


Introduction
Long QT syndrome (LQTS) is an inherited or acquired arrhythmia syndrome that may co-occur with malignant sudden cardiac death. It is characterised by the prolongation of QT interval and an enhanced threat of ventricular arrhythmia (Refs Reference Moss1, Reference Roden2). LQTS is primarily divided into congenital and acquired types. Congenital LQTS is mainly attributed to mutations in ion channels and their accessory proteins. Acquired LQTS is primarily caused by various pharmacological agents (e.g., antibiotics, antidepressants, antihistamines, antineoplastics).
Seventeen genes have been reported to be associated with congenital LQTS. Mutations in three common genes (KCNQ1, KCNH2 and SCN5A) account for more than 75% of clinically validated congenital LQTS cases, whereas less than 5% have been attributed to mutations in other genes (Ref. Reference Tester and Ackerman3). Genetic aetiologies for the remaining approximately 20% of hereditary LQTS are unknown. Intriguingly, a clinical study revealed that the identical LQTS-relevant mutations are not necessarily linked to the same disease phenotype (Ref. Reference Priori, Napolitano and Schwartz4). Currently, effective treatment options for LQTS are beta-blockers (propranolol, nadolol, etc.), sodium channel blockers (e.g., mexiletine, flecainide, etc.), and surgical interventions that include left cardiac sympathetic denervation (LCSD) and implantable cardioverter-defibrillator (ICD) (Ref. Reference Etheridge and Cohen5).
In this review, we introduce the clinical manifestations and diagnosis of LQTS. We discuss the molecular basis of ion channels underlying congenital LQTS and explore the potential of modelling LQTS using patient-specific induced pluripotent stem cell (iPSCs). We then summarise the current treatment strategies for LQTS and speculate on the future of precision medicine in LQTS through utilising patient-specific iPSCs and whole-genome sequencing.
Clinical manifestations and diagnosis of LQTS
Clinical symptoms of LQTS may be present at birth or develop later in life. Severe symptoms may occur because of an irregular heartbeat that can lead to fainting, seizures or sudden cardiac death. LQTS patients show high risks of cardiac arrhythmia events, including ventricular tachycardia and torsades de pointes, which predisposes transient syncope and further deteriorates into ventricular fibrillation (Ref. Reference Schwartz, Crotti and Insolia6). Most symptomatic LQTS patients suffer from primary arrhythmic events with a notable difference in gender, age and health differences (Ref. Reference Schwartz7). An array of stimuli is correlated with LQTS, including running, swimming, unforeseen noises and being scared. Patients with LQTS1 are threatened with upregulated synergistic activities, emotional dysregulation and physical burdens (Refs Reference Schwartz7, Reference Ackerman, Tester and Porter8). LQTS2 patients are susceptible to low plasma potassium levels, with female patients in the postpartum period are vulnerable to sleep disturbance (Ref. Reference Khositseth9). For LQTS3 patients, most cardiac arrhythmic events occur during rest or sleep (Ref. Reference Perez-Riera10).
To monitor the electrical activity within each heartbeat, an electrocardiogram (ECG) of the electrical signal travelling through the heart is recorded. A normal heartbeat has five separate electrical waves (Fig. 1a): P, Q, R, S and T. The P wave corresponds to the process of atrial depolarisation that extends from the SA node throughout the atria. Subsequent QRS wave combination represents the ventricular depolarisation process. The subsequent ST-segment is considered the isoelectric period in which the T wave indicates the process of ventricular repolarisation. The occasionally exhibited U wave is a tiny, circular deflection that follows the waves of depolarisation (QRS) and repolarisation (T) (Ref. Reference Ritsema van Eck, Kors and van Herpen11). U waves reflect the last step of ventricular repolarisation from a functional standpoint. Although the origin of the U wave is still a matter of debate, most clinicians and researchers associate it with ‘afterdepolarisations’ in the ventricle.
