

INTRODUCTION
Tuberculosis (TB) remains a major infectious and deadly disease in many parts of the world. Among the most prevalent Mycobacterium tuberculosis (Mtb) strains worldwide is the Beijing genotype, which has caused large outbreaks of TB in many countries [Reference Bifani1–Reference Mokrousov3] and been highly associated with drug resistance [Reference Singh4]. Currently, Beijing strains are present in >10% of patients with TB worldwide and are highly endemic throughout much of East and South East Asia, where they are present in >50% of TB patients [Reference Brudey5, Reference Filliol6]. Mtb strains belonging to the Beijing genotype might have a selective advantage over other Mtb strains and be associated with hypervirulence and multidrug resistance [Reference Parwati, van Crevel and van Soolingen7–Reference Anh9].
Characteristics facilitating the dissemination of Beijing family strains remain unknown, but they are presumed to have been acquired through evolution of the lineage. Therefore, it is necessary to investigate the phylogeny of the sublineage and to elucidate the prevalence of these characteristics. The mycobacterial interspersed repetitive units-variable number of tandem repeats (MIRU-VNTR) may be phylogenetically informative for the Mtb Beijing family [Reference Chang10]. Phylogenetic application of MIRU-VNTR profiles may be assessed by a graphing method known as the minimum spanning tree (MST) [Reference Schouls11–Reference Boxrud13]. The results of MIRU-VNTR genotyping exhibited concordance with the results of other genotyping techniques [Reference Supply14], including spoligotype clade assignment [Reference Filliol6] and single nucleotide polymorphism (SNP) analyses [Reference Filliol15, Reference Gutacker16] in Mtb. Sublineages of the Beijing family could be classified using 10 synonymous SNPs [Reference Iwamoto17, Reference Wada, Iwamoto and Maeda18]. These SNPs were detected by comparison among the whole-genome sequences, including strain 210, a Beijing family strain belonging to the modern sublineage [Reference Filliol15, Reference Beggs, Eisenach and Cave19].
The Beijing family was originally reported to be a genetically closely related genotype family in 1995 [Reference van Soolingen20]. Later studies showed that these strains were prevalent in East Asia [Reference Wang21], the Former Soviet Union [Reference Mokrousov22], and South Africa [Reference Warren23]. Whole genome analyses have indicated that the evolution of Mtb paralleled human migration and that Mtb expanded due to increases in human population density [Reference Comas24]. Several epidemiological studies have also indicated that Mtb genotype distribution is highly related to geography, human population and ethnicity [Reference Dou25–Reference Dou, Huang and Su27].
This study utilized MIRU-VNTR genotyping and SNPs to analyse the genetic population structure and diversity of the Beijing family. These results were subsequently compared to published profiles of Beijing family Mtb strains isolated from patients in neighbouring geographical areas (e.g. other areas of China and Japan) to assess potential genetic linkages, which refer to phylogenetic relatedness. These analyses may promote a better understanding of the population genetic structure of the Beijing family and extend the information obtained from the MIRU-VNTR genotyping method.
MATERIALS AND METHODS
Study samples
A cross-sectional study was conducted with a total of 163 Beijing strains collected from registered TB patients between 1 June 2009 and 31 November 2010 in Funing County, China, a relatively closed and low socioeconomic county in eastern China with a population of 1·08 million, to explore the genetic diversity of the Beijing family in several regions of East Asia. After obtaining fresh isolates, all these cultures were stored and maintained at –80 °C until required at the molecular laboratory of the School of Public Health, Fudan University. The Beijing family strains assessed in this study were identified on the basis of an RD105 deletion. Further characterization of the modern and ancestral sublineages was performed following the presence of an RD181 deletion, as determined by polymerase chain reaction amplification, which was used to subclassify all of the Beijing family strains in our collection as ancient [RD181(+)] and modern [RD181(–)] types.
The 199 Mtb isolates obtained from the study site consisted of 182 Beijing family strains; of these, 19 were excluded because of insufficient of DNA samples. The Ethics Committee of the School of Public Health of Fudan University approved this study. All participants provided written informed consent to allow their information to be stored and used for research.
