


Introduction
Colorectal cancer (CRC) ranks among the most prevalent tumours of the digestive system and stands as the second leading cause of cancer-related deaths worldwide (Refs Reference Siegel, Miller, Goding Sauer, Fedewa, Butterly, Anderson, Cercek and Smith1, Reference Siegel, Miller, Wagle and Jemal2). Emerging evidence underscores the contribution of diets deficient in dietary fibre, whole grains and other protective nutrients, along with adverse lifestyle patterns, to the rising incidence and mortality associated with CRC, which remains a major public health concern (Ref. Reference Song, Chan and Sun3). The disease develops from benign or precancerous polyps in the majority of cases, which gradually transform over several years, eventually infiltrating the bowel wall, spreading to adjacent lymph nodes or metastasizing to distant organs. This prolonged progression frequently results in delayed diagnosis, whereas early detection and polypectomy of precancerous polyps can prevent CRC progression (Ref. Reference Simon4). Colon polyps can be classified into low-risk and high-risk categories based on their potential to progress to CRC. This classification is essential for determining appropriate screening intervals and prevention strategies (Ref. Reference Shussman and Wexner5). Based on their growth patterns, current histological practices categorize these lesions into two main types: serrated polyps and adenomatous polyps (APs). APs are further divided into three subtypes: villous polyps (VP), tubulovillous polyps (TVP) and tubular adenomas (TA). Similarly, serrated polyps are classified into hyperplastic polyps (HP), sessile serrated adenomas (SSA synonyms: sessile serrated lesions or polyps) and traditional serrated adenomas (TSA) (Refs Reference Jass6–Reference Sninsky, Shore, Lupu and Crockett8). Therefore, it is crucial to identify the risk factors associated with colorectal polyps (CPs) based on their histological characteristics to enhance prevention and management strategies.
CRC risk factors can be classified into non-modifiable and modifiable risk factors. Among the non-modifiable factors are characteristics such as ethnicity, sex and age, alongside hereditary mutations and certain medical conditions like inflammatory bowel diseases (IBD), cystic fibrosis and acromegaly (Refs Reference Rawla, Sunkara and Barsouk9–Reference Ionescu, Gheorghe, Bacalbasa, Chiotoroiu and Diaconu11). Research indicates that about 5–10% of CRC instances are linked to known genetic backgrounds, while the rest occur in individuals without any notable family history or genetic predisposition (Ref. Reference Nagy, Sweet and Eng12). In contrast, modifiable risk factors are related to lifestyle choices, such as physical inactivity, obesity and dietary habits. Additionally, smoking and alcohol use have been associated with an elevated risk of developing CRC (Refs Reference Marchand, Wilkens, Kolonel, Hankin, Lyu LCMarchand, Wilkens, Kolonel, Hankin and Lyu13–Reference Cho, Lee, Oh, Chang, Sohn, Shin and Kim15). Moreover, the gut microbiome’s vast array of bacteria has sparked interest in how infectious agents may contribute to cancer development. This growing body of research underscores the complex interplay between genetic factors, environmental exposures and microbial influences in the pathogenesis of CRC (Ref. Reference Papastergiou, Karatapanis and Georgopoulos16).
Recent research has indicated a possible connection between Helicobacter pylori infection and its role as a risk factor for adenomatous polyps and CRC (Refs Reference Zumkeller, Brenner, Zwahlen and Rothenbacher17, Reference Teimoorian, Ranaei, Hajian Tilaki, Shokri Shirvani and Vosough18). Although H. pylori primarily inhabit the stomach, studies have revealed a notable correlation between H. pylori infection and various non-gastric diseases, such as neurodegenerative disorders, IBD, celiac disease, hepatic disorders and CRC (Refs Reference Gravina, Zagari, Musis, Romano, Loguercio and Romano19–Reference Bravo, Hoare, Soto, Valenzuela and Quest21). Epidemiological evidence underscores a significant association between H. pylori infection and an increased risk and aggressiveness of CRC (Refs Reference Zuo, Jing, Bie, Xu, Hao and Wang20, Reference Kim, Kim, Chang, Kim, Baek, Kim and Hong22), with an odds ratio of 1.32 (95% CI: 1.07–1.61) for any colorectal adenoma and 1.90 (95% CI: 1.05–3.56) for advanced neoplasms and this association remains significant after adjusting for established risk factors such as smoking, alcohol consumption and body mass index (Ref. Reference Zhang, Hoffmeister, Weck, Chang-Claude and Brenner23). Nevertheless, the specific biological mechanisms that contribute to this heightened risk are still not fully understood.
However, the interpretation of epidemiological associations between H. pylori infection and colorectal cancer should consider several potential confounding factors. The prevalence of H. pylori infection is strongly associated with socio-economic status, including sanitation conditions, educational level, healthcare access and population density, which may independently influence cancer risk and screening patterns (Ref. Reference Wang, Wang, Yu and Jin24). In addition, dietary habits such as high consumption of processed or red meat, low intake of fibre, fruits and vegetables as well as lifestyle factors including smoking, alcohol consumption, obesity and physical inactivity may confound the observed relationship (Ref. Reference Avila-Nava, Gutiérrez-Solis, Pacheco-Can, Sagols-Tanoira, González-Marenco, Cabrera-Lizarraga, Castillo-Avila, Aguilar-Franco, Chim-Aké, Rubio-Zapata, Reyes-Sosa, Medina-Vera, Guevara-Cruz, Sánchez-Pozos and Lugo25). Furthermore, concurrent infections and alterations in the gut microbiota may co-exist with H. pylori infection and contribute to colorectal carcinogenesis (Ref. Reference Iino and Shimoyama26). Therefore, since most available studies are observational in nature, they cannot definitively establish causality and residual confounding cannot be fully excluded. Future longitudinal and mechanistic studies are required to clarify whether H. pylori play a direct causal role in colorectal tumorigenesis. While previous reviews have primarily focused on the epidemiological association between H. pylori and colorectal neoplasia, this article is designed as a narrative review that provides a broader and more integrated synthesis of the available evidence. This narrative review summarizes current epidemiological findings, examines the direct and indirect mechanisms through which H. pylori may contribute to colorectal carcinogenesis and discusses the potential role of probiotic interventions in prevention and management strategies. By integrating epidemiological, mechanistic and translational evidence, this review aims to provide a comprehensive overview of the current state of knowledge and highlight important areas for future research.
Epidemiological evidence: connecting H. pylori to colorectal neoplasia
Helicobacter pylori infection
Helicobacter pylori , a gram-negative coccoid-spiral bacterium that has co-existed with humans for over 100,000 years, is a significant player in the global landscape of gastrointestinal health (Ref. Reference Moodley, Linz, Bond, Nieuwoudt, Soodyall, Schlebusch, Bernhöft, Hale, Suerbaum, Mugisha, van der Merwe and Achtman27). Helicobacter pylori colonizes the gastric mucosa of over half the global population, with prevalence varying by region (Ref. Reference Hooi, Lai, Ng, Suen, Underwood, Tanyingoh, Malfertheiner, Graham, Wong, Wu, Chan, Sung, Kaplan and Ng28). While colonization is common, it does not always result in disease; progression to infection and subsequent pathology depends on factors such as bacterial virulence (e.g. CagA, VacA), host immune responses and environmental conditions (Refs Reference Hooi, Lai, Ng, Suen, Underwood, Tanyingoh, Malfertheiner, Graham, Wong, Wu, Chan, Sung, Kaplan and Ng28, Reference Engelsberger, Gerhard and Mejías-Luque29). Emerging microbiome research suggests that H. pylori may act as a long-term gastric colonizer, potentially contributing to early-life gastric and immune development. Consequently, it may not be strictly considered an ‘infection’ unless associated with mucosal damage or symptomatic disease (Refs Reference Blaser and Atherton30, Reference Atherton and Blaser31). However, the transition from colonization to infection is continuous and, in this review, the term ‘infection’ is used to describe this continuous transition from colonization to infection.
Critically, beyond well-established environmental and sanitary determinants, certain modifiable lifestyle factors may influence host susceptibility to H. pylori infection and its persistence (Ref. Reference Kumar, Kumar, Ali, Shaikh, Khokhar and Matilo MA.32). High-salt diets can promote H. pylori growth and survival and exacerbate gastric injury by promoting gastric epithelial hyperplasia, parietal cell loss and increased bacterial load (Refs Reference Balendra, Amoroso, Galassi, Esposto, Bareggi, Luu, Scaramella and Ghidini33, Reference Fox, Dangler, Taylor, King, Koh and Wang34). Cigarette smoking has also been associated with altered gastric histology and a reduced humoral response to H. pylori , which may bacterial persistence and impair eradication (Refs Reference Koivisto, Voutilainen and Färkkilä35, Reference Namiot, Kemona and Namiot36). In addition, obesity may modify host–bacterium interactions through changes in gastric hormones, including reduced ghrelin signalling and obesity-related alterations in leptin pathways, rather than directly increasing bacterial acquisition (Ref. Reference Shiotani, Miyanishi, Uedo and Iishi37).
Helicobacter pylori , often found nestled in the gastric mucosa, actively engages in a complex relationship with its host, contributing to chronic gastritis and serving as a precursor to more severe conditions, such as peptic ulcers and gastric cancer (GC) (Ref. Reference Malfertheiner, Camargo, el-Omar, Liou, Peek, Schulz, Smith and Suerbaum38). Recent genomic studies, including those from the H. pylori Genome Project, have illuminated the intricate genomic structure of this pathogen, revealing its adaptability and the mechanisms it employs to thrive in the acidic environment of the stomach (Ref. Reference Thorell, Muñoz-Ramírez, Wang, Sandoval-Motta, Boscolo Agostini, Ghirotto, Torres, Romero-Gallo, Krishna, Peek, Piazuelo, Raaf, Bentolila, Aftab, Akada, Matsumoto, Haesebrouck, Colanzi, Bartelli, Nunes, Pelosof, Sztokfisz, Dias-Neto, Assumpção, Tishkov, Mabeku, Goodman, Geary, Cromarty, Price, Quilty, Corvalan, Serrano, Gonzalez, Riquelme, García-Cancino, Parra-Sepúlveda, Bernal, Castillo, Goldstein, Hu, Taylor, Bravo, Pazos, Bravo, Wilson, Fox, Ramírez-Mayorga, Molina-Castro, Durán-Bermúdez, Campos-Núñez, Chaves-Cervantes, Tshibangu-Kabamba, Tumba, Tshimpi-Wola, de Jesus Ngoma-Kisoko, Ngoyi, Cruz, Hosking, Abreu, Varon, Benejat, Secka, Link, Malfertheiner, Adinortey, Bockarie, Adinortey, Ofori, Sgouras, Martinez-Gonzalez, Michopoulos, Georgopoulos, Hernandez, Tacatic, Aguilar, Dominguez, Morgan, Harðardóttir, Gunnarsdóttir, Guðjónsson, Jónasson, Björnsson, Ballal, Shetty, Miftahussurur, Sugihartono, Alfaray, Waskito, Fauzia, Syam, Maulahela, Malekzadeh, Sotoudeh, Peretz, Azrad, On, de Re, Zanussi, Cannizzaro, Canzonieri, Shimura, Tokunaga, Osaki, Kamiya, Jadallah, Matalka, Igissinov, Moldobaeva, Rakhat, Choi, Kim, Kim, Song, Leja, Vangravs, Šķenders, Rudzīte, Rūdule, Vanags, Kikuste, Kupcinskas, Skieceviciene, Jonaitis, Kiudelis, Jonaitis, Kiudelis, Varkalaite, Vadivelu, Loke, Vellasamy, Herrera-Goepfert, Alonso-Larraga, Yee, Htet, Matsuhisa, Shrestha, Ansari, Abiodun, Jemilohun, Akande, Olu-Abiodun, Magaji, Omotoso, Osuagwu, Okonkwo, Owoseni, Castaneda, Castillo, Velapatino, Gilman, Krzyżek, Gościniak, Pawełka, Korona-Glowniak, Cichoz-Lach, Oleastro, Figueiredo, Machado, Ferreira, Bordin, Livzan, Tsukanov, Tan, Yeoh, Zhu, Ally, Haas, Montes, Fernández-Reyes, Tamayo, Lizasoain, Bujanda, Lario, Ramírez-Lázaro, Calvet, Brunet-Mas, Domper-Arnal, García-Mateo, Abad-Baroja, Delgado-Guillena, Moreira, Botargues, Pérez-Martínez, Barreiro-Alonso, Flores, Gisbert, Muro, Linares, Martin, Alcoba, Fleitas-Kanonnikoff, Altayeb, Engstrand, Enroth, Keller, Wagner, Pohl, Lee, Liou, Wu, Kocazeybek, Sarıbas, Tasçı, Demiryas, Kepil, Quiel, Villagra, Norton, Johnson, Huang, Hwang, Szymczak, Rajagopalan, Asare, Jacobs, in, Bollag, Lopez, Kruse, White, Graham, Lane, Gao, Fields, Gold, Cruz-Correa, González-Pons, Rodriguez, Tuan, Dung, Binh, Trang, van Khien, Chen, Raley, Kessing, Zhao, Tran, Gutiérrez-Escobar, Wan, Hicks, Zhu, Yu, Zhu, Yeager, Hutchinson, Teshome, Jones, Luo, Jehanne, Katsura, Gonzalez-Hormazabal, Didelot, Sheppard, Tarazona-Santos, Mariño-Ramírez, Loh, Backert, Naumann, Abnet, Smet, Berg, Chiner-Oms, Comas, Martínez-Martínez, Zamudio, Lehours, Megraud, Yahara, Blaser, Vincze, Morgan, Roberts, Chanock, Dekker, Torres, Cover, Noureen, Fischer, Vale, Cherry, Osada, Fukuyo, Arita, Yamaoka, Kobayashi, Uchiyama, Falush, Camargo and Rabkin39). Recent estimates indicate that infections account for approximately 15% of all cancer diagnoses, with H. pylori recognized as the most significant infectious agent associated with cancer. This bacterium is responsible for about 770,000 cancer cases globally each year, primarily due to its strong role in the development of gastric cancer (Ref. Reference Plummer, de Martel, Vignat, Ferlay, Bray and Franceschi40). Helicobacter pylori shows marked geographic variation, with higher prevalence in East Asia, Africa and parts of South America and lower prevalence in the United States, Oceania and Western Europe (Ref. Reference Hooi, Lai, Ng, Suen, Underwood, Tanyingoh, Malfertheiner, Graham, Wong, Wu, Chan, Sung, Kaplan and Ng28). In Latin America, countries like Peru exhibit high H. pylori prevalence, with rates exceeding 70%, driven by factors such as sanitation challenges (Ref. Reference Hooi, Lai, Ng, Suen, Underwood, Tanyingoh, Malfertheiner, Graham, Wong, Wu, Chan, Sung, Kaplan and Ng28). In Asia, China reports prevalence around 40–60% in adults, influenced by socio-economic conditions (Ref. Reference Hooi, Lai, Ng, Suen, Underwood, Tanyingoh, Malfertheiner, Graham, Wong, Wu, Chan, Sung, Kaplan and Ng28). Other Eastern Mediterranean countries, such as Saudi Arabia and Egypt, show rates between 22% and 87.6%, reflecting regional hygiene issues (Ref. Reference Eshraghian41). Similarly, Iran has a notable prevalence, ranging from 54% to over 80% among adults and children, underscoring the impact of local environmental and cultural factors (Refs. Reference Eshraghian41, Reference Moosazadeh, Lankarani and Afshari42).