Characteristic summary of ion channels in LQTS. (a) A representative pattern of normal ECG. (b) A schematic view of APD in normal ventricular cardiomyocytes. (c) A representative pattern of LQTS-specific ECG. (d) A schematic view of APD in LQTS ventricular cardiomyocytes. INa: sodium currents; Ito: transient outward potassium currents; ICa,L: L-type calcium current; IKs: slow delayed rectifier potassium current; IKr: rapid delayed rectifier potassium current; IK1: inward rectifier potassium current.

The QT interval is the overall duration of the ventricular depolarisation and repolarisation processes, which is inversely proportional to the heart rate. Compared to those with a normal heart rhythm, ECGs of LQTS patients show a prolonged QT interval (Fig. 1b). The overall ECG pattern is largely determined by electrophysiological profiles of each ventricular cardiomyocyte in the heart. In a ventricular cardiomyocyte, an action potential (AP) is produced by the coordinated activation (open) and inactivation (close) of several membrane-embedded ion channels (Fig. 1c) (Ref. Reference Kanno and Saffitz12). The initial upstroke of the cardiac AP is orchestrated by the activation and inactivation of sodium channels, followed by the repolarisation that is conducted by many distinctive genres of potassium channels. Initial repolarisation is directed by the efflux of transient outward potassium currents (Ito). Subsequently, a plateau stage is achieved by the flow of outward currents through opening delayed rectifier potassium channels (ultra-rapid: IKur, rapid: IKr, slow: IKs) and the counteracting force driven by L-type calcium channels. The terminal stage of the repolarisation is operated by rapid delayed rectifier potassium currents (IKr) and inward rectifier potassium currents (IK1). The combined dysregulated activities of several ion channels extend the action potential duration (APD), resulting in the QT interval prolongation (Fig. 1d).
Diagnosis of LQTS depends on a broad spectrum of factors including heart rate corrected QT intervals (QTc) and electrocardiographic parameters (Ref. Reference Wilde, Amin and Postema13). The upper values of normal QTc are 440 ms for males and 460 ms for females, respectively (Ref. Reference Schwartz14). To diagnose LQTS, accurate measurements of QTc duration at rest are required (Ref. Reference Schwartz14). Regarding electrocardiographic parameters, the abnormal shape of the T-wave is a common phenomenon in LQTS patients. Precordial leads are particularly instructive in reflection of biphasic or notched T-wave. From clinical perspective, patients with different types of LQTS exhibit distinct patterns of ST-T segment (Refs Reference Moss15, Reference Van Langen16). ECG of LQTS1 is characterised by a broad T wave and prolonged QT interval (Ref. Reference Modi and Krahn17). ECG in LQTS2 present the notched asymmetrical T wave and low amplitude (Ref. Reference Modi and Krahn17). LQTS3 patients are predisposed to encounter cardiac episodes and QT during rest (Ref. Reference Schwartz7). LQTS3 may present as a prolonged isoelectric interval following a normal T wave shape on the ECG (Ref. Reference Modi and Krahn17).
Based on QTc duration, precordial leads and a series of criteria, Schwartz et al. and Priori et al. established the scoring system to stratify the risks associated with LQTS (Refs Reference Schwartz and Ackerman18, Reference Priori19). In addition, genetic screening is beneficial to both diagnosis of asymptomatic LQTS and identification of the proband along with their familial members carrying causative mutations (Ref. Reference Schwartz14). Even acquired LQTS has been shown to be associated with underlying genetic factors (Ref. Reference Itoh20). Therefore, genetic screening is essential for deciphering the relationship between genotype and phenotype, which can potentially explore the unidentified LQTS relevant mutations.
