s predicted outcomes on olanzapine versus haloperidol, or with different levels of weight gain). Given the small sample size, our causal actionable prediction models should not be used clinically. However, they are an early demonstration of the potential of causal actionable prediction models to predict counterfactuals which can guide individualised treatment decisions to achieve true precision medicine, albeit tempered by the feasibility scope of this study and the small sample size.
Interpretation and comparison with prior literature
The period immediately following initial exposure to antipsychotics is generally regarded as an important time during which metabolic changes are most pronounced. Pérez-Iglesias et al reported a mean weight gain of patients treated with antipsychotics 3 years into treatment of 12.1 kg, with 85% of this occurring during the first year of treatment. Reference Perez-Iglesias, Martinez-Garcia, Pardo-Garcia, Amado, Garcia-Unzueta and Tabares-Seisdedos33 In an early study, patients with olanzapine treatment experienced weight gain of 1–5 kg in a period of 4 weeks. This effect was not modulated by baseline BMI or the dosage of the medication and was more pronounced in female patients more than 45 years old. Reference Jain, Bhargava and Gautam34 Similarly, a recent study involving 392 olanzapine-treated participants across various dosages also observed significant early weight gain after 90 days into treatment but did not identify any dosage-dependent pattern. Reference Schoretsanitis, Dubath, Grosu, Piras, Laaboub and Ranjbar35 These findings suggest that olanzapine administration itself is a risk factor for weight gain, irrespective of dosage. Although these previous studies highlighted a critical period of weight gain during the early stages of antipsychotic treatment, Reference Dayabandara, Hanwella, Ratnatunga, Seneviratne, Suraweera and de Silva36 to our knowledge, our study is the first to demonstrate that later antipsychotic-induced weight gain outcomes at 1 year may be causally mediated by early weight gain within the first 12 weeks of treatment. It is also notable that there was no correlation between weight change from 0 to 12 weeks and later weight change between 12 and 52 weeks.
In the context of these findings, we attempted to develop serial causal actionable prediction models to show the potential for counterfactual prediction. The baseline model used antipsychotic choice as the causal actionable predictor of weight gain at 12 weeks, demonstrating some evidence (albeit with poor calibration) that altering medication changes early weight trajectories. However, selecting a less obesogenic antipsychotic may not be clinically indicated for all patients or may not prevent significant weight gain in some, owing to treatment heterogeneity or residual confounding. Therefore, our 12-week model used weight gain during the initial 12 weeks of treatment as the causal actionable predictor of obesity outcomes at 1 year, highlighting the potential for early intervention to alter longer-term outcomes. Given the sample size, our models should not be used clinically.
At present, there has been limited published research modelling ITE and predicting counterfactuals in clinical populations. Prediction models based on traditional statistical methodologies may be the best current possible means to stratify treatments for patients who may be more likely to need them most (e.g. PsyMetRiC, a cardiometabolic risk prediction algorithm for young people with psychosis Reference Perry, Osimo, Upthegrove, Mallikarjun, Yorke and Stochl9 ). Yet, current prediction models may not be accurate and fully actionable at the individual level. In future, causal actionable prediction models modelling the ITE and predicting counterfactuals may enable better approximation of the ideals of precision medicine.
The reason our causal actionable prediction models are genuinely actionable and differ from simple manipulation of variables in a traditional multivariable prediction model is that we have causally specified the actionable intervention variables. In the baseline model, the causal actionable variable, antipsychotic choice, is randomised so we know that manipulating it will actually change the outcome and by how much. Randomisation means the effect should not be subject to confounding bias. For the 12-week model, we causally specify the actionable variable 12-week weight gain by modelling relevant confounders. Therefore, we can say that if it is manipulated, the relationship and its effect size is not biased, so the outcome will alter. However, in the 12-week prediction model, the effect of the antipsychotic is now biased, despite being randomised, as we are conditioning on its main causal predictor outcome mediator (i.e. 12-week weight change). Conditioning on a mediator blocks the path from the antipsychotic intervention to the outcome via the mediator. In a traditional multivariable prediction model, none of the variables is correctly causally specified, so we do not know that manipulating them will actually alter the outcome, as they are subject to bias. The naive assumption for a traditional multivariable prediction model would be that each variable represents a separate causal arrow into the outcome with no more complex causal interrelationships. This assumption is clearly incorrect.