SNP typing
Sequence types (STs) were determined based on the 10 synonymous SNPs, which were sufficient to classify Beijing strains obtained globally [Reference Filliol15]. The chromosomal positions in the whole-genome sequence of H37Rv [Reference Cole28] were 797736, 909166, 1477596, 1548149, 1692069, 1892017, 2376135, 2532616, 2825581, and 4137829. STs were designated as described previously [Reference Filliol15, Reference Iwamoto29].
15-locus MIRU-VNTR
The 15 classical MIRU-VNTR loci were selected and individually amplified in all 163 Beijing isolates as described previously [Reference Supply14, Reference Supply30]. The resulting typing pattern was used to create a 15-digit allelic profile for each isolate. The discrimination ability of the locus combinations was calculated using the Hunter–Gaston discriminatory index (HGDI) [Reference Hunter and Gaston31].
Data retrieved from previous reports
We searched the studies in which the strains were collected from venues in East Asia between 1 January 2000 and 30 December 2010; literature with the absence of ST or 15-MIRU-VNTR data were excluded from the analysis. ST data were retrieved for isolates obtained from patients in Taiwan [Reference Chen32] and Japan [Reference Wada, Iwamoto and Maeda18] to compare population structures. In addition, 15-MIRU-VNTR data and subfamily classifications were retrieved for isolates obtained from patients in Shanghai [Reference Luo33], Taiwan [Reference Chen32], and Japan [Reference Wada, Iwamoto and Maeda18]. Their origins and the years during which the isolates were obtained are as follows: 191 isolates from Shanghai, collected between 2007 and 2010 in the Chongming District [Reference Luo33]; 338 isolates from Taiwan, collected between 2003 and 2007 from four geographical regions of Taiwan (north, east, central, south) [Reference Chen32]; 355 isolates from Japan, collected between 2001 and 2004 in Osaka and Kobe [Reference Wada, Iwamoto and Maeda18]. The MIRU-VNTR data from Funing and these three other East Asian regions were compared by constructing a MST using Bionumerics software v. 7.1 (Applied Maths, Belgium).
RESULTS
Classification of Beijing strains by SNP typing
The 163 Beijing strains could be classified into 13 STs (Fig. 1), with most of the strains classified into four major Beijing sublineages: ST10 (53·4%), ST19 (11·7%), ST22 (10·4%) and ST11 (10·4%). Based on 10-loci SNP typing, the sequence of ST11 was consistent with the standard reference strain H37Rv, suggesting a close genetic distance. By contrast, ST22 and ST10 had many mutations relative to H37Rv, indicating a greater genetic distance. These findings suggest that the order of appearance of the STs in phylogenetic evolution was ST11, ST26, ST3, STK, ST19, ST10 and ST22.
SNP genotyping results of 163 Mtb strains of Beijing genotype in Funing County, China. Alleles indicate SNP positions of H37Rv: 909166, 797736, 2825581, 1892017, 4137829, 1477596, 2532616, 2376135, 1692069, and 1548149.
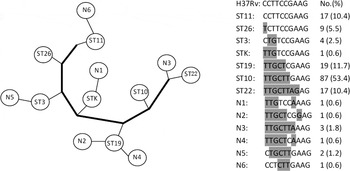
Comparison of ST profiles from Funing, Taiwan, and Japan
To better understand the extent of geographical variations in STs of Mtb isolates, the ST profiles of Beijing strains from the present study were compared with profiles from strains isolated in Taiwan and Japan. The 856 strains isolated in these three studies included 403 ancient and 453 modern Beijing isolates (Table 1). Most of the Beijing genotypes from Funing (78·5%) and Taiwan (73·4%) belonged to the modern subfamily, whereas most of the strains from Japan (78·9%) were of the ancient subfamily. The proportion of strains classified as ST10 was significantly higher in Funing (53·4%) and Taiwan (53·3%) than in Japan (17·2%). Of the strains isolated from Funing and Taiwan, 75·5% and 82·6%, respectively, belonged to three major Beijing sublineages, ST10, ST19 and ST22. By contrast, 68·4% of strains from Japan belonged to the sublineages ST3, ST19 and STK.
Sequence types of Mycobacterium tuberculosis isolates from Funing, Taiwan and Japan

* The proportions of ancient and modern Beijing strains were calculated by determining the presence of the RD181 deletion in strains from Funing and an IS6110 insertion in the NTF chromosomal region in strains from Taiwan and Japan.