Although QT interval prolongation is the defining characteristic of LQTS, roughly 40% of genetically verified LQTS patients have a normal QTc at rest (Ref. Reference Bos21). It is crucial to distinguish individuals with LQTS from those with a normal QTc to properly diagnose the illness and put effective LQTS preventative measures into place. Another confounding factor in diagnosing LQTS is the subjective determination of Q and T wave boundaries. To overcome these above-mentioned obstacles, recent studies using machine learning (ML) and deep learning (DL) have shown an improvement in the diagnosis of LQTS (Refs Reference Bos21, Reference Hermans22). Prifti et al. developed an artificial intelligence (AI)-driven approach to identify ECG patterns alternations that are indicatives of drug-induced LQTS as well as congenital LQTS (Ref. Reference Prifti23). Their algorithm identified ECGs of LQTS2 patients with excellent accuracy; ECGs of LQTS1 and LQTS3 patients were also detected, albeit with lower accuracy than LQTS2. It is possible that rule-based expert systems may be eventually replaced by AI someday since DL methodologies can delineate the intricate patterns of ECGs. One challenging aspect of using DL to implement LQTS diagnosis is the need for large-scale training datasets to generate high-quality and unbiased results. Another major obstacle is the interpretability of the findings as the non-linear characteristics of deep neural networks are often difficult to comprehend.
Identification of disease-specific mutations enables clinicians to accumulate invaluable data for future diagnosis and precision treatment. A clinical trial (NCT04581408) is currently underway to test the mutation-specific treatment for LQTS2. The most common genetic causes of LQTS are associated with mutations in KCNQ1, KCNH2 and SCN5A (Ref. Reference Priori24). Identification of specific locations of genetic mutations within ion channels has proven beneficial in predicting the onset of LQTS (Ref. Reference Moss25). From prognostic studies, clinicians can explicitly educate LQTS patients to avoid exposure to LQTS triggering factors as mentioned before. Therefore, genotyping may aid in devising alternative strategies in combination with other treatment options (Ref. Reference Makita26). Because it is likely to identify irrelevant genetic variants of LQTS-related genes in asymptomatic populations, symptomatic individuals and those with a familial history of LQTS should be prioritised to receive genetic testing. Clinical manifestations have the potential to become a reliable way to classify pathogenic variants in LQTS. For instance, using ECG T waves morphology, we can classify the pathogenic genetic variants through unique pattern recognition. A broad-based T wave is frequently observed in LQTS1 patients carrying KCNQ1 variants. Low-amplitude notched T waves and late-onset peaked/biphasic T waves are ECG features of LQTS2 individuals bearing KCNH2 variants and LQTS3 subjects bearing SCN5A variants, respectively. CALM1-3 mutation carriers exhibit bradycardia or atrioventricular block along with severe QT prolongation, which is seldom observed in other LQTS subtypes. A patient with a loss-of-function variant in TRDN exhibited negative T waves and QT prolongation in precordial leads (Ref. Reference Altmann27). KCNJ2 mutations were found in patients with prominent U-waves without prolonged QT, suggesting a phenotypic manifestation of Andersen-Tawil Syndrome. Extracardiac signs of Andersen-Tawil Syndrome include dysmorphic features, recurrent paralysis and neurocognitive deficits (Ref. Reference Perez-Riera28). Paediatric patients with certain CACNA1C variants demonstrated a spectrum of electrophysiological abnormalities including bradycardia, atrioventricular block, QT prolongation and polymorphic ventricular arrhythmias, which is correlates with Timothy Syndrome (Ref. Reference Splawski29). Nine (AKAP9, ANK2, CAV3, KCNE1, KCNE2, KCNJ2, KCNJ5, SCN4B, SNTA1) of the seventeen genes previously described as pathogenic LQTS variants were recently classified to have insufficient or disputed evidence for disease causation (Ref. Reference Adler30). Due to the complexity of the genetic architecture of LQTS (Refs Reference Adler30, Reference Ingles and Semsarian31, Reference Roberts32), it is not recommended to solely utilise genetic variation for clinical decision-making until they are validated by solid clinical evidence.