The persistent inflammation, mucosal injury, genetic and epigenetic modifications and altered gene expression patterns observed in individuals with H. pylori infection are key factors that contribute to the development of GC over time (Ref. Reference Saruuljavkhlan, Alfaray, Oyuntsetseg, Gantuya, Khangai, Renchinsengee, Matsumoto, Akada, Azzaya, Davaadorj and Yamaoka43). Helicobacter pylori have distinctive characteristics and virulence factors that enable effective infection of the host and manipulation of the immune response. The enzyme urease enables H. pylori to neutralize gastric acid by generating ammonia, which acts as a shield for bacterial survival while concurrently exerting a cytotoxic effect on gastric epithelial cells and disrupting mucosal tight junctions (Ref. Reference Wroblewski, Shen, Ogden, Romero–Gallo, Lapierre, Israel, Turner and Peek44). In addition, adhesion factors such as SabA (sialic acid-binding adhesin), BabA (blood group antigen-binding adhesin) and HopQ (Helicobacter outer membrane protein) play a pivotal role in establishing persistent gastric infection. BabA mediates bacterial attachment to Lewis B blood group antigens on gastric epithelial cells, whereas SabA binds to sialylated glycoconjugates that are up-regulated during gastric inflammation. HopQ further strengthens bacterial adherence through interaction with carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) expressed on host cells (Refs. Reference Engelsberger, Gerhard and Mejías-Luque29, Reference Javaheri, Kruse, Moonens, Mejías-Luque, Debraekeleer, Asche, Tegtmeyer, Kalali, Bach, Sieber, Hill, Königer, Hauck, Moskalenko, Haas, Busch, Klaile, Slevogt, Schmidt, Backert, Remaut, Singer and Gerhard45). Furthermore, bacterial adhesion promotes host–pathogen interactions that trigger downstream intracellular signalling cascades. Specifically, outer membrane proteins and bacterial pathogen-associated molecular patterns (PAMPs) are recognized by host pattern recognition receptors (PRRs), including Toll-like receptors expressed on gastric epithelial cells (Ref. Reference Smith, Mitchell, Li, Ding, Fitzmaurice, Ryan, Crowe and Goldberg46). This recognition event recruits downstream nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, which orchestrate the hyper-secretion of potent pro-inflammatory cytokines and chemokines, most notably interleukin-8 (IL-8), IL-1β and tumour necrosis factor-alpha (TNF-α) (Refs Reference Smith, Mitchell, Li, Ding, Fitzmaurice, Ryan, Crowe and Goldberg46, Reference Hu, Liu, Zhu and Lu47). The pathogenic potential of H. pylori is largely attributed to two key virulence factors: CagA (cytotoxin-associated gene A) and VacA (vacuolating cytotoxin A). The CagA oncoprotein infuses into host T cells via a type IV secretion system (T4SS) that is encoded by cag pathogenicity island (cag PAI), which negatively affects cell growth and the apoptosis process (Ref. Reference Chen, Zhang and Duan48). VacA, when internalized by host T cells, induces various adverse effects, such as cell vacuolation, which enhances the persistence of infection, along with mitochondrial dysfunction, stress and, ultimately, apoptosis (Ref. Reference Ansari and Yamaoka49). This continuous molecular and cellular crosstalk drives a sustained influx of neutrophils, macrophages and lymphocytes into the lamina propria. The persistent activation of these immune cells generates chronic oxidative stress through the excessive release of reactive oxygen and nitrogen species (ROS/RNS), leading to cumulative mucosal damage and maintaining the chronic active gastritis that serves as the foundation for downstream neoplasia (Ref. Reference Sah, Arjunan, Lee and Jung50). Evidence increasingly suggests that H. pylori infection may be linked to CRC (Refs Reference Engelsberger, Gerhard and Mejías-Luque29, Reference Butt, Varga, Blot, Teras, Visvanathan, le Marchand, Haiman, Chen, Bao, Sesso, Wassertheil-Smoller, Ho, Tinker, Peek, Potter, Cover, Hendrix, Huang, Hyslop, Um, Grodstein, Song, Zeleniuch-Jacquotte, Berndt, Hildesheim, Waterboer, Pawlita and Epplein51, Reference Shmuely, Melzer, Braverman, Domniz and Yahav52). Although a single definitive pathway has not been universally established, this oncogenic association is postulated to rely on several inter-related mechanisms. Rather than driving carcinogenesis through a localized event, this correlation appears to involve an extended physiological network that operates beyond the stomach, where the bacterium first interacts with the host. In this context, it is crucial to distinguish the conceptual and pathophysiological boundaries between H. pylori -induced GC and its hypothesized role CRC. In gastric carcinogenesis, the pathogen’s oncogenic role is locally restricted to the gastric niche, occurring within a highly acidic environment and directly driving the well-established, sequential Correa’s cascade (Ref. Reference Cheng, Yang, Yang, Tsai, Chang, Wu, Kuo, Yu, Yang, Cheng, Chen and Sheu53). Conversely, the biological landscape of CRC is fundamentally distinct, presenting a more complex and expanded paradigm; as discussed in detail later in this review, the link to colorectal neoplasia involves a sophisticated combination of both indirect systemic alterations and direct distal actions. Consequently, moving past a simple blanket assumption to closely evaluate how these broad pathways are structured in current literature is essential to appreciate their full clinical significance. This review aims to explore the complex relationship between H. pylori infection and CRC. To illuminate this gastrointestinal field of research, we will first examine in detail the association between H. pylori infection and the development of colorectal polyps and cancer. Next, we will discuss the proposed biological pathways, including both direct and indirect mechanisms, that have been hypothesized to link H. pylori infection with CRC development. Finally, we will investigate the role of probiotics in modulating H. pylori pathogenesis and their potential to improve CRC management.
Association between H. pylori infection and colorectal neoplasia
Table 1 summarizes the available evidence on the association between H. pylori infection and colorectal neoplasia, including crude and adjusted odds ratios extracted from the individual studies included in this review. The evidence was organized according to major colorectal outcome categories, including neoplasms, advanced neoplasms, CRC, overall colorectal polyps, adenoma polyps, advanced adenoma polyps, conventional adenoma polyps, hyperplastic polyps and SSAs. These categories reflect the polyp-to-CRC progression model and the histological classifications commonly used in the literature and recent meta-analyses. Although some overlap exists across sub-groups, this structure allows a clearer synthesis of the available evidence. Associations according to polyp number and size were also reviewed. Because the covariates included in adjusted models varied across publications, adjusted odds ratios are presented descriptively and should be interpreted within the context of each individual study rather than compared directly across studies. These findings are summarized next according to outcome category and, where available, crude and adjusted estimates.
Reported crude and adjusted odds ratios for the association between Helicobacter pylori infection and colorectal neoplasia across individual studies

Table 1. Long description
The table is organized into 11 columns: Study design, Outcome category, Sample size (Cases, Control), Crude (O R, Lower, Upper), Adjusted (O R, Lower, Upper), and Reference.
Key data points include:
* Neoplasms (adenoma polyp plus C R C): Four studies listed. A case-control study (Ref. 54) shows an adjusted O R of 1.90 (1.23 to 2.93). A cross-sectional study (Ref. 52) shows a crude O R of 4.07 (2.26 to 7.35).
* Advanced neoplasms: Five studies listed. A cross-sectional study (Ref. 52) shows a high crude O R of 9.75 (4.31 to 21.20).
* C R C: Twenty-four studies listed. A large retrospective study (Ref. 58) with 82,420 cases and over 47 million controls shows an adjusted O R of 1.89 (1.69 to 2.10). A case-control study (Ref. 61) shows a high adjusted O R of 10.60 (2.70 to 41.30).
* Overall colorectal polyps: Eight studies listed. A retrospective study (Ref. 81) shows a crude O R of 5.23 (3.60 to 7.70).
* Adenoma polyps: Seventeen studies listed. A cross-sectional study (Ref. 87) shows an adjusted O R of 3.24 (2.18 to 4.80).
* Advanced adenoma polyps: Five studies listed. A case-control study (Ref. 67) shows a crude O R of 1.80 (1.69 to 1.92).
* Hyperplastic polyp: Four studies listed. A retrospective cohort (Ref. 94) shows an adjusted O R of 1.23 (1.18 to 1.28).
* Single vs. Multiple polyps: Multiple polyps generally show higher O R values, such as Ref. 68 showing an adjusted O R of 1.98 for single polyps versus 2.41 for two or more polyps.
* Polyp size: Ref. 68 shows an adjusted O R of 2.33 for polyps greater than or equal to 10 mm and 2.15 for polyps less than 10 mm.
Note: Adjusted odds ratios were extracted as reported in the original publications. The covariates included in the multivariable models differed across studies and were not standardized. Therefore, adjusted estimates should be interpreted within the context of each individual study and should not be directly compared across publications as equivalent effect estimates.
Neoplasms (adenoma polyp + CRC)
Several studies evaluated the association between H. pylori and the broad category of colorectal neoplasms, which includes adenoma polyps and CRC (Refs Reference Shmuely, Melzer, Braverman, Domniz and Yahav52, Reference Nam, Baeg, Kwon, Cho, Na and Choi54–Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56). Crude ORs reported by Nam et al. (Ref. Reference Nam, Baeg, Kwon, Cho, Na and Choi54) (OR = 1.94, 95% CI: 1.28–2.95), Shmuely et al. (Ref. Reference Shmuely, Melzer, Braverman, Domniz and Yahav52) (OR = 4.07, 95% CI: 2.26–7.35) and Lee et al. (Ref. Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56) (OR = 1.33, 95% CI: 1.19–1.48) indicate an increased risk of colorectal neoplasia among individuals with H. pylori infection. Several studies also reported adjusted estimates above 1, including Nam et al. (Ref. Reference Nam, Baeg, Kwon, Cho, Na and Choi54) (OR = 1.90, 95% CI: 1.23–2.93), Fujimori et al. (Ref. Reference Fujimori, Kishida, Kobayashi, Sekita, Seo, Nagata, Tatsuguchi, Gudis, Yokoi, Tanaka, Yamashita, Tajiri, Ohaki and Sakamoto55) (OR = 1.66, 95% CI: 1.27–2.05) and Lee et al. (Ref. Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56) (OR = 1.22, 95% CI: 1.09–1.36). However, these adjusted estimates were derived from study-specific models with different covariate sets and should therefore be interpreted as within-study findings rather than directly comparable measures across publications.
However, the magnitude of association varies considerably across studies, ranging from modest associations to stronger estimates, suggesting underlying heterogeneity (Refs Reference Shmuely, Melzer, Braverman, Domniz and Yahav52, Reference Nam, Baeg, Kwon, Cho, Na and Choi54–Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56). This variability may reflect differences in study design (case–control vs. cohort), geographic distribution (e.g. East Asia vs. Western populations) and baseline prevalence of both H. pylori infection and colorectal neoplasia, which can influence effect estimates and limit direct comparability.
Advanced neoplasms (advanced adenoma + CRC)
Advanced neoplasms, including high-risk adenomas and CRC, showed mixed findings across studies (Refs Reference Kim, Kim, Chang, Kim, Baek, Kim and Hong22, Reference Shmuely, Melzer, Braverman, Domniz and Yahav52, Reference Nam, Baeg, Kwon, Cho, Na and Choi54, Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56, Reference Park, Kim, Park, Baik, Youn, Kim and Park57). Nam et al. (Ref. Reference Nam, Baeg, Kwon, Cho, Na and Choi54) reported an OR of 2.40 (95% CI: 0.64–8.97) and Park et al. (Ref. Reference Park, Kim, Park, Baik, Youn, Kim and Park57) reported an OR of 1.10 (95% CI: 0.02–2.26); however, both confidence intervals were wide and crossed 1, indicating substantial statistical uncertainty and no clear evidence of increased risk in these studies. In contrast, Shmuely et al. (Ref. Reference Shmuely, Melzer, Braverman, Domniz and Yahav52) reported a markedly increased risk of advanced neoplasms among individuals with H. pylori infection (OR = 9.75, 95% CI: 4.31–21.20). Adjusted estimates were also reported by Kim et al. (Ref. Reference Kim, Kim, Chang, Kim, Baek, Kim and Hong22) (OR = 1.90, 95% CI: 1.05–3.56), Lee et al. (Ref. Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56) (OR = 1.34, 95% CI: 1.04–1.72) and Nam et al. (Ref. Reference Nam, Baeg, Kwon, Cho, Na and Choi54) (OR = 2.39, 95% CI: 0.69–9.21). Because these models did not account for an identical set of covariates, they provide study-specific evidence rather than directly comparable adjusted contrasts.
Overall, the evidence for advanced neoplasms should be interpreted cautiously because some estimates were imprecise, with wide confidence intervals that crossed 1, while stronger positive findings were reported only in selected studies (Refs Reference Kim, Kim, Chang, Kim, Baek, Kim and Hong22, Reference Shmuely, Melzer, Braverman, Domniz and Yahav52, Reference Nam, Baeg, Kwon, Cho, Na and Choi54, Reference Lee, Park, Choi, Lee, Koo, Chung, Chang, Choe, Yang, Myung, Jung, Yang and Byeon56, Reference Park, Kim, Park, Baik, Youn, Kim and Park57).
Colorectal cancer
Several studies have assessed the association between H. pylori infection and CRC, with heterogeneous findings across populations and study designs (Refs Reference Butt, Varga, Blot, Teras, Visvanathan, le Marchand, Haiman, Chen, Bao, Sesso, Wassertheil-Smoller, Ho, Tinker, Peek, Potter, Cover, Hendrix, Huang, Hyslop, Um, Grodstein, Song, Zeleniuch-Jacquotte, Berndt, Hildesheim, Waterboer, Pawlita and Epplein51, Reference Boustany, Onwuzo, Almomani and Asaad58, Reference Butt, Jenab, Pawlita, Tjønneland, Kyrø, Boutron-Ruault, Carbonnel, Dong, Kaaks, Kühn, Boeing, Schulze, Trichopoulou, Karakatsani, la Vecchia, Palli, Agnoli, Tumino, Sacerdote, Panico, Bueno-de-Mesquita, Vermeulen, Gram, Weiderpass, Borch, Quirós, Agudo, Rodríguez-Barranco, Santiuste, Ardanaz, van Guelpen, Harlid, Imaz, Perez-Cornago, Gunter, Zouiouich, Park, Riboli, Cross, Heath, Waterboer and Hughes60). Crude ORs reported by Jones et al. (Ref. Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63) (OR = 8.13, 95% CI: 1.40–46.99), Shmuely et al. (Ref. Reference Shmuely, Melzer, Braverman, Domniz and Yahav52) (OR = 7.98, 95% CI: 3.16–20.16) and Sonnenberg et al. (Ref. Reference Sonnenberg and Genta67) (OR = 2.35, 95% CI: 1.98–2.80) indicate an increased risk of CRC among individuals with H. pylori infection. Adjusted analyses show similar results, with Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68) (OR = 3.05, 95% CI: 2.33–3.99), Shmuely et al. (Ref. Reference Shmuely, Passaro, Figer, Niv, Pitlik, Samra, Koren and Yahav61) (OR = 10.60, 95% CI: 2.70–41.30) and Pan et al. (Ref. Reference Pan, Zhang, Fang, Chen, He, Zheng and Li72) (OR = 1.34, 95% CI: 1.17–1.53) indicating an increased risk of CRC in the corresponding study populations. Several studies reported ORs above 1 for the association between H. pylori infection and CRC; however, the magnitude of association varied substantially across studies, and the adjusted estimates were based on non-uniform covariate sets (Refs Reference Boustany, Onwuzo, Almomani and Asaad58, Reference Butt, Jenab, Pawlita, Tjønneland, Kyrø, Boutron-Ruault, Carbonnel, Dong, Kaaks, Kühn, Boeing, Schulze, Trichopoulou, Karakatsani, la Vecchia, Palli, Agnoli, Tumino, Sacerdote, Panico, Bueno-de-Mesquita, Vermeulen, Gram, Weiderpass, Borch, Quirós, Agudo, Rodríguez-Barranco, Santiuste, Ardanaz, van Guelpen, Harlid, Imaz, Perez-Cornago, Gunter, Zouiouich, Park, Riboli, Cross, Heath, Waterboer and Hughes60, Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63, Reference Sonnenberg and Genta67, Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68).
However, this apparent robustness must be interpreted cautiously, as effect sizes vary markedly across studies, ranging from null or weak associations to very strong associations, indicating substantial heterogeneity (Refs Reference Nam, Baeg, Kwon, Cho, Na and Choi54, Reference Boustany, Onwuzo, Almomani and Asaad58, Reference Epplein, Pawlita, Michel, Peek, Cai and Blot66, Reference Sonnenberg and Genta67). Such discrepancies may arise from differences in exposure assessment (e.g. serology reflecting past exposure vs. urea breath test reflecting active infection), outcome definition and population risk profiles.
Overall colorectal polyps
Crude ORs for overall colorectal polyps indicate an increased risk of polyp formation among individuals with H. pylori infection, as reported by Ciftel et al. (Ref. Reference Ciftel81) (OR = 5.23, 95% CI: 3.60–7.70), Tongtawee et al. (Ref. Reference Tongtawee, Kaewpitoon, Kaewpitoon, Dechsukhum, Leeanansaksiri, Loyd, Matrakool and Panpimanmas80) (OR = 2.26, 95% CI: 1.32–3.86) and Brim et al. (Ref. Reference Brim, Zahaf, Laiyemo, Nouraie, Pérez-Pérez, Smoot, Lee, Razjouyan and Ashktorab79) (OR = 1.30, 95% CI: 1.00–1.60). Adjusted analyses confirm these findings, with Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68) (OR = 2.19, 95% CI: 1.96–2.44) and Brim et al. (Ref. Reference Brim, Zahaf, Laiyemo, Nouraie, Pérez-Pérez, Smoot, Lee, Razjouyan and Ashktorab79) (OR = 1.50, 95% CI: 1.20–1.90) showing statistical significance. These findings support the hypothesis that H. pylori infection may contribute to early-stage colorectal neoplasia by promoting polyp formation. Nevertheless, these estimates arose from different analytic models and should be interpreted within the methodological context of each study.
Adenoma polyps
The relationship between H. pylori and adenoma polyps was also evident. Crude ORs reported by Nam et al. (Ref. Reference Nam, Baeg, Kwon, Cho, Na and Choi54) (OR = 1.96, 95% CI: 1.27–3.01), Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86)(OR = 2.86, 95% CI: 1.97–4.15) and Inoue et al. (Ref. Reference Inoue, Mukoubayashi, Yoshimura, Niwa, Deguchi, Watanabe, Enomoto, Maekita, Ueda, Iguchi, Yanaoka, Tamai, Arii, Oka, Fujishiro, Takeshita, Iwane, Mohara and Ichinose91) (OR = 2.26, 95% CI: 1.44–3.55) indicate an increased risk of adenoma polyps among individuals with H. pylori infection. Adjusted analyses reinforce this, with Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86) (OR = 3.23, 95% CI: 2.18–4.79) and Inoue et al. (Ref. Reference Inoue, Mukoubayashi, Yoshimura, Niwa, Deguchi, Watanabe, Enomoto, Maekita, Ueda, Iguchi, Yanaoka, Tamai, Arii, Oka, Fujishiro, Takeshita, Iwane, Mohara and Ichinose91) (OR = 2.52, 95% CI: 1.57–4.05) showing strong correlations. The data indicate that H. pylori infection is a significant risk factor for adenoma polyps, a crucial precursor in colorectal carcinogenesis. Because the multivariable models differed between studies, these estimates should be viewed as separate study-level findings rather than directly comparable measures.
However, variability in effect sizes and study populations suggests that these associations may not be uniform across different demographic or geographic contexts.