Comparison of MIRU-VNTR from Funing, Shanghai, Taiwan and Japan
To determine whether certain MIRU-VNTR loci could be used to discriminate between Beijing strains, we analysed published MIRU-VNTR profiles and calculated the HGDI of Beijing strains from three other East Asian regions – Shanghai, Taiwan and Japan (Table 2). We found that the HGDI of Qub11b was >0·6 across the four areas, while the HGDIs of MIRU26, Mtub21, Qub26, and Mtub04 were each >0·3 in all four regions.
HGDI values of Mycobacterium tuberculosis isolates from Funing, Shanghai, Taiwan and Japan

HGDI, Hunter–Gaston discriminatory index.
MST based on 15-MIRU-VNTR genotyping and ST profiles of Beijing family isolates from Funing
To determine the consistency between MIRU-VNTR genotypes and STs classified by SNP, we constructed a MST based on 15-MIRU-VNTR genotyping and identified the SNP information by colour coding (Fig. 2 a). The isolates ST10 and ST22 almost overlapped in the tree, indicating their close genetic relationship. By contrast, ST11 and ST26 were positioned away from the major MIRU-VNTR genotypes and had greater genetic distances to ST10. The entire structure of the MST was consistent with the sublineages defined by SNP typing. However, some of the MIRU-VNTR clusters contained multiple SNP subtypes, suggesting that there was homoplasy within the Beijing strains examined with 15-MIRU-VNTR.
Minimum spanning trees based on 15-MIRU-VNTR genotyping of (a) 163 Mtb strains of Beijing genotype in Funing, and (b) Mtb isolates from Funing, Shanghai, Taiwan and Japan. Each circle indicates a different MIRU-VNTR genotype, with the size of the circle indicating the quantity of the isolate. Heavy lines connecting two MIRU-VNTR types denote single-locus variants; thin lines denote double-locus variants; dashed lines indicate triple-locus variants; and dotted lines indicate variants at more than three loci. The coloured symbols in (a) denote different sequence types of Mtb isolates, whereas the coloured symbols in (b) denote Mtb isolates from Funing, Shanghai, Taiwan and Japan.
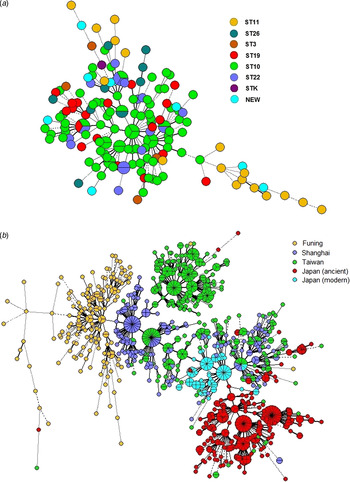
Phylogenetic analysis of Beijing family isolates from Funing, Shanghai, Taiwan and Japan
Figure 2 b shows the genotype distributions in Funing, Shanghai, Taiwan and Japan. The genetic distance of strains from Funing and Shanghai was relatively close, whereas the distances of strains from Funing compared to strains from Taiwan and Japan were longer. However, there were overlaps for genotypes from Shanghai, Taiwan and Japan. Specifically, strains from the modern subfamily in Japan overlapped with strains from Taiwan and Shanghai, whereas strains from the ancient subfamily were crowded together and located at some distance from the strains isolated in other regions.
DISCUSSION
To our knowledge, this study is the first to assess the genetic diversity of Mtb from different regions in East Asia. This study showed that the modern subfamily in Shanghai overlapped with strains from other countries, whereas the ancient subfamily was genetically differentiated across several countries. In addition, strains of the modern subfamily, especially ST10, showed especially wide distribution in populations in these East Asian countries. Qub11b, with an HGDI >0·6, and four other loci, MIRU 26, Mtub21, Qub26, and Mtub04, each with an HGDI >0·3, were able to be distinguished among Beijing strains in a given geographical region.