Molecular basis of ion channel abnormalities underlying congenital LQTS
Major LQTS genes
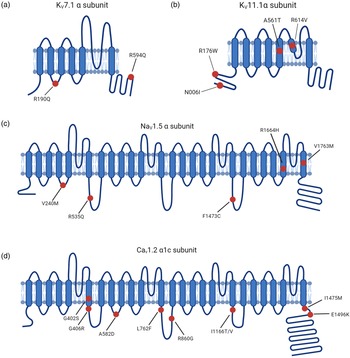
LQTS1 is the most common type of congenital LQTS, accounting for 30%-35% of all LQTS cases. KCNQ1 encodes α-subunit of voltage-gated potassium channel Kv7.1, generating slow delayed rectifier potassium currents (IKs) (Ref. Reference Chen, Sampson and Kass33). Missense mutations in Kv7.1 lead to reduced IKs, thus prolonging cardiac repolarisation. Mutations in voltage-sensing domains of KCNQ1 are frequently observed in LQTS1 (Ref. Reference Huang34). The dynamic balance of this membrane protein is achieved by forward trafficking and retrieval transport. Due to lack of proper forward trafficking, KCNQ1 mutants exhibit reduced presence on the plasma membrane (Fig. 2a) (Ref. Reference Moretti35). Moreover, incomplete removal of the extracellular loop and helix S6 by heterozygous exon 7 deletion mutations in KCNQ1 leads to defective trafficking and APD prolongation (Ref. Reference Ma36). IKs are upregulated in response to beta-adrenergic stimulation through the activation of protein kinase A (PKA). Missense mutant A341 V-Kv7.1 exhibited attenuated upregulation of IKs after stimulation due to insensitivity to PKA activation (Ref. Reference Heijman37). Furthermore, missense variants (G189R, R190Q, R243C and V254 M) located in the cytoplasmic loops were found to disrupt PKA regulation of Kv7.1 activity in the context of beta-adrenergic stimulation (Ref. Reference Barsheshet38). The extremely rare autosomal recessive Jervell and Lange-Nielsen Syndrome 1 is caused by homozygous or compound heterozygous mutations in KCNQ1 and is characterised by prominent QT prolongation and bilateral sensorineural hearing loss, which is a consequence of complete loss of IKs (Refs Reference Giudicessi and Ackerman39, Reference Splawski40).
LQTS pathogenic hotspots in the subunits of representative ion channels including Kv7.1, Kv11.1, Nav1.5 and Cav1.2. (a) Predicted topology of Kv7.1 α subunit with the location of mutations associated with LQTS1. (b) Predicted topology of Kv11.1 α subunit with mutation sites related to LQTS2. (c) Predicated topology of Nav1.5 α subunit with the location of genetic mutations associated with LQTS3. (d) Predicted topology of Cav1.2 α1c subunit with the mutation positions related to LQTS8. Red dots indicate potential mutation positions in the respective ion channel proteins.

LQTS2 accounts for 25–30% of all LQTS cases and is caused by mutations in KCNH2, which encodes α-subunit of voltage-gated potassium channel Kv11.1. KCNH2 contributes to the rapid delayed rectifier potassium currents (IKr) (Ref. Reference Chen, Sampson and Kass33). KCNH2 mutations are categorised according to the molecular mechanism by which they reduce IKr (Ref. Reference Delisle41). Class 1 is comprised of nonsense mutations that impair the production of the Kv11.1 channel subunits by nonsense mediated RNA decay (Ref. Reference Gong42). Class 2 consists of missense mutations that disrupt the intracellular transport of full-length Kv11.1 to the cell plasma membrane (Ref. Reference Anderson43). Abnormal Kv11.1 gating kinetics are identified in class 3 mutations, which are predicted to decrease the open probability in repolarisation phase (Ref. Reference Chen44). Permeation of Kv11.1 is impaired in class 4 mutations due to a presumable blockade of K+ selectivity pore, leading to a diminished IKr (Ref. Reference Anderson43). Heterozygous KCNH2 mutations exert a dominant-negative effect on KCNH2 channel-associated IKr by interfering with trafficking and impairing the heterodimer assembly (Fig. 2b) (Ref. Reference Mehta45). Mutations within the Kv11.1 lead to reduced IKr, thus prolonging the repolarisation process.