Advanced adenoma polyps
Crude ORs for advanced adenomas varied across studies. While Hong et al. (Ref. Reference Hong, Lee, Kim, Lee, Kim, Choe, Lee, Cheon, Sung, Park and Shim89) (OR = 2.19, 95% CI: 1.40–3.42) reported an increased risk of advanced adenomas among individuals with H. pylori infection, other studies reported weaker or non-significant findings, such as Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86) (OR = 1.26, 95% CI: 0.09–1.77). This inconsistency highlights the potential influence of methodological heterogeneity, including differences in polyp classification criteria and diagnostic protocols, which may affect comparability across studies. Similarly, Sonnenberg et al. (Ref. Reference Sonnenberg and Genta67) (OR = 1.80, 95% CI: 1.69–1.92) reported an increased risk of advanced adenomas, whereas Bae et al. (Ref. Reference Bae, Jeon, Cho, Jung, Kweon and Kim88) (OR = 0.59, 95% CI: 0.31–1.12) showed no significant relationship. Study-specific adjusted estimates also showed mixed findings. Nam et al. (Ref. Reference Nam, Hong, Kim, Shin, Ryu, Park, Kim, Sohn, Han, Kim and Lee77) (OR = 1.84, 95% CI: 1.25–2.70) and Hong et al. (Ref. Reference Hong, Lee, Kim, Lee, Kim, Choe, Lee, Cheon, Sung, Park and Shim89) (OR = 1.48, 95% CI: 1.41–2.21) reported increased risk estimates in their respective adjusted models. However, these adjusted estimates were derived from study-specific multivariable analyses with different covariate sets and should not be directly contrasted across publications. In contrast, Bae et al. (Ref. Reference Bae, Jeon, Cho, Jung, Kweon and Kim88) reported an adjusted OR of 0.59 (95% CI: 0.31–1.12), which did not indicate a statistically significant increased risk of advanced adenomas. Overall, the evidence for advanced adenoma polyps is mixed; some studies reported increased risk estimates, whereas others showed non-significant findings with confidence intervals crossing 1.
Conventional adenoma polyps
Pan et al. (Ref. Reference Pan, Zhang, Fang, Chen, He, Zheng and Li72) reported a modestly increased risk of conventional adenoma polyps among individuals with H. pylori infection (OR = 1.11, 95% CI: 1.05–1.17). However, the available evidence for this outcome is limited and the observed effect size was small. Therefore, additional well-designed prospective studies are needed to determine whether this association is reproducible across different populations and whether it remains significant after standardized adjustment for major confounders, including age, sex, body mass index, smoking, alcohol use, dietary factors, socio-economic status, medication use and colorectal screening history. Future studies should also distinguish active H. pylori infection from past exposure and examine whether bacterial virulence factors, such as CagA status, modify the risk of conventional adenoma formation. These studies are needed because conventional adenomas are important precursors of CRC, but the current evidence is insufficient to determine whether H. pylori has an independent role in their development or whether the observed association reflects residual confounding, exposure misclassification or differences in study design.
Sessile serrated adenomas/ polyps
The role of H. pylori in SSAs/polyps remains unclear because the available studies reported inconsistent results. Dent et al. (Ref. Reference Dent, Ripley, Chan, Rickard, Keshava, Stewart and Chapuis94) reported a crude OR close to the null value (OR = 1.02, 95% CI: 0.96–1.10), suggesting little or no association between H. pylori infection and SSAs. In contrast, Pan et al. (Ref. Reference Pan, Zhang, Fang, Chen, He, Zheng and Li72) reported an adjusted estimate indicating a modest but statistically significant increased risk of SSAs (OR = 1.41, 95% CI: 1.22–1.63). These contradictory findings may be explained by differences in study design, population characteristics, outcome definition, sample size and statistical adjustment. In particular, Dent et al. reported a crude estimate, whereas Pan et al. reported an adjusted estimate derived from a retrospective cohort model. Therefore, these results should not be interpreted as directly comparable. Differences in colorectal screening practices, histopathological classification of serrated lesions and the method used to define H. pylori exposure may also have contributed to the inconsistent findings. Therefore, the evidence for SSAs remains limited and further studies using standardized definitions of serrated lesions and uniform adjustment for confounders are needed.
Hyperplastic polyps
The relationship between H. pylori infection and hyperplastic polyps is inconsistent across studies. Sonnenberg et al. (Ref. Reference Sonnenberg and Genta67) reported a modest increased risk of hyperplastic polyps (OR = 1.24, 95% CI: 1.18–1.30) and Selgrad et al. (Ref. Reference Selgrad, Bornschein, Kandulski, Hille, Weigt, Roessner, Wex and Malfertheiner95) also reported an increased risk (OR = 2.26, 95% CI: 1.23–5.74). Similarly, Dent et al. (Ref. Reference Dent, Ripley, Chan, Rickard, Keshava, Stewart and Chapuis94) reported a modest increased risk in an adjusted analysis (OR = 1.23, 95% CI: 1.18–1.28). In contrast, Tongtawee et al. (Ref. Reference Tongtawee, Kaewpitoon, Kaewpitoon, Dechsukhum, Leeanansaksiri, Loyd, Matrakool and Panpimanmas80) reported an inverse association (OR = 0.14, 95% CI: 0.03–0.66). These conflicting findings may reflect important differences between the studies, including geographic setting, study design, sample size, patient selection, indication for colonoscopy, background prevalence of H. pylori infection and diagnostic criteria for hyperplastic polyps. The studies also differed in whether they reported crude or adjusted estimates, and the covariates included in adjusted models were not uniform. In addition, variation in H. pylori detection methods may have contributed to exposure misclassification, particularly when tests reflected previous exposure rather than active infection. Therefore, the evidence does not support a consistent conclusion regarding hyperplastic polyps, and future studies should use standardized histological definitions, clearly defined H. pylori exposure assessment and comparable multivariable adjustment.
Role of number and size of polyps
Data on polyp number and size were extracted from the individual studies summarized in Table 1, particularly Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68), Yan et al. (Ref. Reference Boustany, Onwuzo, Almomani and Asaad58), Basmaci et al. (Ref. Reference Basmaci, Karataş, Ergin and Dumlu96) and Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86). No new pooled or secondary statistical analysis was performed for these variables in this review. Instead, we descriptively summarized the crude and adjusted odds ratios reported in the original publications. Polyp number was categorized as single polyp or two or more polyps, while polyp size was categorized as <10 mm or ≥10 mm, according to the definitions used in the included studies (Refs Reference Smith, Mitchell, Li, Ding, Fitzmaurice, Ryan, Crowe and Goldberg46, Reference Shmuely, Melzer, Braverman, Domniz and Yahav52, Reference Boustany, Onwuzo, Almomani and Asaad58, Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63). Because the original studies differed in study design, population characteristics, H. pylori assessment and covariate adjustment, these estimates should be interpreted as study-level findings rather than directly comparable pooled estimates.
Single polyp
The association between H. pylori infection and single polyp formation was evaluated in several studies, with mixed findings. Crude estimates from two studies showed odds ratios above 1, suggesting a possible increased occurrence of single polyps among individuals with H. pylori infection (Refs Reference Yan, Chen, Zhao, Chen, Lin, Jin, Pan and Wu85, Reference Basmaci, Karataş, Ergin and Dumlu96). In contrast, another study reported no statistically significant association (OR = 1.08, 95% CI: 0.94–1.24) (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86). Wang et al. reported an adjusted odds ratio of 1.98 (95% CI: 1.74–2.25) (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68); however, this estimate was extracted directly from the original publication and was not re-analysed or re-adjusted in this review. Therefore, the evidence regarding single polyp formation should be interpreted cautiously as study-level findings rather than as a definitive or pooled association.
Two or more polyps
A stronger association was observed when multiple polyps were considered. Crude ORs from Basmac et al. (Ref. Reference Basmaci, Karataş, Ergin and Dumlu96) (OR = 2.10, 95% CI: 1.74–2.58) and Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86) (OR = 1.15, 95% CI: 1.00–1.32) demonstrate a higher risk of polyp multiplicity in H. pylori -infected individuals. Adjusted ORs from Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68) (OR = 2.41, 95% CI: 2.12–2.74) further support this finding.
Polyps ≥ 10 mm
Crude ORs for polyps ≥10 mm showed non-significant results. Yan et al. (Ref. Reference Yan, Chen, Zhao, Chen, Lin, Jin, Pan and Wu85) (OR = 0.94, 95% CI: 0.50–1.75) and Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86) (OR = 1.33, 95% CI: 0.95–1.85) found no significant association. Adjusted analyses from Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68) (OR = 2.33, 95% CI: 1.98–2.74) reported an increased risk of polyps ≥10 mm among individuals with H. pylori infection.
Polyps < 10 mm
For smaller polyps, crude ORs showed mixed results. Yan et al. (Ref. Reference Yan, Chen, Zhao, Chen, Lin, Jin, Pan and Wu85) (OR = 1.35, 95% CI: 1.09–1.66) reported an increased risk of small polyp formation among individuals with H. pylori infection, while Chen et al. (Ref. Reference Chen, Zhou, Fang, Zhang, Lin, Feng, Wang, Wu, Wang and Lin86) (OR = 1.10, 95% CI: 0.98–1.22) did not find a statistically significant increase. Wang et al. (Ref. Reference Wang, Kong, Zhang, Lu, Hui, Liu, Kang and Gao68) (OR = 2.15, 95% CI: 1.92–2.41) reported an increased risk of polyps <10 mm among individuals with H. pylori infection.
The increased risk estimates reported in several crude and adjusted analyses suggest that H. pylori infection may be associated with a higher occurrence of colorectal neoplastic outcomes beyond its well-established impact on gastric pathology; however, these findings reflect epidemiological associations rather than direct evidence that H. pylori play a causal role in colorectal carcinogenesis. Several studies reported increased odds ratios for the association between H. pylori infection and advanced colorectal neoplasms, particularly high-risk adenomas and CRC; however, the strength of this association varied across studies and should be interpreted cautiously because of heterogeneity in study design, exposure assessment, outcome definition and confounder adjustment. However, while some sub-categories of colorectal polyps, such as SSAs and HPs, exhibited moderate or inconsistent associations with H. pylori infection, the overall trend points to an increased risk of neoplastic progression. Importantly, the magnitude and direction of these associations vary substantially across studies, reflecting significant heterogeneity in study populations, including differences in ethnicity, geographic region, baseline risk and healthcare access, all of which may influence both exposure prevalence and CRC detection. The differences observed in adjusted odds ratios across studies further emphasize the need for standardized methodologies in future research to control for these confounders effectively. Moreover, although many studies report adjusted estimates, the range and quality of confounder adjustment are inconsistent and key factors such as diet, antibiotic exposure, screening practices and gut microbiota composition are not uniformly accounted for, leaving the possibility of residual confounding. Variability in ORs may stem from methodological differences, such as diverse H. pylori detection methods (e.g. serology vs. urea breath test), study designs (case–control vs. cohort) or population-specific factors like antibiotic resistance and dietary habits, necessitating standardized protocols to ensure comparable results (Ref. Reference Wu, Yang, Xu, Gao and Fan97). In particular, the use of serological assays in some studies may reflect past exposure rather than active infection, potentially leading to exposure misclassification and biasing the observed associations. Further investigations, particularly prospective longitudinal studies, are needed to establish a causal relationship and elucidate the precise biological mechanisms underlying H. pylori -induced colorectal carcinogenesis. Given the high H. pylori prevalence in Middle Eastern and developing countries, region-specific studies are critical to assess whether unique risk factors, like dietary patterns or socio-economic conditions, modulate the H. pylori -CRC association in these populations (Refs Reference Eshraghian41, Reference Moosazadeh, Lankarani and Afshari42). Additionally, studies examining the impact of H. pylori eradication on CRC risk reduction could provide valuable insights into potential preventive strategies. Emerging evidence suggests H. pylori eradication may reduce CRC risk, as a Taiwanese study found a threefold higher colorectal adenoma incidence in patients with persistent infection compared to those with successful eradication, though data remain limited (Ref. Reference Hu, Wu, Chu, Wang, Lin, Liu, Su, Liao, Chen, Liu and Shih98). At the same time, the presence of both strong positive associations and null findings across the literature indicates that this relationship is not entirely consistent and may be context dependent or influenced by study design limitations and unmeasured biases. Understanding these interactions will be crucial in developing targeted interventions for individuals at high risk of CRC, particularly those with a history of H. pylori infection. Ultimately, these findings support the growing recognition of H. pylori infection as a possible factor associated with colorectal neoplasia, while also highlighting that current epidemiological evidence remains insufficient to establish a definitive causal relationship.
Despite the consistent associations observed across multiple studies, these findings should be interpreted with caution due to the inherent limitations of observational epidemiological designs. Most of the included studies are case–control or cohort studies, which are susceptible to residual confounding and cannot establish causality. In addition, the covariates included in adjusted analyses differed across studies, limiting the direct comparability of multivariable estimates summarized in Table 1. Several potential confounders may influence the observed relationship between H. pylori infection and colorectal neoplasia, including dietary patterns (e.g. high red meat intake, low fibre consumption), socio-economic status (affecting sanitation, healthcare access and screening practices), lifestyle factors such as smoking, alcohol use and obesity, as well as the presence of concurrent infections and underlying alterations in gut microbiota. Furthermore, heterogeneity in diagnostic methods for H. pylori detection and variability in study populations may contribute to inconsistent findings across studies. Therefore, while the epidemiological evidence suggests a possible association, a direct causal relationship remains unproven and requires confirmation through well-designed longitudinal and mechanistic studies.
Controversy in the literature: A balanced view
Although prior sections have presented substantial evidence supporting an association between H. pylori infection and CRC or colorectal polyps, some studies report no significant link, highlighting the complexity of this relationship. Notably, Luo et al. (Ref. Reference Luo, Zhou, Ran, Gu and Zhou99) conducted a bidirectional Mendelian randomization study and found no causal association between H. pylori infection and CRC (OR = 1.12, 95% CI: 0.98–1.27, P = 0.08). Similarly, Blase et al. (Ref. Reference Blase, Campbell, Gapstur, Pawlita, Michel, Waterboer and Teras65) reported no association between pre-diagnostic H. pylori antibodies and CRC risk in an elderly Caucasian population (OR = 1.17, 95% CI: 0.91–1.50). Additionally, Guo et al. (Ref. Reference Guo and Li100) found no statistical association with CRC in East Asian populations (OR = 1.08, 95% CI: 0.89–1.68), though an association with colorectal adenomas was noted. Boyuk et al. (Ref. Reference Boyuk, Ozgur, Atalay, Celebi, Ekizoglu and Aykurt101) also reported no significant relationship between H. pylori infection and colorectal neoplasms, including CRC and polyps (P > 0.05). These findings may be limited by factors such as reliance on genetic or serological methods that may not fully capture active infections or by population-specific characteristics that reduce generalizability. Inconsistencies in the literature likely arise from variations in study design, population demographics, detection methods for H. pylori and colorectal outcomes, and unaccounted confounders like diet or lifestyle. Despite these dissenting studies, the broader scientific consensus, supported by extensive epidemiological and mechanistic evidence, suggests that H. pylori infection contributes to an increased risk of CRC and colorectal polyps, emphasizing the need for further research to resolve these discrepancies and confirm the association.
These discrepancies further highlight the limitations of observational evidence and the potential impact of unmeasured or residual confounding. Collectively, current data highlight the need for cautious interpretation of epidemiological associations in the absence of robust causal inference.
Proposed mechanistic pathways linking H. pylori infection to colorectal carcinogenesis
The mechanisms discussed in this section are proposed pathways derived from experimental, observational and pre-clinical evidence. While these findings provide biologically plausible links between H. pylori infection and colorectal carcinogenesis, most remain hypothetical and require further validation in human studies (Figures 1 and 2).
Proposed flowchart illustrating the relationship between Helicobacter pylori infection and CRC. The blue pathway represents direct mechanisms and the orange pathway represents indirect mechanisms through which H. pylori may contribute to colorectal carcinogenesis. Despite their different origins, these pathways converge at intestinal inflammation, which promotes chronic infection, immune response disruption and the establishment of a pro‑inflammatory, tumour‑promoting intestinal microenvironment that ultimately facilitates CRC development.

Figure 1. Long description
A flowchart titled H. pylori Infection at the top center.
* The left side, labeled Direct mechanisms in blue, leads to a box containing Virulence factors and Hypergastrinemia. A blue arrow continues from this box down to the final outcome, C R C.
* The right side, labeled Indirect mechanisms in orange, leads to a box containing Morphological change, Intracellular lifestyle, Extracellular vesicle release, Autophagic process, H. pylori–n c R N A interaction, and Gut microbiota changes. An orange arrow continues from this box down to the final outcome, C R C.
* A central red pathway connects both side boxes to a vertical series of red ovals. The sequence is as follows:
1. Inflammation.
2. Chronic infection and Immune response disruption (side-by-side ovals).
3. Pro-inflammatory intestinal environment.
4. Tumor-promoting microenvironment.
All pathways converge at the bottom box labeled C R C.
An overview of Helicobacter pylori ’s mechanisms contributing to CRC pathogenesis. Helicobacter pylori contributes to CRC development through multiple mechanisms, including genotoxic effects and hypergastrinaemia induced by the bacterium’s direct presence and secreted toxins. Additionally, H. pylori modulates immune responses, creating an inflammatory microenvironment and promoting tumorigenesis in colorectal tissue. These effects are mediated through morphological alterations, intracellular survival, release of OMVs, modulation of host autophagy, impacts on non-coding RNAs and changes in the intestinal microbiota. Created by biorender.com.

Figure 2. Long description
The diagram is divided into three horizontal sections.
Top Section: A human silhouette shows the digestive tract. Arrows indicate a decrease in gastric acid and an increase in Gastrin. To the right, H. pylori bacteria interact with the intestinal epithelium, releasing O M V s and causing microbiota dysbiosis. A legend identifies components like the Type 4 secretion system, vac A, cag A, L P S, and various immune cells.
Middle Section: A zoomed-in view of the epithelial membrane. On the left, a purple box lists Inflammation, E M T, and Tumor growth, influenced by m i R N A s like m i R-21 and m i R-146 a. In the center, the Type 4 secretion system and other receptors trigger pathways leading to R O S production and D N A damage. On the right, a T naive cell differentiates into T H 1, T H 17, or T reg cells, releasing cytokines like I F N gamma, I L-17, and I L-10.