The need to develop a prediction model and causally model the intervention variable is perhaps less obvious in our example as, in the 12-week model, the intervention variable is the same as the outcome variable (i.e. weight in kg). It clearly follows that change in weight at 12 weeks will affect weight at 1 year. However, it is only by causally modelling in this way that we can say that changing the amount of weight by X will prevent obesity Y, given confounding variables Z, i.e. the ITE (assuming homogeneity of treatment effect). Reference Hoogland, IntHout, Belias, Rovers, Riley and Harrell37 It would become more clear that a causal prediction model is required for an outcome different to the intervention, e.g. if the intervention was a medication or a dietary intervention.
Limitations and future work
Our study had several important limitations. Primarily, it was designed as a feasibility study, and the causal actionable prediction models developed are not intended for clinical use. The limited sample size will affect the stability and generalisability of the models; thus, there is a need for larger-scale counterfactual prediction studies. Further, external validation and regulatory approval are required before application of any model in clinical practice. Reference Steyerberg and Harrell38 However, external validation only tests overall model performance for factual predictions, as counterfactuals are unobserved for the individual. After excluding those with missing outcomes (as outcome imputation in prediction studies is controversial Reference Steyerberg19 ), there were no missing data for the 12-week development and causal mediation analysis sample. However, we did exclude six participants with missing height information from the baseline model development sample. Although this represented less than 5% of the total sample, and the potential for introducing bias was therefore low, a better solution may be multiple imputation. Reference Steyerberg19 From the existing literature, it is unclear how multiple imputation could be combined with counterfactual prediction; this will be a focus for future research. The choice of binary outcomes for the prediction models, although often mirroring clinical decision boundaries, does make somewhat artificial divisions which do not reflect underlying biology and loses information and statistical power. Our future work will explore use of linear regression for prediction and other biologically meaningful binary outcomes including cardiometabolic disease or death.
The estimation of the effects of weight change in the causal mediation and 12-week prediction models may have been influenced by unmeasured residual confounders, such as diet and exercise, and pre-existing insulin resistance. Reference Perry, McIntosh, Weich, Singh and Rees39 The impact of diet and exercise is likely to have been minimal, as much of their explained variance would have been accounted for by the inclusion of baseline BMI. The impact of insulin resistance will be the subject of future research. Residual confounders are unlikely to have varied systematically between participants in each of the randomised treatment arms during the short follow-up period compared with before randomisation. However, our analysis was robust to unobserved residual confounding on sensitivity analysis. If there is unmeasured residual confounding of the weight change mediator and the outcome, conditioning on weight change also risks introducing collider bias on the effect of the antipsychotic (or any other baseline predictor mediated though weight change). This is because the mediators are colliders for the predictor of interest (i.e. antipsychotic choice) and every other possible cause of the mediator. If the causes of the mediator are also causes of the outcome, conditioning on the mediator introduces collider bias by creating a conditional dependency between its causes, biasing the apparent effect of the predictor of interest on the outcome.
The randomised controlled trial data used in this study showed a significant gender imbalance; male participants represented 82.6% of those remaining at 12 weeks and 82.5% at 1-year follow-up, far outnumbering female participants at both time points; this may also have affected the generalisability of our findings. In addition, a higher proportion of participants in the haloperidol arm compared with the olanzapine arm did not complete follow-up both at 12 weeks excluding those with missing height data (59.1% v. 71.8%) and at 1 year (67.4% v. 58.8%), although only the 12-week proportions were significantly different. Such differential attrition may introduce bias if dropout is related to both treatment and outcomes. An earlier study found that significantly fewer olanzapine-treated patients reported extrapyramidal side-effects in self-report assessments compared with those treated with haloperidol, and haloperidol-treated patients reported more use of anticholinergic drugs. Reference Tran, Dellva, Tollefson, Beasley, Potvin and Kiesler40 This suggests that the lower discontinuation rates in the olanzapine group could be partially attributed to the side-effect profile of olanzapine. Clinically, rather than a change in antipsychotic, a dose reduction would probably be considered first, if a patient develops side-effects. Further, although the original trial found no significant difference between the haloperidol and olanzapine treatment arms for the primary outcome (Positive and Negative Syndrome Scale total score) at any time point tested during follow up (12, 24, 52 and 104 weeks), Reference Green, Lieberman, Hamer, Glick, Gur and Kahn41 this is the ATE, which may be heterogenous, and clinically olanzapine may be preferred over haloperidol.