LQTS3 is attributed to gain-of-function mutations in SCN5A, which encodes α-subunit of cardiac sodium channel Nav1.5 and mediates the sodium current underlying the depolarisation phase of the cardiac AP (Ref. Reference Fatima46). A broad spectrum of auxiliary proteins acts as part of a macromolecular complex that controls Nav1.5 activity (Ref. Reference Abriel47). Gain-of-function mutations in SCN5A primarily impair cardiac sodium channel inactivation and are associated with LQTS3 (Fig. 2c) (Ref. Reference Ma48).
Minor LQTS genes
A-kinase anchoring proteins (AKAPs) are scaffolding proteins that propagate signalling molecules and deliver them to downstream sites for modulating phosphorylation. AKAP9-S1570L mutation has been identified in LQTS patients and interferes with KCNQ1 phosphorylation and reduces the sensitivity of IKs channel to cAMP stimulation (Ref. Reference Chen49). In the heart, macromolecular complexes controlled by AKAPs coordinate the phosphorylation status of L-type calcium channel (Ref. Reference Hulme50) and slow delayed rectifier IKs channel (Ref. Reference Marx51).
CACNA1C mutations outside of exon 8 have been reported in non-syndromic LQTS patients (Ref. Reference Boczek52). Substitutions at positions 582, 762 and 860 slow the inactivation process of Cav1.2, thus increasing the amplitude of ICa,L (Fig. 2d) (Refs Reference Fukuyama53, Reference Landstrom54). Changes in the activation of Cav1.2 triggered by mutations at 1166, 1475 and 1496 positions also enlarge the ICa,L (Fig. 2d) (Ref. Reference Wemhoner55). Gain-of-function mutations upregulate Cav1.2 activities and delay the repolarisation phase, leading to prolonged APD in LQTS.
CAV3 is responsible for caveolin 3 biosynthesis and mutations in CAV3 are seen in LQTS. Caveolin 3 is a vital constituent of caveolae that are embedded in the plasma membrane and involved in endocytosis and lipid homoeostasis. Caveolin 3 is coupled to the α subunit of Nav1.5, which regulates the depolarising INa current. Disruption of the association between caveolin 3 and Nav1.5 leads to CAV3-LQTS and sudden infant death (Refs Reference Cronk56, Reference Vatta57, Reference Arnestad58). Previous studies on the downregulation of KCNH2-mediated IKr under low extracellular K + suggest that caveolin proteins are required in the endocytosis of KCNH2 (Ref. Reference Massaeli59).
Mutations in KCNE1 account for less than 1% of known LQTS cases (Ref. Reference Splawski60). KCNE1 regulates both the voltage-gated slowly activating potassium channel and the rapidly activating potassium channel (Refs Reference Sanguinetti61, Reference Barhanin62). KCNE1 mutations may cause sensorineural hearing loss in Jervell and Lange-Nielsen Syndrome 2 (Ref. Reference Tyson63). A common variant (D85N) in KCNE1 (Ref. Reference Tesson64) delays IKs channel opening in Xenopus oocytes and displays severe loss-of-function effects on IKs and IKr in CHO cells (Ref. Reference Nishio65). A considerable extension of QT interval is also associated with the KCNE1 variant D85N. Moreover, the D85N variant is found in severe LQTS3 patients and some individuals with drug-induced LQTS (Ref. Reference Westenskow66).
KCNE2 encodes an accessory subunit (β-subunit) that acts as a modulator of a variety of voltage-gated potassium channels (Ref. Reference Abbott67). Recent studies indicate that rare loss-of-function KCNE2 variants are likely to confer proarrhythmic susceptibility instead of inducing LQTS6 phenotype (Ref. Reference Roberts68).
KCNJ5 encodes cardiac G-protein-coupled inward rectifier potassium channel 4 (Kir3.4) (Ref. Reference Yang69). LQTS patients with KCNJ5 mutations show peak QT interval prolongation, a right-skewed T wave and abnormal repolarisation durations (Ref. Reference Wang70). Kir3.4 forms a heterodimer with Kir3.1 to maintain the resting membrane potential and contributes to the last repolarisation phase. Intriguingly, loss-of-function in atria-specific Kir3.4 induces atrial fibrillation and supraventricular arrhythmia (Ref. Reference Calloe71).