Bottom Section: Two distinct panels. The left panel (blue background) details the Autophagy process from Initiation to Autophagosome formation, involving P I 3 K, m T O R, and A T G complexes. This process leads to H. pylori survival and cancer progression. The right panel (yellow background) summarizes the immune effects: Inflammation, Promotes tumor immunity, and Immune suppression.
Direct mechanisms
Immunohistochemical analyses have detected H. pylori in TA, TVP, VP polyps and adenocarcinomas (Refs Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63, Reference Soylu, Ozkara, Alıs, Dolay, Kalaycı, Yasar and Kumbasar102). Additionally, a notable association exists between H. pylori gastritis and various types of polyps – including HP, APs, advanced adenomas and high-grade dysplasia – which may indicate a potential role for this pathogen in CRC (Refs Reference Sonnenberg and Genta67, Reference Liu, Yang, Jiang and Jiao103). The detection of H. pylori DNA and antigens in colorectal tissues and cancers prompts a deeper exploration into its potential direct role in the mechanisms underlying tumorigenesis (Ref. Reference Butt and Epplein104). Two main routes have been proposed for the direct mechanisms by which H. pylori contribute to CRC: (1) the direct secretion of bacterial toxins that affect colorectal tissue and (2) systemic alterations, such as hypergastrinaemia, that create a favourable environment for carcinogenesis in the gastrointestinal tract.
Secreted toxins of H. pylori
The presence of H. pylori or its active molecules, such as secreted toxins, within the colorectal tissue signifies a direct effect. The possibility that H. pylori or its components can migrate through the colon is substantiated by multiple lines of evidence. Grahn et al. (Ref. Reference Grahn, Hmani-Aifa, Fransén, Söderkvist and Monstein105) reported that H. pylori was found in 22%–27% of analysed CPs or cancers. In a separate case–control study comparing cancer patients to healthy controls, researchers found that H. pylori were present in the tumour tissues of 19 out of 118 individuals diagnosed with cancer. In contrast, only 1 out of 58 healthy individuals showed evidence of H. pylori in their colon tissue (Ref. Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63).
CagA: An animal study employing mouse models of intestinal inflammation has provided evidence that certain pathogenic factors of H. pylori can influence areas beyond the stomach (Ref. Reference Luther, Owyang, Takeuchi, Cole, Zhang, Liu, Erb-Downward, Rubenstein, Chen, Pierzchala, Paul and Kao106). Studies indicate that the CagA protein from H. pylori , which necessitates direct contact with host T cells through the T4SS, is associated with an increased risk of CRC and heightened inflammatory responses in individuals infected with CagA-positive strains (Refs Reference Shmuely, Passaro, Figer, Niv, Pitlik, Samra, Koren and Yahav61, Reference Hong, Lee, Kim, Lee, Kim, Choe, Lee, Cheon, Sung, Park and Shim107).
VacA: Unlike CagA, the virulence factor VacA appears to disseminate more easily beyond the stomach. Additionally, antigens such as (outer membrane protein) OMP (HP 1564), chaperonin GroEl and (Helicobacter cysteine-rich protein) HcpC have also been linked to an increased likelihood of CRC and are known to be released into the extracellular environment, much like VacA (Refs Reference Butt, Varga, Blot, Teras, Visvanathan, le Marchand, Haiman, Chen, Bao, Sesso, Wassertheil-Smoller, Ho, Tinker, Peek, Potter, Cover, Hendrix, Huang, Hyslop, Um, Grodstein, Song, Zeleniuch-Jacquotte, Berndt, Hildesheim, Waterboer, Pawlita and Epplein51, Reference Dunn, Vakil, Schneider, Miller, Zitzer, Peutz and Phadnis108, Reference Snider, Voss, McDonald and Cover109).
Lipopolysaccharides (LPS): A recent in vitro experimental study has revealed that the LPS of H. pylori , a principal component of its outer membrane, may disrupt the DNA repair mechanisms that are responsible for maintaining genomic stability in colonic cells. This disruption could lead to genotoxicity and subsequently influence the development of CRC (Ref. Reference Cavallo, Cianciulli, Mitolo and Panaro110). Nitric oxide (NO) plays a crucial role in the stomach, acting as a messenger molecule that regulates blood flow, mucus production and gastric motility. It also contributes to protecting the gastric mucosa from ulcers and other damage. However, elevated levels of nitric oxide (NO) are produced by cells in response to H. pylori LPS, which is implicated in the genotoxicity associated with this bacterium. In colon cells, the carcinogenic potential of NO may be mediated by the inhibition of DNA repair enzymes and its interaction with pro-apoptotic proteins. These effects result the nitrosylation of specific cysteine and tyrosine residues, as demonstrated in other gastrointestinal cell types (Refs Reference Török, Higuchi, Bronk and Gores111, Reference Jaiswal, LaRusso, Burgart and Gores112). However, the specific biological roles of these antigens in the development of cancer, as well as the underlying mechanisms linking antibody responses to this group with an increased risk of CRC, have yet to be clarified.
Hypergastrinaemia
Hypergastrinaemia is characterized by elevated levels of the peptide hormone gastrin in the bloodstream, often resulting from various gastrointestinal conditions, including H. pylori infection (Ref. Reference Chuang, Sheu, Yang, Kao, Cheng and Yao113). The bacterium’s presence in the gastric epithelium can lead to chronic inflammation and damage to the gastric mucosa, which in turn affects gastric acid secretion (Ref. Reference Butt and Epplein104). The chronic infection of H. pylori can inhibit parietal cell function, leading to decreased gastric acid secretion and a compensatory increase in gastrin release (Ref. Reference Park, Yoo, Lee, Yoon and Cha114). The rise in gastrin levels has important consequences for gastrointestinal health. Elevated gastrin can enhance gastric acid secretion and promote the proliferation of gastric epithelial cells (Ref. Reference Jain and Samuelson115). Studies have suggested that chronic hypergastrinaemia due to H. pylori infection may lead to changes in colonic mucosa and potentially increase susceptibility to colorectal neoplasia (Ref. Reference Lu, Wang, Ke, Wang, Wang, Li, Wang and Wang116). Recent research has established a significant association between gastrin levels and the up-regulation of inflammatory mediators, particularly cyclooxygenase 2 (COX-2) and IL-8 (Refs Reference Hartwich, Konturek, Pierzchalski, Zuchowicz, Labza, Konturek, Karczewska, Bielanski, Marlicz, Starzynska, Lawniczak and Hahn117, Reference Chao and Hellmich118). Moreover, the up-regulation of the inducible enzyme COX-2 following H. pylori infection has been implicated in the increased risk of CRC (Ref. Reference Konturek, Konturek, Hartwich and Hahn119). This association is supported by evidence linking COX-2 expression and tissue activity to the initiation and progression of CRC and various other neoplasms (Refs Reference Fournier and Gordon120, Reference Fosslien121). These elevations ultimately increase angiogenesis within the proliferating and mutagenic mucosal cells of the colon. This process is accompanied by reduced apoptosis, both of which are closely linked to carcinogenesis (Refs Reference Fosslien121, Reference Tsujii, Kawano, Tsuji, Sawaoka, Hori and DuBois122).
Indirect mechanisms
Helicobacter pylori may also influence host physiology through indirect mechanisms. These mechanisms do not directly engage with the host T cell but instead initiate cascades of physiological or immune responses that facilitate bacterial survival and pathogenesis. Indirect interactions often involve complex host–pathogen dynamics, including alterations in the host T-cell morphology, immune system modulation and changes to the gut microbiota. Each of these processes plays a critical role in shaping the infection outcome, and understanding them is crucial for developing therapeutic strategies.
Morphological shift in H. pylori : spiral to coccoid
The transformation of H. pylori from its spiral to coccoid form plays a significant role in its ability to evade the host’s immune system and resist antibiotic treatment, both of which may contribute to the development of CRC. One of the primary mechanisms by which H. pylori escape immune detection is through its coccoid form, known as the VBNC (viable but non-cultivable) state. This form can persist in adverse conditions, allowing the bacteria to survive despite the host’s immune response (Refs Reference Chaput, Ecobichon, Cayet, Girardin, Werts, Guadagnini, Prévost, Mengin-Lecreulx, Labigne and Boneca123, Reference Sadeghloo, Fatemi, Raeisi, Azizmohammad Looha and Sadeghi124). Studies indicate that coccoid forms can induce a humoral immune response similar to that of bacillary forms; however, they are less detectable by standard serological methods, which may complicate the identification of ongoing infections (Refs Reference Sarem and Corti125, Reference Figueroa, Faúndez, Troncoso, Navarrete and Toledo126).
A study by Jones et al. examined 176 colorectal specimens and found positive staining for H. pylori exclusively in the coccoid form, with no evidence of the typical spiral shape normally found in the stomach. Specifically, positive staining was observed in 1.7% of normal samples, 15.3% of adenomas and 16.9% of adenocarcinomas (Ref. Reference Jones, Helliwell, Pritchard, Tharakan and Mathew63).
The potential ability of H. pylori to evade the immune response might allow it to maintain chronic infections. These infections could lead to long-term mucosal inflammation, fostering a microenvironment that may be conducive to adenomas and malignant tumours in the colorectal region (Refs Reference Butt and Epplein104, Reference Sarem and Corti125, Reference Fan, Zhu and Xu127). Moreover, the coccoid form’s reduced ability to adhere to gastric epithelium and the lower antigenicity of these small particles may facilitate its survival. When antibiotics are administered, the bacillary forms are often targeted and eliminated; however, the coccoid forms may endure, potentially leading to the recurrence of infection. This persistence has been suggested to create a cycle of chronic inflammation and immune evasion that heightens the risk for CRC development, though more longitudinal clinical data are needed to confirm this mechanistic link (Refs Reference Sarem and Corti125, Reference Guo, Zhang, Jiang, Wang, Chen, Zhang, Zhou, Zhang and Leung128, Reference Ierardi, Losurdo, Mileti, Paolillo, Giorgio, Principi and di Leo129).
Intracellular life of H. pylori
Although H. pylori is predominantly regarded as an extracellular pathogen, experimental evidence suggests it may possess the ability to penetrate epithelial cells (Ref. Reference Chu, Wang, Wu and Lei130), macrophages (Ref. Reference Wang, Wu and Lei131) and dendritic cells (Ref. Reference Wang, Gorvel, Chu, Wu and Lei132), allowing it to survive and multiply within these cells. In adverse environmental conditions, it has been proposed that H. pylori may enter host T cells and remain dormant, possibly re-emerging when the environment becomes favourable again. This proposed intracellular life cycle may help explain how H. pylori could contribute to CRC. In vitro studies have shown that H. pylori can enter AGS cell lines (Refs Reference Segal, Falkow and Tompkins133–Reference Petersen, Blom, Andersen and Krogfelt135) and persist in either spiral or coccoid forms within specialized membrane-bound vacuoles in the cytoplasm (Ref. Reference Segal, Falkow and Tompkins133). Numerous pathogenic bacteria such as Legionella (Ref. Reference Linder136), Mycobacterium (Ref. Reference Miltner and Bermudez137) and Pseudomonas (Ref. Reference Michel, Burghardt and Bergmann138) can endure adverse conditions by inhabiting eukaryotic cells. The ability of prokaryotes to live intracellularly is regarded as a crucial evolutionary development, facilitating their adaptation to diverse environmental habitats (Ref. Reference Ruiz-Lozano and Bonfante139).
An intriguing yet underexplored interaction between H. pylori and the yeast Candida has been identified, where the bacterium resides within the vacuoles of the yeast. Siavoshi et al. (Ref. Reference Siavoshi and Saniee140) observed the rapid movement of bacterial-like bodies (BLBs) within the vacuoles of oral, gastric and vaginal yeasts (Ref. Reference Heydari, Siavoshi, Jazayeri, Sarrafnejad and Saniee141), suggesting that these yeasts may act as carriers for this pathogen. Given the ubiquitous presence of yeast throughout the digestive system, it is suggested that they might influence gut health (Refs Reference Salmanian, Siavoshi, Akbari, Afshari and Malekzadeh142, Reference Kreulen, de Jonge, van den Wijngaard and van Thiel143). Although studies have linked Candida albicans overabundance to IBD and CRC risk (Refs Reference Caetano, Gaspar, Martinez-de-Oliveira, Palmeira-de-Oliveira and Rolo144, Reference Kaźmierczak-Siedlecka, Dvořák, Folwarski, Daca, Przewłócka and Makarewicz145), the direct role of yeast as a ‘Trojan Horse’ for H. pylori transmission to the colon is an emerging concept that requires more robust in vivo validation. Because yeasts are more resilient to environmental stresses than bacteria, they may help protect H. pylori in extra-gastric environments and facilitate its transfer to the human colon (Refs Reference Siavoshi and Saniee140, Reference Lin, Xu and Yu146). The intracellular life of H. pylori within yeast vacuoles may not only allow for nutrient acquisition but also promote its transmission within human populations. Similar to how amoebae have been proposed as training grounds for intracellular pathogens such as Legionella and Mycobacterium spp., yeast vacuoles might play a role in selecting important pathogenic features of H. pylori and enhancing its invasion capabilities. This interaction could contribute to the bacterium’s pathogenicity and its potential involvement in CRC, highlighting the need for further research into these complex relationships (Refs Reference Molmeret, Horn, Wagner, Santic and Abu Kwaik147–Reference Ramezani, Sadeghloo, Azizmohammad Looha and Sadeghi149).
Helicobacter pylori -derived outer membrane vesicles
Helicobacter pylori -derived outer membrane vesicles (OMVs) represent a potential mechanism linking gastric infection to distal inflammatory and tumour-promoting effects in the colorectal microenvironment. The OMVs of H. pylori , characterized by their bilayer lipid membrane nanostructures, encapsulate a diverse array of bacterial components, including peptidoglycan (PG), LPS, outer membrane proteins, periplasmic proteins, various enzymes, DNA and toxins (Ref. Reference Kaparakis-Liaskos and Ferrero150). Recent studies suggest that OMVs from H. pylori may serve as an important aspect of virulence linked to the bacterium’s pathogenic mechanisms (Refs Reference Chew, Chung, Lin, Wu, Huang and Kao151–Reference Lekmeechai, Su, Brant, Alvarado-Kristensson, Vallström, Obi, Arnqvist and Riesbeck153).
Evidence has shown that OMVs produced by H. pylori are implicated in disease progression by triggering inflammatory responses at both local and systemic levels, such as the modulation of neutrophil migration in areas beyond the stomach, thereby fostering a pro-inflammatory environment (Refs Reference Ansari and Yamaoka154–Reference Qiang, Hu, Tian, Li, Ren, Deng and Jiang156). They disrupt the regulation of immune responses by delivering biologically active substances into host T cells (Refs Reference Ansari and Yamaoka154, Reference González, Díaz, Sandoval-Bórquez, Herrera and Quest157). Helicobacter pylori -derived OMVs are detected by PRRs, such as (nucleotide-binding oligomerization domain-containing protein) NOD1, which may be involved in various host T-cellular processes, including invasion, autophagy, apoptosis and tumorigenesis (Refs Reference Sadeghloo, Saffarian, Hakemi-Vala, Sadeghi and Yadegar152, Reference Gonciarz, Krupa, Hinc, Obuchowski, Moran, Gajewski and Chmiela158, Reference Greenfield and Jones159). The absorption of H. pylori OMVs by epithelial cells can occur through either clathrin-dependent endocytosis or a clathrin-independent mechanism involving lipid rafts, and it significantly relies on the adhesins present in the OMVs.
Research has demonstrated that the presence of the VacA toxin may enhance the uptake of OMVs by host T cells (Refs Reference Parker, Chitcholtan, Hampton and Keenan160, Reference Kaparakis, Turnbull, Carneiro, Firth, Coleman, Parkington, le Bourhis, Karrar, Viala, Mak, Hutton, Davies, Crack, Hertzog, Philpott, Girardin, Whitchurch and Ferrero161). The elements found in H. pylori OMVs can stimulate the release of both pro-inflammatory and anti-inflammatory cytokines, such as IL-6 and IL-10, and trigger an oxidative burst (Refs Reference Winter, Letley, Rhead, Atherton and Robinson162, Reference Parker and Keenan163). This process is facilitated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which becomes activated by various components of H. pylori OMVs, particularly HP-NAP (neutrophil-activating protein). This protein plays a crucial role in increasing β2 integrin expression and drawing leukocytes towards epithelial cells (Refs Reference Satin, del Giudice, Della Bianca, Dusi, Laudanna, Tonello, Kelleher, Rappuoli, Montecucco and Rossi164, Reference Unemo, Aspholm-Hurtig, Ilver, Bergström, Borén, Danielsson and Teneberg165). Additionally, these OMVs have the potential to trigger the degranulation of human eosinophils, resulting in the release of eosinophil cationic protein (ECP). This process raises the levels of intracellular adhesion molecule (ICAM)-1 on epithelial cells and enhances the expression of CD11b integrin on eosinophils (Refs Reference Winter, Letley, Rhead, Atherton and Robinson162, Reference Ko, Jeon, Kim, Yoon, Kim, Kim, Kim and Kim166). Secreted OMVs from H. pylori can influence host transcription regulators, including NF-κB, MAPK and extracellular signal-regulated kinase (ERK), resulting in changes in gene expression that contribute to the secretion of cytokines such as IL-8, TNF-α and IL-1β, along with promoting cell growth and cancer progression (Refs Reference Suzuki, Murata-Kamiya, Yanagiya, Suda, Hattori, Kanda, Bingo, Fujii, Maeda, Koike and Hatakeyama167, Reference Maeda, Akanuma, Mitsuno, Hirata, Ogura, Yoshida, Shiratori and Omata168).