The Beijing strain is one of the predominant Mtb lineages in East Asia as well as in other countries throughout the world [Reference Hanekom34, Reference Schurch35]. Beijing strains can be divided into several sublineages that may differ in their ability to spread and cause disease. Methods to subdivide Beijing strains are important for exploration of their phylogenetic evolution and propagation. Although Mtb phylogenetic evolution was first assessed by 15-MIRU-VNTR and 24-MIRU-VNTR in China [Reference Allix-Beguec36], these methods yielded uncertain results with relatively large errors [Reference Luo33]. Mtb gene sequencing showed the advantages and accuracy of SNP typing in phylogenetic analyses [Reference Dan-Dan and Qian37]. Although the discriminatory index of SNPs is not very high [Reference Cardoso38], SNP typing is regarded as an optimal method for phylogenetic research [Reference Comas39], allowing Beijing strains to be subdivided into different STs. ST11, ST26, ST3, STK, ST19 and ST25, each of which possesses intact RD181 and NTF regions, correspond to ancient sublineages; whereas ST10 and ST22 correspond to modern sublineages, with all possessing an RD181 region deletion and some having an IS6110 insertion on the right side of the NTF region [Reference Filliol15, Reference Iwamoto17, Reference Dou25, Reference Dou40]. The order of appearance of the STs in phylogenetic evolution was consistent from previous studies, implying the direction of genetic mutations in Mtb [Reference Filliol15].
This study used 15-MIRU-VNTR genotyping to construct an MST of Beijing strain, with this MST validated by profiling of SNPs. Each MIRU-VNTR genotype was roughly concordant with an ST classification. However, some strains belonging to different ST sublineages were found to be mixed and not to be classified correctly in the same MIRU-VNTR clusters. Other highly discriminatory loci are required to make MIRU-VNTR results agree better with SNP information. Due to the high homoplasy of Beijing strains, the MIRU-VNTR method is less than optimal in genotyping these strains. The MST suggested that combining SNP and MIRU-VNTR typing may be superior to either method alone in assessing Mtb epidemiology in areas dominated by Beijing strains.
This study found that strains of the modern subfamily, especially ST10, were highly prevalent. Interestingly, ST10 was also dominant in Taiwan (53·3%) [Reference Wada, Iwamoto and Maeda18] and Thailand (57·7%) [Reference Faksri41], suggesting that ST10 may be epidemiologically successful, with Asian populations being especially susceptible to infection by, and transmission of, these strains. The genotypes of Beijing strains from Shanghai, Taiwan and Japan showed overlaps and relatively short genetic distances. The reason for the apparent global success of the Beijing strains is not yet fully understood, but may be related to a variety of host-related factors, including movements of human populations [Reference Mokrousov22]. The modern Beijing sublineages are more predominant than the ancient sublineages throughout Asian countries, including South Korea, Vietnam, Malaysia, Thailand and China [Reference Bifani1, Reference Chen32, Reference Faksri41], whereas most of the Beijing strains in Japan belong to the ancient subfamily [Reference Wada and Iwamoto42]. We found that strains of the modern subfamily in Japan overlapped with strains from Taiwan and Shanghai, while ancient subfamily strains were genetically unequal across countries. These findings suggest that the modern genotype has a greater ability to expand throughout East Asia, even around the world, compared to the ancient genotype. However, caution should be exercised when drawing conclusions because the sampling period and study population were different in the four studies. Interestingly, the phylogenetic analysis according to SNPs supported the monophyletic clade structure and suggests different Beijing STs across the different Asian sites. However, the order in which all of the above evolutionary events occurred remains to be determined, in particular we did not include an analysis of temporal evolution of the strains across the study sites within such a short period.
CONCLUSION
In conclusion, we found that Beijing strains of the modern subfamily, especially ST10, were highly prevalent. Qub11b, with an HGDI >0·6, and four other loci, MIRU 26, Mtub21, Qub26, and Mtub04, each with an HGDI >0·3, were able to discriminate among Beijing strains within a given geographical region. The combination of SNP and MIRU-VNTR typing was better than either alone for genotyping in areas dominated by Beijing strains.
ACKNOWLEDGEMENTS
We thank department of Tuberculosis Control, Yinzhou Center for Disease Control and Prevention, Ningbo, China for providing the samples. This study was funded by the Chinese National Science Foundation (30800937), the China-UK Global public health research programme (GHSP-CS-OP 302) and the programme of Key Discipline Development of Public Health of Shanghai (grant no. 12GWZX0101).
DECLARATION OF INTEREST
None.