Our causal actionable prediction models predict counterfactual outcomes assuming that the treatment effect is homogeneous across individuals. In other words, the population-level ATE approximates each patient’s ITE in our study population. However, the treatment effect is likely to be heterogeneous, such that the ITE for specific individuals varies based on their personal characteristics. Indeed, a criticism of randomised controlled trials is that the population is highly selected, for example, often excluding those with severe mental illness, such that the measured ATE may differ from the true effect when the intervention is actually applied. Reference Humphreys, Blodgett and Roberts42 Modelling treatment effect heterogeneity requires consideration of treatment covariate interactions, Reference Krishnadas, Leighton and Jones11 which in turn requires larger sample sizes. This will be the subject of future work.
Our causal actionable prediction models involve two serial prediction models; a better solution would be dynamic prediction, in which past predictions are incorporated into future predictions. Further, the models only correctly specify one causal actionable predictor as the potential intervention (i.e. exposure of interest). Correctly specifying more than one exposure of interest in a causal model would be considerably more difficult; this will also be the subject of future work.
We provide counterfactual explanations that would change the predicted outcome class from obesity to no obesity at a threshold of 50%. However, counterfactual generation could be combined with Vicker’s decision curve analysis such that counterfactuals are generated which lower the predicted probability below a clinician-determined treatment threshold. Here, a probability threshold for recommendation of an intervention on the basis of a clinical prediction model is determined by the balance of the relative harms of the intervention (e.g. side-effects or cost) versus the benefits (e.g. disease prevention). Reference Vickers and Elkin27 Finally, there is growing recognition of the importance of understanding the uncertainty surrounding risk prediction estimates provided by prediction models. This could help with critical evaluation of the model and affect shared decision-making. Future work will explore providing uncertainty around factual and counterfactual risk estimates, including use of bootstrap and Bayesian approaches. Reference Jain, Bhargava and Gautam34
Finally, although our results are hypothesis-generating and not yet clinically deployable, we hope this work represents an initial statement of intention for developing future causal prediction work to enable precision psychiatry.
Supplementary material
The supplementary material is available online at https://doi.org/10.1192/bjp.2026.10561
Data availability
Vivli is the source of the data analysed during the current study. The data are available on request from https://vivli.org/.
Acknowledgements
We thank the three people with lived experience who reviewed the proposed analysis plan, and the Scottish Mental Health Research Network who put us in touch with two of them.
Author contributions
S.P.L.: conceptualisation, methodology, formal analysis, visualisation, writing – original draft, writing – review and editing. I.L.L.: writing – original draft, writing – review and editing. D.M.: methodology, writing – review and editing. B.I.P.: writing – review and editing. S.A.T.: methodology, writing – review and editing. F.D.: methodology, writing – original draft, writing – review and editing. S.M.L.: writing – review and editing. R.K.: conceptualisation, methodology, writing – review and editing.
Funding
All research at the Department of Psychiatry in the University of Cambridge is supported by the National Institute for Health and Care Research (NIHR) Cambridge Biomedical Research Centre (NIHR203312) and the NIHR Applied Research Collaboration East of England. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. We acknowledge the support of the UKRI AI programme, and the Engineering and Physical Sciences Research Council (EPSRC), for the Causality in Healthcare AI Hub (grant number EP/Y028856/1).
Declaration of interest
S.M.L. and R.K. are editorial board members of the British Journal of Psychiatry. They did not take part in the review or decision-making process for this paper.







 Open access
Open access
eLetters
No eLetters have been published for this article.