Several studies have examined the role of H. pylori -derived OMVs in extra-gastric diseases. For instance, previous research has suggested that H. pylori -derived OMVs can traverse biological barriers, reach brain tissue and contribute to the development of cognitive disorders. Astrocytes treated with OMVs secreted interferon-γ, which was dependent on NF-κB activation, leading to neurotoxicity (Refs Reference Xie, Cools, van Imschoot, van Wonterghem, Pauwels, Vlaeminck, de Witte, el Andaloussi, Wierda, de Groef, Haesebrouck, van Hoecke and Vandenbroucke169, Reference Gesualdo, Fagoone, Saracco and Pellicano170). Another investigation focused on the effects of H. pylori OMVs on liver cells, revealing that exosomes derived from OMV-contaminated hepatocytes showed increased expression of markers associated with fibrosis and stellate cell activation, including β-catenin, vimentin, α-SMA (smooth muscle actin) and (tissue inhibitor of metalloprotease) TIMP-1 (Ref. Reference Zahmatkesh, Jahanbakhsh, Hoseini, Shegefti, Peymani, Dabin, Samimi and Bolori171). Findings from an in vitro study by Hock et al. (Ref. Reference Hock, McKenzie and Keenan172) demonstrated that OMVs from H. pylori markedly stimulate COX-2 expression in peripheral blood mononuclear cells (PBMCs). This stimulation results in significantly increased levels of prostaglandin E2 and IL-10, which contribute to the inhibition of human T-cell responses. Emerging research suggests that CagA-positive H. pylori , unlike CagA-negative strains, contributes to the development of atherosclerosis by generating ROS through CagA-containing OMVs via the (Janus kinase signal transducer and activator of transcription) JAK-STAT3 signalling pathway (Refs Reference Xia, Zhang, Wu, Chen, Liu, Xu, Cui, Zhu, Wang, Hao, Li, Fay, Martinez-Lemus, Hill, Xu and Liu173, Reference Tahmina, Hikawa, Takahashi-Kanemitsu, Knight, Sato, Itoh and Hatakeyama174). The transmission of CagA proteins by OMVs to distant sites from the initial infection is associated with the promotion of Claudin-2 expression, alterations in cell structure and intestinal epithelial barrier dysfunction in chronic colitis and CRC (Refs Reference Guo, Xu, Gong, Hu, Zhang, Xie, Chi, Li, Xia and Liu175, Reference Baker, Cross, Curtius, al Bakir, Choi, Davis, Temko, Biswas, Martinez, Williams, Lindsay, Feakins, Vega, Hayes, Tomlinson, McDonald, Moorghen, Silver, East, Wright, Wang, Rodriguez-Justo, Jansen, Hart, Leedham and Graham176).
These findings suggest that OMVs may also contribute to CRC pathogenesis. Further investigation is needed to clarify the role of H. pylori -derived OMVs in colorectal tumorigenesis.
Autophagy process
The role of the autophagy pathway in CRC is complex, exhibiting both oncogenic and tumour-suppressive properties. Its effects are modulated by a variety of biological factors, such as genetic alterations, the specific type of tumour and its stage of development (Refs Reference Dong, Wu, Zhao, Ye, Xing and Yang75, Reference Alves, Castro, Fernandes, Francisco, Castro, Priault, Chaves, Moyer, Oliveira, Seruca, Côrte-Real, Sousa and Preto177, Reference Singh, Vats, Chia, Tan, Deng, Ong, Arfuso, Yap, Goh, Sethi, Huang, Shen, Manjithaya and Kumar178). Autophagy is a vital biological process characterized by the degradation and recycling of cellular components, enabling cells to maintain homeostasis and respond effectively to stressors. The formation of an autophagosome, characterized by its double-membrane structure, represents an essential step in the autophagy process. This structure arises from various morphological changes to the phagophore and associated membrane sources. When the autophagosome fuses with a lysosome, it leads to the degradation of targeted macromolecules and organelles (Refs Reference Li, He and Ma179, Reference Sadeghloo, Nabavi-Rad, Zali, Klionsky and Yadegar180).
Autophagy regulation involves several signalling pathways, primarily 5′AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin (MTOR). Together, they inhibit ‘self-eating’ under favourable conditions and activate it during stressors like low energy, nutrient scarcity, hypoxia and the presence of pathogens (Ref. Reference Kroemer, Mariño and Levine181). AMPK facilitates the restoration of low energy levels within cells by suppressing the MTOR complex 1 (MTORC1) and activating UNC-like autophagy-activating kinase 1 (ULK1) through phosphorylation (Ref. Reference Li and Chen182). In stressful conditions, the inactivation of MTORC1 leads to the phosphorylation of ULK1 complex proteins, which collaborate with the class III phosphatidylinositol 3-kinase complex to initiate phagophore formation (Refs Reference Park, Jung, Seo, Otto, Grunwald, Kim, Moriarity, Kim, Starker, Nho, Voytas and Kim183–Reference Chen, Jiang, Li, Zhong, Wang, Yuan and Yan185). The (antithymoglobulin) ATG5-ATG12-ATG16L1 complex facilitates the conversion of (microtubule-associated protein light chain) LC3-I to LC3-II, a critical step in selective autophagy that recruits damaged cargo via the adaptor sequestosome protein SQSTM1 (Refs Reference Russell, Yuan and Guan186, Reference Koustas, Sarantis, Kyriakopoulou, Papavassiliou and Karamouzis187). Ultimately, autophagosomes mature through fusion with lysosomes, a process supported by proteins such as (lysosome-associated membrane protein) LAMP2 and RAB7. This fusion allows lysosomal enzymes to degrade the contents, recycling essential substrates back into the cytoplasm for cellular use (Refs Reference Chen, Jiang, Li, Zhong, Wang, Yuan and Yan185, Reference Debnath, Gammoh and Ryan188).
Different intracellular pathogens, such as Yersinia pseudotuberculosis, Legionella pneumophila and H. pylori , have developed various strategies to manipulate autophagy to their advantage. They do this by co-opting the membranes of autophagosomes to further their survival and proliferation (Ref. Reference Upadhyay and Philips189). Research has shown that chronic infection with H. pylori can lead to autophagy modulation that contributes to gastric cancer progression; this may also provide insights relevant to CRC (Refs Reference Zhang, Chen, Hu, Su, Zhang, Han, Chen and Li190, Reference Yang, Shu and Xie191). Specifically, H. pylori can induce defective autophagy or inhibit it altogether, allowing the bacterium to thrive within epithelial cells and facilitate carcinogenesis (Refs Reference Yang, Shu and Xie191, Reference Courtois, Haykal, Bodineau, Sifré, Azzi-Martin, Ménard, Mégraud, Lehours, Durán, Varon and Bessède192).
Autophagy impairment has been linked to aging (Ref. Reference Levine and Kroemer193), neurodegenerative diseases (Ref. Reference Maiese194), cardiovascular disorders (Ref. Reference Lai, Tsai, Kuo, Ho, Day, Pai, Chung, Huang, Wang, Liao and Huang195), infections (Ref. Reference Koepke, Hirschenberger, Hayn, Kirchhoff and Sparrer196) and cancers (Ref. Reference Rajendran and Oon197). Numerous studies have indicated that the dysregulated expression of autophagy-related markers is linked to the advancement in CRC (Refs Reference Wu, Sun, Tian, Li, Gao, He and Li198, Reference Yang and Deng199). A study involving CRC patients revealed a significant correlation between LC3B expression and aggressive tumour phenotypes, indicating that autophagy may play a role in tumour promotion (Ref Reference Zheng, Zhang, Wang and Sun70). In a separate study involving 155 CRC patients, the overexpression of Beclin1 – a key scaffold in autophagosome formation – was associated with increased tumour aggressiveness (Ref. Reference Koukourakis, Giatromanolaki, Sivridis, Pitiakoudis, Gatter and Harris200). It is also revealed that ATG7 is essential for the survival of colon cancer cells, and its inhibition triggers cell death by activating apoptotic pathways (Ref. Reference Scherr, Jassowicz, Pató, Elssner, Ismail, Schmitt, Hoffmeister, Neukirch, Gdynia, Goeppert, Schulze-Bergkamen, Jäger and Köhler201).
Research has demonstrated that H. pylori , along with the OMVs it produces (Refs Reference Chew, Chung, Lin, Wu, Huang and Kao151, Reference Sadeghloo, Saffarian, Hakemi-Vala, Sadeghi and Yadegar152), which are known to carry virulence factors, can disrupt T-cellular mechanisms (Refs Reference Li, Tang, Jia, Zhu, Zhuang, Fang, Li, Wang, Zhang, Guo, Wang, Feng, Qiao, Mao and Zou202, Reference Terebiznik, Raju, Vázquez, Torbricki, Kulkarni, Blanke, Yoshimori, Colombo and Jones203). In the investigation by Yang et al. (Ref. Reference Yang and Deng199), it was demonstrated that H. pylori significantly up-regulates Beclin1 expression and increases the LC3-II/I ratio while concurrently reducing p62 protein levels in human colon carcinoma cells. Furthermore, the activation of autophagy in colon cells leads to elevated levels of the cell proliferation marker Ki67 (discovered in Kiel, Germany) and the apoptosis inhibitor (B-cell lymphoma-2) Bcl-2, indicating that H. pylori infection is associated with enhanced cellular proliferation and suppressed apoptosis in CRC (Ref. Reference Yang and Deng199). Recent studies have highlighted the potential interaction between H. pylori infection, autophagy and apoptosis, suggesting that this connection occurs through the binding of Bcl-2 and Bcl-xL to Beclin1, thereby influencing autophagic processes (Ref. Reference Zhou, Yang and Xing204). During H. pylori infection, VacA leads to an elevated autophagic flux index, particularly reflected in increased levels of LC3-II (Refs Reference Kim, Lee, Oh, Yoon, Jang, Holland, Reno, Hamad, Maeda, Chung and Chen184, Reference Courtois, Haykal, Bodineau, Sifré, Azzi-Martin, Ménard, Mégraud, Lehours, Durán, Varon and Bessède192, Reference Zhu, Xue, Zhang, Jia, Tong, Han, Li, Xiang, Mao and Tang205). Findings from Lai et al. (Ref. Reference Jan, Chen, Yang, Ong, Chang, Muthusamy, Abera, Wu, Gervay-Hague, Mong and Lin206) suggest that cholesterol glucosyltransferase (CGT) from H. pylori , which converts host cholesterol to cholesterol-α-glucosides, enhances the survival of this bacterium in J774A.1 macrophage cells promote autophagy by increasing the levels of LC3-II, ATG12 and Beclin1. Additionally, CagA-positive strains can alter autophagy through the c-Met-PI3K/Akt–mTOR signalling pathway, resulting in the release of pro-inflammatory cytokines such as IL-1β, TNF-α and IL-8 (Ref. Reference Li, Tang, Jia, Zhu, Zhuang, Fang, Li, Wang, Zhang, Guo, Wang, Feng, Qiao, Mao and Zou202).
Further research is needed to clarify how H. pylori -induced autophagy dysregulation contributes to colorectal tumour biology. Understanding these mechanisms may support the development of therapeutic strategies aimed at restoring autophagic balance and potentially reducing CRC-related pathogenic effects.
Intestinal immune responses induced by H. pylori
Helicobacter pylori modulates both innate and adaptive immune responses, contributing to the progression of infection-associated disease (Ref. Reference Balani, dos Santos Cortez, da Silveira Freitas, de Melo, Zarpelon-Schutz and Teixeira207). Through its array of virulence factors, H. pylori not only evade initial immune responses but also subtly modulates T-cell activity and antigen presentation (Ref. Reference Marzhoseyni, Mousavi and Ghotloo208). This allows it to persist within the host and establish chronic infection, all while circumventing the immune defences. This evasion strategy is a hallmark of H. pylori ’s ability to manipulate host immune processes, leading to chronic infection and the promotion of a pro-inflammatory environment, which may increase the risk of CRC (Refs Reference Engelsberger, Gerhard and Mejías-Luque29, Reference Jackson, Britton, Lewis, McKeever, Atherton, Fullerton and Fogarty209).
One key aspect of H. pylori infection is its influence on T-cell populations, particularly CD4+ and CD8+ T cells, which contribute to inflammation. The bacterium induces a mixed Th1 and Th17 response in the gastric mucosa, with the release of pro-inflammatory cytokines like interferon gamma (IFN-γ) and IL-17A, which are critical to the pathology of gastritis (Ref. Reference Hitzler, Kohler, Engler, Yazgan and Müller210). Dendritic cells (DCs) that encounter H. pylori up-regulate markers such as CD83 and CD86, which in turn drive a strong Th1 response. IL-23-producing DCs also stimulate Th17 responses, thereby intensifying inflammation (Refs Reference Bimczok, Clements, Waites, Novak, Eckhoff, Mannon, Smith and Smythies211, Reference Khamri, Walker, Clark, Atherton, Thursz, Bamford, Lechler and Lombardi212). Experimental studies have shown that both H. pylori -specific CD4+ T cells and the sustained secretion of IFN-γ are essential for the development of gastritis, as mice lacking these components fail to exhibit significant gastric inflammation. Furthermore, regulatory T cells (Tregs) are crucial in maintaining immune homeostasis, counter-acting the inflammatory Th1/Th17 response and enabling H. pylori ’s persistence in the host (Refs Reference Gray, Fontaine, Poe and Eaton213, Reference Kao, Zhang, Miller, Mills, Wang, Liu, Eaton, Zou, Berndt, Cole, Takeuchi, Owyang and Luther214).
Research has also indicated that Peyer’s patches (PPs) in the small intestine are vital for priming naive CD4+ T cells against H. pylori antigens. PPs can engulf the coccoid form of H. pylori , allowing DCs to capture its antigens and subsequently activate CD4+ T cells (Ref. Reference Nagai, Mimuro, Yamada, Baba, Moro, Nochi, Kiyono, Suzuki, Sasakawa and Koyasu215). Studies have shown that mice with mutations in the adenomatous polyposis coli (APC) gene develop a higher tumour burden when infected with H. pylori compared to non-infected controls. This increased tumorigenesis is associated with an imbalance between pro-inflammatory and regulatory T cells within the intestinal epithelium (Ref. Reference Ralser, Dietl, Jarosch, Engelsberger, Wanisch, Janssen, Middelhoff, Vieth, Quante, Haller, Busch, Deng, Mejías-Luque and Gerhard216). Furthermore, H. pylori infection has been linked to the activation of pro-carcinogenic signalling pathways such as STAT3 in both intestinal and colonic epithelium. The persistent pro-inflammatory immune response driven by H. pylori infection contributes to the development of CRC through mechanisms that include increased gut barrier permeability and subsequent antigen influx (Refs Reference Corvinus, Orth, Moriggl, Tsareva, Wagner, Pfitzner, Baus, Kaufman, Huber, Zatloukal, Beug, Öhlschläger, Schütz, Halbhuber and Friedrich217, Reference Balic, Saad, Dawson, West, McLeod, West, D’Costa, Deswaerte, Dev, Sievert, Gough, Bhathal, Ferrero and Jenkins218).
Recent findings have highlighted a dynamic role for CD8+ T cells in H. pylori infection. At the early stages of infection, tissue-resident CD8+ T cells respond to the bacterial CagA antigen by producing IFN-γ, contributing to the initial immune response. As illustrated in Figure 3, the transition from early to chronic H. pylori infection is accompanied by a decline in CD8+ T-cell populations and a progressive predominance of CD4+ T cells, indicating a shift in the immune landscape during persistent infection (Refs Reference Quiding-Järbrink, Lundin, Lönroth and Svennerholm219, Reference Koch, Gong, Friedrich, Engelsberger, Kretschmer, Wanisch, Jarosch, Ralser, Lugen, Quante, Vieth, Vasapolli, Schulz, Buchholz, Busch, Mejías-Luque and Gerhard220). Interestingly, studies have shown that CD8+ T-cell infiltration increases in the gastric mucosa when CD4+ T cells are absent. Such changes correlate with more severe disease manifestations. This suggests that CD4+ T cells may play a regulatory role in controlling CD8+ T-cell activity during H. pylori infection (Refs Reference Tan, Pedersen, Zhan, Lew, Pearse, Wijburg and Strugnell221, Reference Fukui, Nishio, Okazaki, Kasahara, Saga, Tanaka, Uza, Ueno, Kido, Ohashi, Asada, Nakase, Watanabe and Chiba222). In terms of cancer prognosis, tumours with higher levels of CD8+ T-cell infiltration are associated with better outcomes due to the role of these cells in controlling tumour growth and their response to immunotherapies (Refs Reference Mlecnik, Tosolini, Charoentong, Kirilovsky, Bindea, Berger, Camus, Gillard, Bruneval, Fridman, Pagès, Trajanoski and Galon223–Reference Piroozkhah, Gholinezhad, Piroozkhah, Shams and Nazemalhosseini-Mojarad225). However, the specific impact of H. pylori on CD8+ T-cell function during carcinogenesis remains under investigation. There is evidence suggesting that the bacterium’s presence might suppress tumour-specific CD8+ T-cell responses through mechanisms involving increased numbers of (forkhead box protein) FoxP3+/IL-17+ T cells in the intestines, which could impair effective anti-tumour immunity (Ref. Reference Ma and Dong226).
Dynamics of CD4+ and CD8+ T-cell responses during Helicobacter pylori infection. Helicobacter pylori trigger immune responses by activating dendritic cells (DCs), leading to CD4+ T-cell differentiation and CD8+ T-cell activation. The acute phase features a robust CD8+ T-cell response, while the chronic phase is marked by CD4+ T-cell dominance, facilitating immune evasion and persistent inflammation, which drives disease progression and may contribute to the development of CRC. Created by biorender.com.

Figure 3. Long description
The diagram is divided into two main sections.
Top section:
At the top, H. pylori bacteria and red dots labeled Virulence factor are in the gastric lumen. An arrow shows a virulence factor passing between pink columnar epithelial cells to reach a D C (dendritic cell) below. The D C points to a row of blue T naive cells. From these, arrows branch into C D 4 (green) and C D 8 (light blue) cells. The C D 4 cell further differentiates into three types: Th 1 (purple), Th 17 (brown), and F O X P 3 T reg (orange). Each of these three cells releases signaling molecules represented by small colored dots. A flat-headed inhibitory line extends from the F O X P 3 T reg cell toward the C D 8 cell.
Bottom section:
A rectangular box titled C D 4 and C D 8 T cell dynamics is split diagonally. The left side, shaded green and labeled Acute phase, shows a high density of light blue C D 8 cells and fewer green C D 4 cells. The right side, shaded pink and labeled Chronic phase, shows a shift to a high density of green C D 4 cells and fewer light blue C D 8 cells.
Recent studies have also demonstrated that H. pylori can dampen systemic CD8+ T-cell responses, potentially reducing the effectiveness of cancer immunotherapies. In models such as MC38 colon adenocarcinoma and azoxymethane/DSS-induced tumours, H. pylori infection was found to increase tumour size and reduce the effectiveness of treatments targeting (cytotoxic T-lymphocyte-associated protein) CTLA-4 and (programmed cell death ligand) PD-L1 pathways. This is attributed to a reduction in the activation and proliferation of tumour-specific CD8+ T cells, driven by altered dendritic cell function in infected mice (Refs Reference Koch, Gong, Friedrich, Engelsberger, Kretschmer, Wanisch, Jarosch, Ralser, Lugen, Quante, Vieth, Vasapolli, Schulz, Buchholz, Busch, Mejías-Luque and Gerhard220, Reference Oster, Vaillant, Riva, McMillan, Begka, Truntzer, Richard, Leblond, Messaoudene, Machremi, Limagne, Ghiringhelli, Routy, Verdeil and Velin227).
Overall, H. pylori have a profound influence on the immune system, particularly through its modulation of CD4+ and CD8+ T cells. By affecting CD4+ T-cell differentiation, the bacterium plays a critical role in disease pathogenesis, including the heightened risk of CRC. Additionally, H. pylori may dampen tumour-specific CD8+ T-cell responses, thus hindering the effectiveness of cancer immunotherapy and potentially influencing colon carcinogenesis through immune cell dynamics.
Helicobacter pylori and non-coding RNAs interactions
For decades, scientific inquiry has predominantly centred on the mere 2% of the human genome responsible for encoding proteins (Refs Reference Deveson, Hardwick, Mercer and Mattick228, Reference Adams, Parsons, Walker, Zhang and Slack229). However, recent advancements have illuminated the significance of the remaining 98%, previously dismissed as non-functional ‘junk’ DNA. This vast segment of our genetic material encompasses non-coding RNAs (ncRNAs), which are now recognized as pivotal players in a multitude of biological processes, including growth, development and organ function. Non-coding RNAs are categorized into two main types: small non-coding RNAs, which include microRNAs (miRNA), and long non-coding RNAs (lncRNAs) (Refs Reference Hombach and Kretz230). The miRNA are small, single-stranded non-coding RNAs, approximately 22 nucleotides long, encoded by genes within the genome. They play a crucial role in regulating gene expression by binding to target messenger RNAs (mRNAs) and specifically interacting with the 3′-untranslated region (UTR). This interaction can lead to the degradation of the mRNA or inhibition. Consequently, it reduces the expression of specific target genes (Refs Reference Ritchie231, Reference Mellis and Caporali232). The regulation of gene expression by miRNAs is essential for various biological processes, including tumour development, metastasis and drug resistance (Refs Reference Xu, Zheng, Wang and Shao233–Reference Li, Gao, Xie, Zhang, Ning, Liu and Gu235). On the other hand, lncRNAs are defined as RNA molecules longer than 200 nucleotides (Ref. Reference Liu, Ao, Wang, Li and Wang236). While they do not have the capacity to encode proteins, lncRNAs fulfil a variety of complex regulatory roles (Ref. Reference Nojima and Proudfoot237). They can impact a wide array of biological processes, such as transcription, translation, cellular architecture, cell cycle progression, apoptosis and the maintenance of stem cell pluripotency (Refs Reference Liu, Ao, Wang, Li and Wang236, Reference Martianov, Ramadass, Serra Barros, Chow and Akoulitchev238–Reference Zhou, Ao, Jia, Li, Kuang, du, Zhang, Wang and Liu242).
Helicobacter pylori influence on miRNAs is well-established in gastric oncogenesis (Ref. Reference Xie, Li, Li, Wu, Zhao, Zhang, Zhang, Shi, Wang, Yin, Deng and Tao243); however, its impact on the colonic ncRNA profile is an emerging area of research. The study by Zhong et al. (Ref. Reference Zhong, Chen, Zhou, Lü and Wan82) demonstrated that CagA-positive H. pylori promotes autophagy in colon cancer cells. This occurs through the down-regulation of miR-125b-5p, which in turn affects LC3B-II/LC3B-I and Beclin-1 levels. The suppression of miR-125b-5p, a recognized tumour suppressor, contributes to increased cell proliferation, invasion and enhanced autophagic processes – key mechanisms in cancer development. The findings reveal that overexpression of the CagA protein from H. pylori leads to decreased levels of miR-125b-5p, while restoration of miR-125b-5p expression effectively reverses these effects, inhibiting both autophagy and cell invasion. These results suggest a potential link where H. pylori infection might not only play a direct role in the initiation of CRC but also modulate critical non-coding RNAs like miR-125b-5p, thereby linking microbial infection to tumorigenesis (Ref. Reference Zhong, Chen, Zhou, Lü and Wan82). Several other miRNAs have also been implicated in H. pylori -associated cancer progression, supporting a role for this bacterium in gene regulation. For instance, miR-21 is frequently up-regulated during H. pylori infection and has been proposed to promote cancer progression by targeting tumour suppressor genes such as RECK (reversion-inducing-cysteine-rich protein with kazal motifs), TGF-β1 and TGF-βR2 (transforming growth factor), thereby facilitating cell proliferation and invasion (Refs Reference Zhang, Li, Gao, Chen, Chen, Liu, Xiao and Lu244, Reference Shiotani, Uedo, Iishi, Murao, Kanzaki, Kimura, Kamada, Kusunoki, Inoue and Haruma245). Similarly, miR-155 is up-regulated in response to the infection, potentially contributing to inflammation and tumorigenesis by regulating cytokine production and targeting the (Sma- and Mad- related protein) SMAD2 gene, which is involved in tumour growth and immune response modulation (Ref. Reference Xiao, Liu, Li, Tang, Li, Guo, Shi, Wang, Wu, Tong, Guo, Mao and Zou246). In contrast, H. pylori down-regulate specific tumour-suppressive miRNAs, such as let-7b, which normally inhibits pathways critical for inflammation and cancer development. The reduction of let-7b expression is hypothesized to lead to enhanced activity of pro-inflammatory genes like TLR4, NF-κB, COX-2 and IL1B, promoting a chronic inflammatory environment that facilitates tumorigenesis (Refs Reference Isomoto, Matsushima, Inoue, Hayashi, Nakayama, Kunizaki, Hidaka, Nakayama, Hisatsune, Nakashima, Nagayasu, Nakao and Hirayama247, Reference Teng, Wang, Dai, Wang, Chu and Li248). Another important miRNA related to H. pylori infection, miR-146a, inhibits components of the immune response, such as (interleukin 1 receptor associated kinase) IRAK1 and (caspase recruitment domain) CARD10, while promoting tumour-supportive pathways, including COX2 and PTGS2, further linking chronic inflammation with cancer progression (Refs Reference Liu, Xiao, Tang, Li, Li, Zhu, Guo, Gu, Zhuang, Liu, Ding, Zhao, Guo, Mao and Zou249, Reference Liu, Wang, Hu, Zhou, Zhu, Yu, Chi, Cao, Jia and Zou250). Moreover, miR-200b and miR-200c, which are also up-regulated, play crucial roles in promoting epithelial–mesenchymal transition (EMT), a process essential for cancer metastasis. By down-regulating targets like (zinc-finger E-box-binding homeobox) ZEB1 and IL6, these miRNAs enable cancer cells to gain migratory and invasive properties (Refs Reference Isomoto, Matsushima, Inoue, Hayashi, Nakayama, Kunizaki, Hidaka, Nakayama, Hisatsune, Nakashima, Nagayasu, Nakao and Hirayama247, Reference Baud, Varon, Chabas, Chambonnier, Darfeuille and Staedel251). ZEB1 is closely associated with cagPAI+ H. pylori strains (Ref. Reference Baud, Varon, Chabas, Chambonnier, Darfeuille and Staedel251) and has been linked to poor prognostic outcomes in CRC, highlighting its role in promoting EMT. By facilitating the invasive properties of cancer cells through pathways influenced by H. pylori , ZEB1 may serve as a pivotal player in the progression of CRC (Refs Reference Lindner, Paul, Eckstein, Hampel, Muenzner, Erlenbach-Wuensch, Ahmed, Mahadevan, Brabletz, Hartmann, Vera and Schneider-Stock252, Reference Zhang, Zhou, Tian, Liu and Xia253). Xie et al. (Ref. Reference Xie, Ge, Wang, Sun and Kang254), revealed a significant association between lncRNA ZFAS1 and H. pylori infection. This link extends to lymph node metastasis, advanced TNM stage and poor overall survival in CRC patients. Additionally, functional assays demonstrated that inhibiting (zinc finger antisense) ZFAS1 markedly diminished both the proliferation and invasion capabilities of CRC cells in vitro and in vivo, ultimately leading to a reduction in tumour growth. Bioinformatics assessments and luciferase reporter assays confirmed a direct interaction between ZFAS1 and miR-484. Furthermore, the study indicated that employing a miR-484 inhibitor could reverse the tumour-suppressing effects observed with ZFAS1 knockdown. Consequently, ZFAS1 is characterized as an oncogene associated with H. pylori , is regulated by the miR-484 pathway, which plays a pivotal role in CRC tumorigenesis (Ref. Reference Xie, Ge, Wang, Sun and Kang254).
Collectively, the alterations in miRNAs, their target genes and lncRNA profiles induced by H. pylori infection are increasingly recognized as a possible molecular bridge between initiation of tumorigenic pathways and CRC, highlighting their significance in the pathogen’s oncogenic mechanisms. However, further clinical studies are necessary to confirm whether these non-coding RNA dysregulations observed in experimental models translate directly to the human colonic landscape during chronic H. pylori infection.
Changes in gut microbiota communities induced by H. pylori infection
Early research has established a significant connection between microorganisms and the development of CRC, with studies dating back to the 1960s exploring how gut microbiota influences CRC susceptibility. Notably, a pivotal study from 1967 demonstrated that intestinal microorganisms were essential in mediating the carcinogenic effects of cycasin in conventional rats, while germ-free rats exhibited no such cancer development (Ref. Reference Laqueur, McDaniel and Matsumoto255). Further investigations revealed that when both germ-free and conventional rats were exposed to the carcinogen 1,2-dimethylhydrazine, a staggering 93% of conventional rats developed colonic tumours compared to only 21% of their germ-free counterparts (Refs Reference Reddy, Weisburger, Narisawa and Wynder256, Reference Reddy, Narisawa, Wright, Vukusich, Weisburger and Wynder257). The role of gut microbiota in colorectal carcinogenesis has gained renewed attention, particularly in relation to H. pylori infection. Table 2 summarizes the reported effects of H. pylori infection on gut microbiota composition. Recent findings indicate that H. pylori infection not only disrupts the balance of commensal bacterial species within the gastric mucosa but also induces substantial changes in intestinal microbial communities. This shift towards dysbiosis is associated with various gastrointestinal disorders and may play a role in carcinogenesis (Refs Reference Benavides-Ward, Vasquez-Achaya, Silva-Caso, Aguilar-Luis, Mazulis, Urteaga and del Valle-Mendoza259, Reference Frost, Kacprowski, Rühlemann, Bang, Franke, Zimmermann, Nauck, Völker, Völzke, Biffar, Schulz, Mayerle, Weiss, Homuth and Lerch262, Reference Dash, Khoder, Nada and al Bataineh267–Reference Ternes, Karta, Tsenkova, Wilmes, Haan and Letellier272).
The effects of Helicobacter pylori infection on gut microbiota compositions

Table 2. Long description
The table is organized into eight columns: Study design, Study model, Sample size, Methodology, Statistical methods used, Microbiota changes, Potential functional implications, and References.
Key findings include:
* Animal models (Mongolian gerbils and mice) show increases in E. coli, Enterococcus, Akkermansia, and Ruminococcus, linked to intestinal inflammation and tumor progression.
* Human studies (children and adults) frequently report increases in Prevotella, Proteobacteria, and Firmicutes, while often showing decreases in Subdoligranulum and Bacteroidetes.
* Methodologies primarily involve 16S r R N A sequencing, P C R, and meta-genomic sequencing.
* Functional implications across studies highlight gut homeostasis disruption, promotion of systemic low-grade inflammation, impaired metabolic function, and potential mucosal barrier impairment.
* Statistical methods include Mann-Whitney U tests, Student's t tests, A N O V A, and P E R M A N O V A.
* Sample sizes range from small groups of 17 participants to larger cohorts of 428 participants.
A study by Heimsat et al. (Ref. Reference Heimesaat, Fischer, Plickert, Wiedemann, Loddenkemper, Göbel, Bereswill and Rieder258) on Mongolian gerbils infected with H. pylori revealed significant changes in the large intestinal microbiota after 14 months of infection. Notably, there was an increase in Escherichia coli , Enterococcus spp. and Bacteroides/Prevotella spp., along with a rise in Akkermansia, a mucus-degrading species linked to compromised intestinal barrier function. Alongside these microbiota changes, a pro-inflammatory response was observed, characterized by an increase in CD3+ T-cell infiltration within the colonic tissue. These microbiota changes may be driven by hypochlorhydria and hypergastrinaemia induced by H. pylori , which facilitate the passage of acid-sensitive bacteria into the distal gastrointestinal tract. H. pylori ’s virulence factor CagA also plays a role in promoting inflammation and disrupting the epithelial barrier, further contributing to microbiota imbalances (Ref. Reference Heimesaat, Fischer, Plickert, Wiedemann, Loddenkemper, Göbel, Bereswill and Rieder258).
In wild-type mice, H. pylori infection induced microbial shifts in faecal, cecal and ileal samples, with significant changes in Turicibacteraceae, Erysipelotrichaceae and Desulfovibrionaceae families (Ref. Reference Kienesberger, Cox, Livanos, Zhang, Chung, Perez-Perez, Gorkiewicz, Zechner and Blaser273). A study on Apc-mutant mice infected with H. pylori showed an overgrowth of mucosa-destroying species, particularly Akkermansia and Ruminococcus spp., leading to intestinal barrier disruption and potential tumour promotion. Interestingly, faecal transplantation from H. pylori -infected tumour-prone mice to germ-free recipients caused reduced Treg levels and enhanced STAT3 activation, speeding up tumour development compared to non-infected stool recipients (Refs Reference Ralser, Dietl, Jarosch, Engelsberger, Wanisch, Janssen, Middelhoff, Vieth, Quante, Haller, Busch, Deng, Mejías-Luque and Gerhard216, Reference Li, Kundu, Seow, de Matos, Aronsson, Chin, Kärre, Pettersson and Greicius274). These findings underscore the critical role of H. pylori in reshaping the gut microbial community and its implications for CRC development.
In children’s studies infected with H. pylori , there was an increase in various bacterial populations within their gut microbiota, including Proteobacteria, Clostridium, Firmicutes, Prevotella and Proteus (Refs Reference Benavides-Ward, Vasquez-Achaya, Silva-Caso, Aguilar-Luis, Mazulis, Urteaga and del Valle-Mendoza259, Reference Kakiuchi, Tanaka, Ohno, Matsuo and Fujimoto260). He et al. reported that patients infected with H. pylori exhibited an increased frequency of Alistipes, along with a decrease in Lachnoclostridium (Ref. Reference He, Peng, Wang, Ouyang, Zhu, Shu, Zhu and Lu261). Also, in a large cohort study, individuals infected with H. pylori showed an increase in Prevotella, Parasutterella, Holdemanella, Betaproteobacteria, Alisonella and Howardella. The level of H. pylori antigen had a stronger impact on changes in gut bacteria than factors like age or sex. It was also negatively correlated with beneficial bacteria such as Bacteroides, Fusicatenibacter, Alistipes and Barnesiella (Ref. Reference Frost, Kacprowski, Rühlemann, Bang, Franke, Zimmermann, Nauck, Völker, Völzke, Biffar, Schulz, Mayerle, Weiss, Homuth and Lerch262). Using shotgun meta-genomic sequencing, Wang et al. (Ref. Reference Wang, Li, Zhong, Ding, Lin, Tang, Zong, Wang, Zhang, Yang, Wang and Liu265) found a correlation between H. pylori and the increased proliferation of pathogenic bacteria, such as Prevotella copri, Enterobacter cloacae and Klebsiella pneumoniae. In contrast, they observed a decrease in the levels of Sutterella wadsworthensis , Bacteroides (Phocaeicola) vulgatus and E. coli associated with H. pylori infection, suggesting that these bacteria may play a role in modulating immune responses.
Additionally, H. pylori infection significantly impacts the fungal and viral populations in the gut (Refs Reference Luo, Ru, Mirzaei, Xue, Peng, Ralser, Mejías-Luque, Gerhard and Deng69, Reference Dash, Khoder, Nada and al Bataineh267). In the research conducted by Dash et al. (Ref. Reference Dash, Khoder, Nada and al Bataineh267), a microbial profiling analysis utilizing 16S rRNA and ITS2 was performed on stool samples from adults infected with H. pylori . The analysis revealed not only changes in bacterial populations (increase of Succinivibrio, Coriobacteriaceae, Enterococcaceae, Rikenellaceae) but also a significant increase in Candida glabrata levels and various unclassified fungi. Luo et al. (Ref. Reference Luo, Ru, Mirzaei, Xue, Peng, Ralser, Mejías-Luque, Gerhard and Deng69) observed a striking transformation in the intestinal viral community of mice with H. pylori -induced CRC. Specifically, the infection led to an increased frequency of viral operational taxonomic units (vOTUs) from the Caudoviricetes, Malgrandaviricetes and Faserviricetes classes. These changes in viral populations suggest an intricate interplay between viral dynamics and cancer progression (Ref. Reference Luo, Ru, Mirzaei, Xue, Peng, Ralser, Mejías-Luque, Gerhard and Deng69). The connection between these microbial shifts and disease progression is further illuminated by studies linking temperate phages (which can replicate either lysogenically as prophages or lytically by producing new viral particles) to the worsening of dysbiosis-related diseases such as IBD and CRC. It is thought that the rise in these phages contributes to the disruption of the gut microbiome through immune responses triggered by bacterial lysis and the release of phage progeny (Refs Reference Luo, Ru, Mirzaei, Xue, Peng, Ralser, Mejías-Luque, Gerhard and Deng69, Reference Clooney, Sutton, Shkoporov, Holohan, Daly, ’Regan O, Ryan, Draper, Plevy, Ross and Hill275, Reference Duerkop, Kleiner, Paez-Espino, Zhu, Bushnell, Hassell, Winter, Kyrpides and Hooper276). This process can lead to the release of PAMPs and damage-associated molecular patterns (DAMPs), which in turn trigger inflammatory cytokine production, including IL-12, IL-6, IL-10 and IFN-γ-induced protein 10. These inflammatory signals can exacerbate intestinal inflammation and dysbiosis, contributing to the progression of diseases like CRC and IBD (Refs Reference Kumar, Kawai and Akira277–Reference Braumüller, Mauerer, Andris, Berlin, Wieder and Kesselring279).
Together, these findings indicate that H. pylori infection can alter gut microbial communities in ways that may influence intestinal inflammation and CRC risk. Effective management of H. pylori may therefore have implications beyond gastric health, though this possibility requires further clinical investigation.
Translational implications: probiotics as a therapeutic and preventive strategy
While the protective effects of H. pylori eradication against gastric cancer are well documented, the impact of this intervention on the risk of subsequent CRC remains ambiguous (Ref. Reference Wang, Yang, Moore, Nimmo, Lightfoot and Huycke280). Currently, there is a scarcity of studies investigating the relationship between H. pylori eradication and CRC development, highlighting a significant gap in our understanding of this potential association. In this regard, the study conducted by Guo et al. (Ref. Reference Guo, Zhang, Jiang, Wang, Chen, Zhang, Zhou, Zhang and Leung128), which involved a follow-up of about 10 years, found that treatment for H. pylori significantly reduces the rate of CRC among patients. This suggests that treating H. pylori may provide additional benefits in lowering the risk of CRC. A follow-up investigation of a randomized controlled trial focused on the treatment of H. pylori and its impact on gastric cancer revealed that eradicating H. pylori is linked to a decreased long-term risk of death specifically from CRC. This association remained significant even after adjusting for initial gastric tissue conditions, with a hazard ratio: 0.25, 95% CI: 0.07–0.89 (Ref. Reference Li, Zhang, Ma, Li, Zhang, Zhang, Guo, Zhou, Li, Shen and Liu281). Research has demonstrated that antibiotic treatments for H. pylori lead to a decrease in the alpha diversity of gut microbiota. Following the successful eradication of H. pylori , there is an increase in bacteria that produce short-chain fatty acids, which have been associated with anti-cancer effects against CRC (Refs Reference Dash, Khoder, Nada and al Bataineh267, Reference Guo, Zhang, Gerhard, Gao, Mejias-Luque, Zhang, Vieth, Ma, Bajbouj, Suchanek, Liu, Ulm, Quante, Li, Zhou, Schmid, Classen, Li, You and Pan282, Reference Hou, Chen, Zhang, Zhang, Liu, Wang, Dai, Wang, Zhong and Cao283).
The standard treatment for H. pylori infection conventionally consists of antibiotic-based eradication therapy combined with acid suppression, commonly using a proton pump inhibitor together with two antibiotics, such as clarithromycin plus amoxicillin or metronidazole (Refs Reference Parreira, Fátima Duarte, Reis and Martins284, Reference Sorbara and Pamer285). Although eradication may provide important clinical benefits, it also has potential drawbacks. Antibiotic-based therapy can temporarily disturb gut microbiota composition, reduce beneficial commensal bacteria and cause gastrointestinal side effects such as diarrhoea, abdominal discomfort and antibiotic-associated dysbiosis (Refs Reference Dash, Khoder, Nada and al Bataineh267, Reference Guo, Zhang, Gerhard, Gao, Mejias-Luque, Zhang, Vieth, Ma, Bajbouj, Suchanek, Liu, Ulm, Quante, Li, Zhou, Schmid, Classen, Li, You and Pan282). In addition, widespread or repeated eradication therapy may contribute to antibiotic resistance, particularly resistance to clarithromycin and metronidazole, which can reduce treatment success and limit future therapeutic options (Refs Reference Parreira, Fátima Duarte, Reis and Martins284, Reference Sorbara and Pamer285). Therefore, eradication should not be viewed as a universally risk-free intervention. Treatment decisions should consider the clinical indication, individual cancer risk, previous antibiotic exposure, local resistance patterns and possible microbiome-related consequences.
The rise of antibiotic resistance in H. pylori and other pathogens has led to increased interest in alternative approaches. This has intensified interest in microbiome-based strategies for disease prevention and treatment. Studies have highlighted the potential of beneficial microorganisms to promote health and protect against various conditions (Refs Reference Pandey, Naik and Vakil286, Reference Nemati, Omrani, Ebrahimi and Montazeri-Najafabady287). Figure 4 summarizes the proposed probiotic-related mechanisms relevant to CRC prevention and management, including immunomodulation, anti-inflammatory effects and suppression of tumour-promoting processes. Importantly, much of this evidence is derived from preclinical models and extrapolation to human CRC prevention requires careful consideration.
Schematic of probiotic mechanisms in modulating Helicobacter pylori infection and gut microbiota in CRC prevention. Certain probiotics inhibit H. pylori infection by directly targeting both the spiral and coccoid forms of the bacterium and its secreted components. Other probiotics mitigate the harmful effects of H. pylori by restoring dysregulated microbiota and enhancing the integrity of intestinal epithelial tight junctions. Created by biorender.com.

Figure 4. Long description
The diagram is divided into two main sections.
On the left, an anatomical cross-section of a gastric crypt shows the transition from a healthy state to DYSBIOSIS, indicated by a green-to-red arrow at the bottom. The crypt is lined with epithelial cells and contains a central area labeled Gut microbiota. At the top left, H. pylori spiral forms, coccoid forms, and O M V s are shown. A group of PROBIOTICS is shown entering the crypt to interact with these pathogens and the resident microbiota.
On the right, a flowchart titled Possible mechanisms lists specific probiotic strains and their actions:
* Lactobacillus gasseri O L L 2 7 1 6 leads to Conversion of H. pylori, which Prevents coccoid proliferation.
* Lactococcus lactis subsp. lactis A 1 6 4 produces Lacticin, which Reduces H. pylori population and Damages coccoid surface.
* Lactobacillus reuteri produces Reuterin, which Inhibits H. pylori colonization and adhesion.
* Lactobacillus gasseri A T C C 3 3 3 2 3 Modulates H. pylori-induced autophagy.
* Limosilactobacillus fermentum G R-3 produces S C F A, which, along with Lactobacillus plantarum, Lactobacillus salivarius, and Lactobacillus casei, leads to Restoration of gut microbiota balance and Anti-tumor effects.
* Lactobacillus rhamnosus and Escherichia coli Nissle 1 9 1 7 act on tight junction proteins J A M, Occludin, and Claudin to Enhances intestinal integrity.
Fujimura et al. (Ref. Reference Fujimura, Watanabe, Kimura and Kaji288) showed that simultaneous cultivation of H. pylori and the probiotic Lactobacillus gasseri OLL2716 for 24 hours resulted in a noticeable transformation of the bacteria from their spiral form to a coccoid shape. Furthermore, attempts to re-cultivate the coccoid form on sheep blood agar plates were unsuccessful. In another study, researchers induced the coccoid form of H. pylori and examined the effects of Lactococcus lactis subsp. lactis A164, a probiotic isolated from kimchi. They discovered that this probiotic produces lacticin and not only reduces the microbial population of H. pylori but also damages the surface of its coccoid form (Ref. Reference Kim, Hur, Yu, Cheigh, Kim, Hwang and Pyun289). The shift towards a non-cultivable state suggests functional attenuation of H. pylori and underscores the relevance of targeting this persistent morphology, given its proposed role in chronicity and microenvironmental alterations associated with colorectal carcinogenesis. Mukai et al. (Ref. Reference Mukai, Asasaka, Sato, Mori, Matsumoto and Ohori290) emphasize the potential of Lactobacillus (Limosilactobacillus) reuteri to inhibit H. pylori by preventing its adhesion to gastric glycolipid receptors. By interfering with this early step of bacterial attachment, Lactobacillus reuteri may also have the potential to influence infection-associated inflammatory pathways; however, whether such effects could extend beyond the stomach and contribute to colorectal carcinogenesis remains speculative and requires further investigation. In vivo investigations have demonstrated that the intake of yogurt enriched with diverse strains of L. gasseri can alleviate symptoms associated with dyspepsia, mitigate inflammation of the gastric mucosa and enhance the efficacy of conventional triple therapy in individuals infected with H. pylori (Refs Reference Deguchi, Nakaminami, Rimbara, Noguchi, Sasatsu, Suzuki, Matsushima, Koike, Igarashi, Ozawa, Fukuda and Takagi291–Reference Takagi, Yanagi, Ozawa, Uemura, Nakajima, Inoue, Kawai, Ohtsu and Koga293). Lactobacillus gasseri ATCC 33323 strains affect the expression of several factors, including IL-8, β-catenin and BCL-2. They also modulate the integrin subunit α5β1 in epithelial cells exposed to clinical strains of H. pylori . These strains also inhibit autophagy through the ATG16L1-NOD1 pathway, which is triggered by H. pylori ’s OMVs, thereby helping to protect host T-cells from the activity of H. pylori (Refs Reference Sadeghloo, Saffarian, Hakemi-Vala, Sadeghi and Yadegar152, Reference Yarmohammadi, Yadegar, Ebrahimi and Zali294). Although these findings are primarily derived from gastric models of H. pylori infection, the observed modulation of inflammatory signalling, apoptosis-related pathways and β-catenin may suggest a potential, albeit indirect, relevance to CRC.
Probiotics, in addition to their advantageous effects on H. pylori – an organism linked to CRC development – demonstrate significant potential in restoring gut dysbiosis associated with CRC through a variety of mechanisms. For instance, Limosilactobacillus fermentum GR-3, sourced from traditional fermented foods, has been shown to effectively reduce colitis and lower tumour occurrence in a mouse model of CRC induced by azoxymethane and dextran sulphate sodium (AOM/DSS). This strain enhances the modulation of the gut microbiome by promoting the growth of beneficial short-chain fatty acid (SCFA) producers, such as the Lachnospiraceae NK4A136 group, while simultaneously inhibiting pro-inflammatory bacteria like Bacteroides (Ref. Reference Zhou, Wu, Khan, Hu, Wang, Salama, Su, Han, Jin and Li295). Probiotics such as Lactobacillus (Lacticaseibacillus) rhamnosus and E. coli Nissle 1917 play a vital role in microbiota composition. They strengthen intestinal epithelial integrity by promoting the expression of tight junction proteins, such as claudin-1 and occludin, to prevent CRC. These beneficial microorganisms contribute to maintaining gut barrier function, effectively reducing inflammation and protecting against oncogenic pathogens such as H. pylori , whose translocation can lead to cancer development (Refs Reference Fong, Li and Yu296, Reference Miftahussurur, Yamaoka and Graham297). In the research conducted by Hadicka et al. (Ref. Reference Hradicka, Beal, Kassayova, Foey and Demeckova298), a combination of Lactobacillus strains such as Lactobacillus plantarum VD23, C28 and MS18 and Lactobacillus (Ligilactobacillus) salivarius MS3, MS6 and MS16 demonstrated significant anti-tumour effects while restoring microbial balance in a rat model of CRC. In the investigation by Jacouton et al. (Ref. Reference Jacouton, Chain, Sokol, Langella and Bermúdez-Humarán299), the findings revealed that Lactobacillus (Lacticaseibacillus) casei BL23 altered the gut microbiome’s composition, fostering the proliferation of beneficial bacteria and diminishing the presence of pathogenic microbes. Furthermore, this probiotic influenced immune cell populations (Tregs/Th17) and cytokine production (IL-6, IL-17, IL-10 and TGF-β) within colonic tissues, highlighting its immunomodulatory capabilities. These outcomes suggest promising avenues for developing new preventive strategies against CRC.
Several limitations temper the interpretation of probiotics in CRC prevention and H. pylori management. Several limitations temper the interpretation of probiotics in CRC prevention and H. pylori management. The effects of probiotics are highly strain-specific, and results observed with one strain cannot be generalized to others, even within the same species. Additionally, factors such as dosage, formulation and bacterial viability during storage and gastrointestinal transit significantly influence their efficacy (Ref. Reference Sanders, Merenstein, Reid, Gibson and Rastall300). Furthermore, inter-individual variability in gut microbiome composition can lead to heterogeneous responses to probiotic interventions, limiting their reproducibility and clinical predictability (Ref. Reference Zmora, Zilberman-Schapira, Suez, Mor, Dori-Bachash, Bashiardes, Kotler, Zur, Regev-Lehavi, Brik, Federici, Cohen, Linevsky, Rothschild, Moor, Ben-Moshe, Harmelin, Itzkovitz, Maharshak, Shibolet, Shapiro, Pevsner-Fischer, Sharon, Halpern, Segal and Elinav301). Importantly, much of the current evidence is derived from in vitro studies or animal models, with relatively limited large-scale, well-controlled clinical trials in humans. Consequently, while probiotics hold potential as adjunctive strategies, their clinical translation for CRC prevention should be approached with caution and further standardized, strain-specific clinical investigations are required to establish efficacy, optimal dosing and long-term safety.
Conclusions and future prospects
The association between H. pylori infection and CRC highlights the need to better define microbial contributions to colorectal carcinogenesis. This review highlights the multifaceted mechanisms through which H. pylori may contribute to CRC risk, including direct effects via secreted toxins, alterations in gastric physiology leading to hypergastrinaemia and indirect pathways mediated by changes in gut microbiota and immune responses. The evidence suggests that H. pylori infection may be associated with CRC-related outcomes beyond gastric disease; however, its direct contribution to colorectal carcinogenesis remains to be fully established. However, because much of the current human evidence is observational, causal inferences should be made cautiously in the presence of potential confounding and bias. Given the reported associations between H. pylori infection and CRC-related outcomes, future research should prioritize several key areas. First, it is hypothesized that H. pylori -derived OMVs serve as long-distance vehicles that transport CagA and VacA specifically to colonic crypt stem cells, triggering early epigenetic shifts before visible dysbiosis occurs. Testing this could clarify whether and how a gastric pathogen may contribute to distal colorectal tumour-promoting processes. Second, we propose a ‘synergistic niche’ hypothesis: the morphological shift to coccoid forms in the colon may alter the local pH and metabolite profile, specifically favouring the growth and establishment of pro-tumorigenic bacteria. Investigating these molecular interactions will move the field from observational associations to mechanistic certainty.
Furthermore, there is a pressing need for longitudinal studies that assess the impact of H. pylori eradication on CRC incidence and progression. Such studies could provide valuable insights into whether early intervention can mitigate CRC risk in populations with high prevalence rates of H. pylori infection. Additionally, exploring the gut microbiome’s role in modulating the effects of H. pylori is essential. As emerging evidence suggests that dysbiosis may influence CRC development, understanding how H. pylori interacts with other microbial communities could reveal novel therapeutic targets for prevention and treatment. Finally, public health strategies should consider the implications of these findings by integrating screening for H. pylori into CRC prevention programmes, especially in high-risk populations. By addressing both microbial and lifestyle factors associated with CRC, we can develop more effective strategies for reducing its incidence and improving patient outcomes.
In summary, while significant progress has been made in understanding the relationship between H. pylori and CRC, much remains to be discovered. Continued interdisciplinary research efforts will be vital in unravelling these complex interactions and translating findings into clinical practice to combat one of the leading causes of cancer mortality worldwide. A deeper understanding of H. pylori –host interactions could revolutionize CRC management by enabling precision diagnostics, such as microbial and host genetic profiling to stratify high-risk patients, and targeted therapies disrupting virulence factors like CagA or VacA to halt tumorigenesis. These personalized strategies, integrating microbiome dynamics and host susceptibility, hold transformative potential to reduce CRC burden and improve outcomes in high-prevalence populations.
Abbreviations
- AP
-
adenomatous polyp
- APC
-
adenomatous polyposis coli
- ATG
-
antithymoglobulin
- Bcl-2
-
B-cell lymphoma-2
- CARD10
-
caspase recruitment domain 10
- CagA
-
cytotoxin-associated gene A
- Cag PAI
-
cag pathogenicity island
- CI
-
confidence interval
- COX-2
-
cyclooxygenase 2
- CP
-
colorectal polyp
- CRC
-
colorectal cancer
- CTLA-4
-
cytotoxic t-lymphocyte-associated protein 4
- DC
-
dendritic cell
- ECP
-
eosinophil cationic protein
- EMT
-
epithelial–mesenchymal transition
- ERK
-
extracellular signal-regulated kinase
- FoxP3
-
Forkhead box protein 3
- GC
-
gastric cancer
- HcpC
-
Helicobacter cysteine-rich protein
- HP
-
hyperplastic polyp
- IBD
-
inflammatory bowel disease
- ICAM-1
-
intracellular adhesion molecule 1
- IFN-γ
-
interferon-gamma
- IL
-
interleukin (e.g. IL-1β, IL-6, IL-8, IL-10, IL-17)
- IRAK1
-
interleukin 1 receptor-associated kinase 1
- JAK-STAT3
-
janus kinase signal transducer and activator of transcription 3
- LAMP2
-
lysosome-associated membrane protein 2
- LC3
-
microtubule-associated protein light chain 3
- lncRNA
-
long non-coding RNA
- LPS
-
lipopolysaccharide
- MAPK
-
mitogen-activated protein kinase
- miRNA
-
MicroRNA
- MTOR
-
mechanistic target of rapamycin
- MTORC1
-
mechanistic target of rapamycin complex 1
- NADPH
-
nicotinamide adenine dinucleotide phosphate
- NF-κB
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
-
nitric oxide
- NOD1
-
nucleotide-binding oligomerization domain-containing protein 1
- OMP
-
outer membrane protein
- OMV
-
outer membrane vesicle
- OR
-
odds ratio
- PAMP
-
pathogen-associated molecular pattern
- PD-L1
-
programmed cell death ligand 1
- PG
-
peptidoglycan
- PP
-
Peyer’s patch
- PRR
-
pattern recognition receptor
- RAB7
-
Ras-related protein 7
- RECK
-
Reversion-inducing-cysteine-rich protein with Kazal motifs
- ROS
-
reactive oxygen species
- SabA
-
sialic acid-binding adhesin
- SCFA
-
short-chain fatty acid
- SMAD2
-
Sma- and Mad-related protein 2
- SQSTM1
-
sequestosome protein 1
- SSA
-
sessile serrated adenoma
- T4SS
-
type IV secretion system
- TA
-
tubular adenoma
- TGF-β
-
transforming growth factor beta
- TGF-βR2
-
transforming growth factor Beta receptor 2
- TIMP-1
-
tissue inhibitor of metalloprotease 1
- TLR4
-
Toll-like receptor 4
- TNM
-
tumour, node, metastasis
- Treg
-
regulatory T-cell
- TSA
-
traditional serrated adenoma
- TVP
-
tubulovillous polyp
- ULK1
-
UNC-like autophagy-activating kinase 1
- VacA
-
vacuolating cytotoxin A
- VBNC
-
viable but non-cultivable
- vOTU
-
viral operational taxonomic unit
- VP
-
villous polyp
- ZEB1
-
zinc-finger E-box-binding homeobox 1
- ZFAS1
-
zinc finger antisense 1.
Acknowledgements
We used ChatGPT-5 to assist with paraphrasing and improving the clarity of the English language in this manuscript; no scientific content was generated by the tool.
Authors contribution
Z.S. and E.N.H.: conceptualization, writing original draft, visualization (figures and tables), critical revision of manuscript. M.A.L. and M.E.: involved in data collection and analysis. G.C., S.R.: writing – review and editing, visualization (figures and tables), critical suggestions. A.S.: writing – review and editing. All authors have read and approved the final version of the manuscript.
Funding statement
The authors have not disclosed any funding.
Competing interests
The authors declare that they have no competing interests.
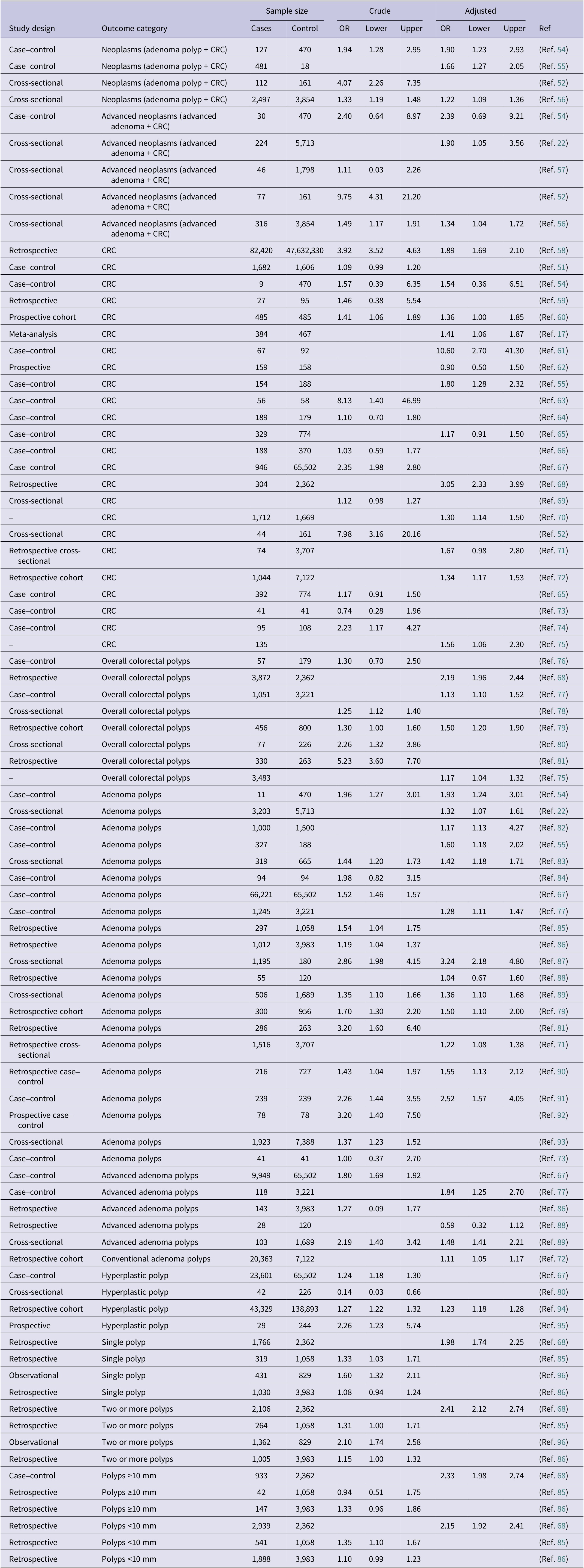
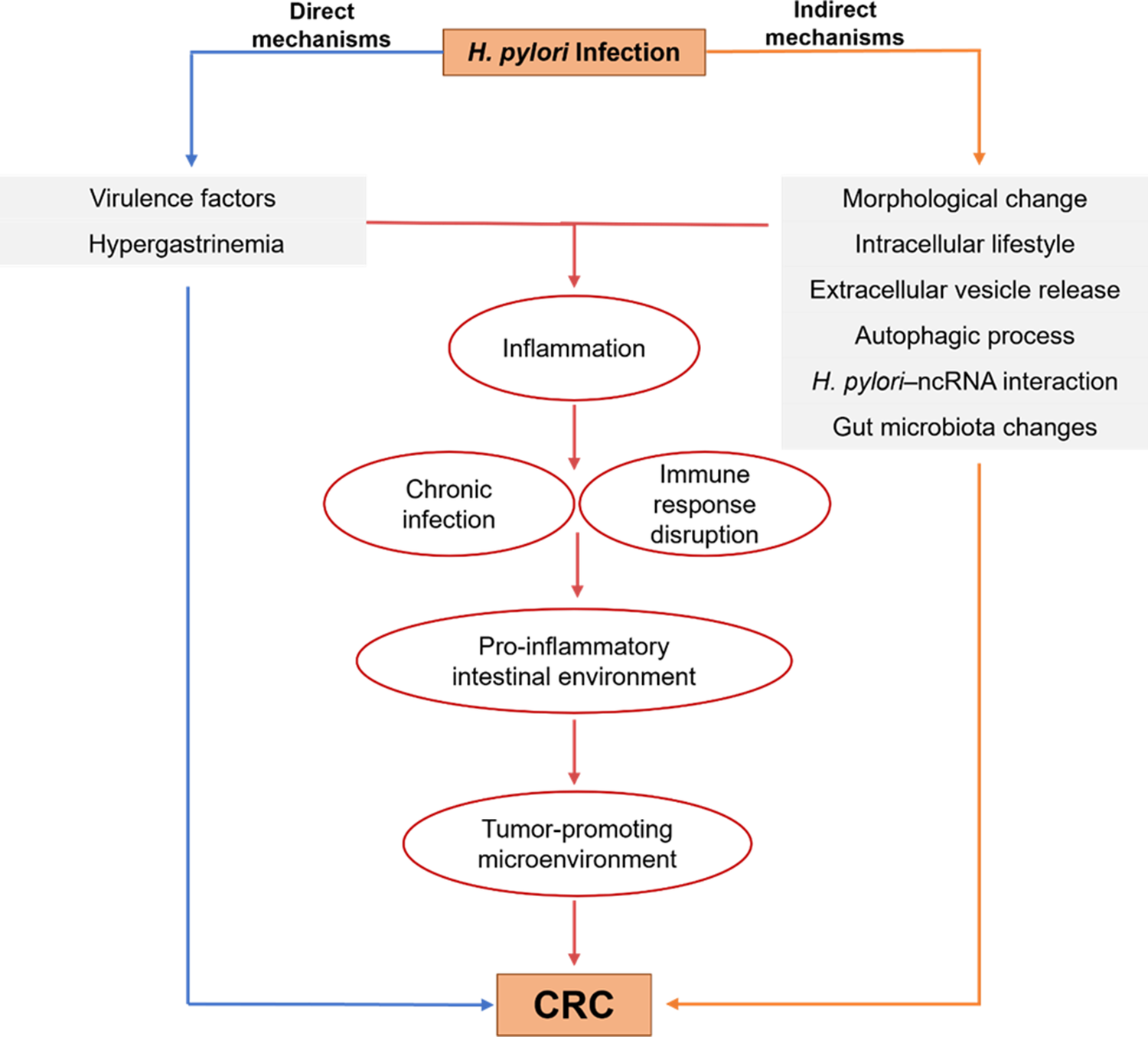
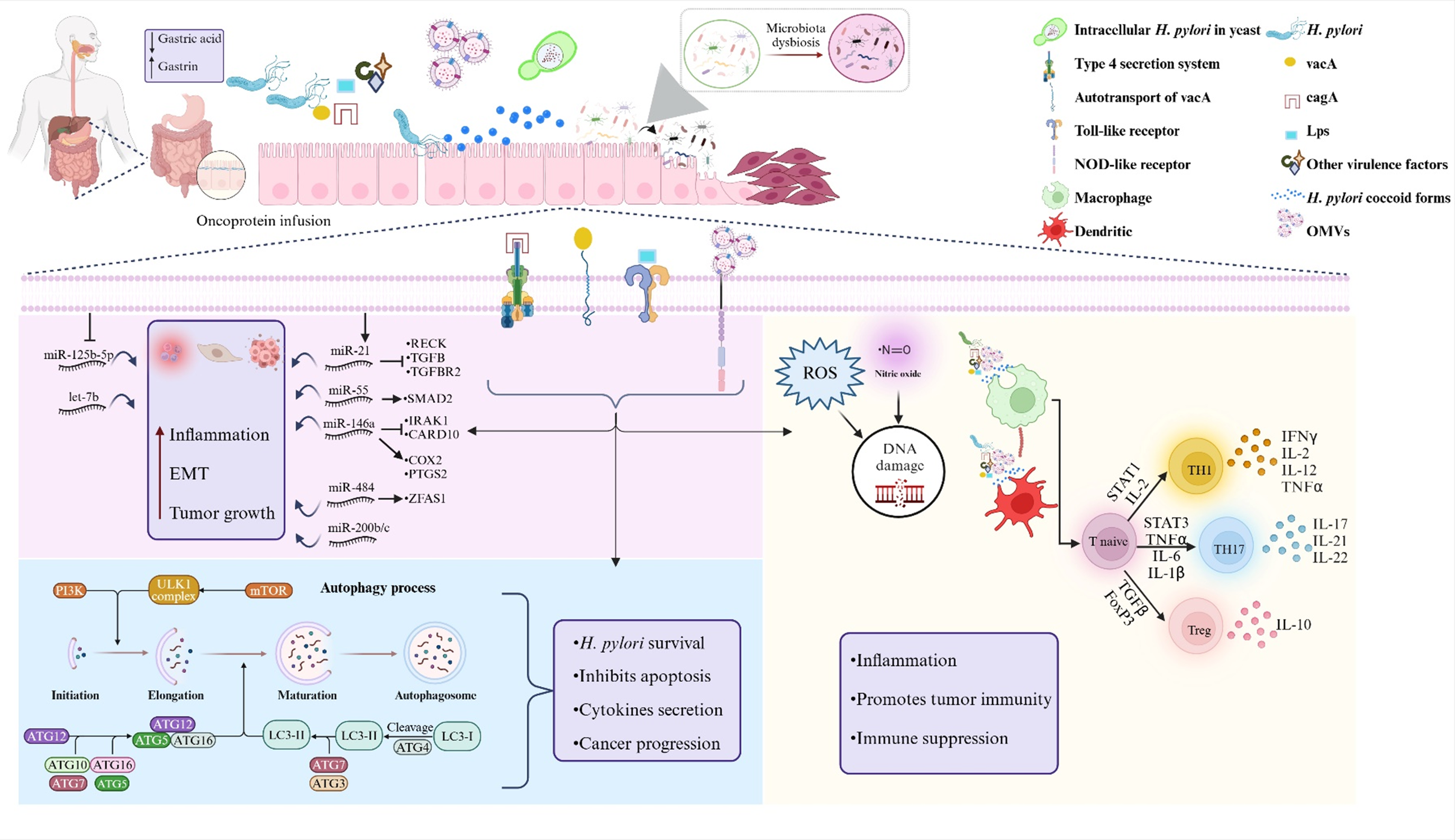
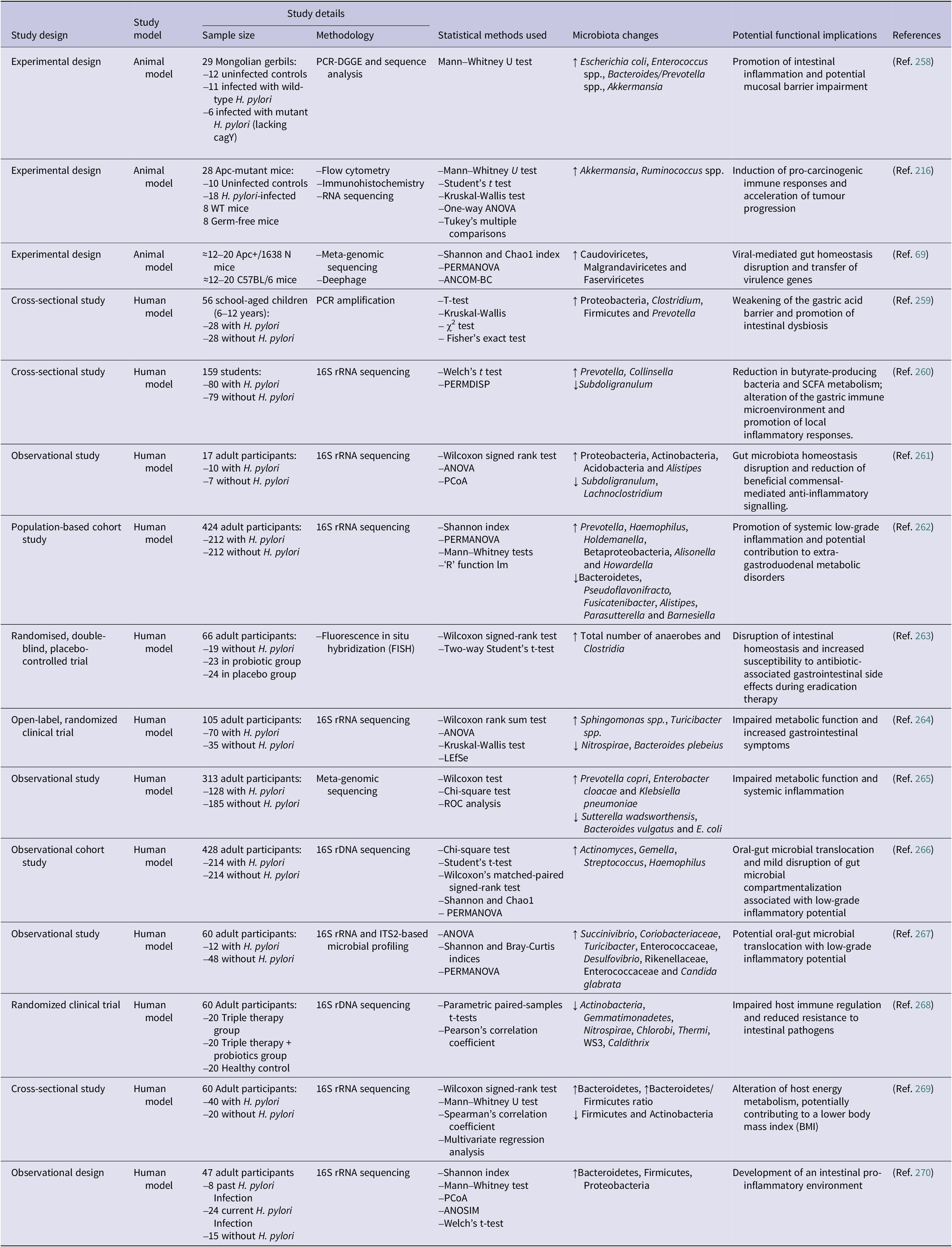

 Open access
Open access