SCN4B encodes the sodium voltage-gated channel β-subunit 4, which is required for the regulation of Nav1.5 sarcolemmal expression and gating. SCN4B mutations in LQTS can significantly increase the late sodium current in vitro (Ref. Reference Medeiros-Domingo72).
SNTA1 encodes the membrane adaptor protein α-1-syntrophin. Two SNTA1 mutants (A390 V and A257G) can increase the late sodium current and are associated with LQTS phenotypes (Refs Reference Ueda73, Reference Wu74). Nav1.5 is regulated by a dystrophin complex that involves the α-1-syntrophin (Ref. Reference Gavillet75). Early studies uncover a functional complex including α-1-syntrophin, neuronal nitric oxide synthase (nNOS) and calcium ATPase subtype 4b (PMCA4b) (Ref. Reference Williams76). The following studies demonstrate that α-1-syntrophin connects nNOS and PMCA4b to Nav1.5 in the heart (Ref. Reference Oceandy77). These studies indicate that α-1-syntrophin is a novel regulator of sodium current.
TRDN encodes Triadin that is involved in calcium release regulation and excitation-contraction coupling in cardiomyocytes. Loss-of-function mutations in TRDN are linked to LQTS phenotypes in five subjects with homozygous or compound heterozygous frameshift mutations (Ref. Reference Altmann27).
Multisystem LQTS genes
Ankyrin-B syndrome is caused by heterozygous loss-of-function mutations in ANK2 (Ref. Reference Mohler78) which encodes the ubiquitous ankyrin-B adaptor protein. Ankyrins are essential for the cellular organisation of various ion channels and transporters (Ref. Reference Mohler, Gramolini and Bennett79). Mutations in ANK2 that cause both QT prolongation and sinus bradycardia are found in both large families and sporadic patients with LQTS3 (Refs Reference Mohler80, Reference Mohler81).
Andersen-Tawil Syndrome is associated with mutations in KCNJ2, which encodes Kir2.1 and contributes to IK1 (Ref. Reference Plaster82). The majority of KCNJ2 mutations in Andersen-Tawil Syndrome result in dominant-negative inhibition of Kir2.1 and diminished IK1 amplitude (Refs Reference Preisig-Müller83, Reference Ai84).
Timothy Syndrome results from rare single amino acid substitutions in the exon 8 or alternatively spliced exon 8A of Cav1.2 which is encoded by CACNA1C. Specifically, mutations that replace either glycine residue 402 or 406 with large bulky side chains interfere with normal deactivation process (Fig. 2d) (Ref. Reference Depil85).
Severe recurrent infantile cardiac arrest syndrome is associated with mutations in calmodulin. Calmodulin is a sensor for the intracellular calcium level, which is encoded by three genes: CALM1, CALM2 and CALM3. Calmodulin activities play a significant role in the modulation of Cav1.2 function. Dysfunctional calmodulin activities caused by mutations in CALM genes are associated with a type of arrhythmogenic syndromes in LQTS (Ref. Reference Boczek86). Patients with LQTS were shown to have a variety of mutations (D130G, F142L and E141 G in CALM1; D96 V, N98I, N98S, D130G, D130 V, D132E, D134H and Q136P in CALM2; and D130 G and N138 K in CALM3) (Refs Reference Boczek86, Reference Crotti87, Reference Kato88). Most mutations are located in calmodulin's C-terminal Ca2 + -binding domains (EF-hands III and IV), particularly in the residues directly involved in Ca2 + binding. A plethora of evidence indicate that LQTS relevant calmodulin mutations attenuate Ca2 + binding affinity and result in a defective Ca2 + -dependent inactivation of Cav1.2, causing an increased and uncontrolled Ca2 + influx (Refs Reference Kato88, Reference Su89). The mutation locations in CALM1-3 reveals special topological domains that are susceptible to genetic variation and imply the significance of Ca2 + -binding affinity for proper function of calmodulin. Table 1 summarises the key findings for all-abovementioned LQTS associated genes.
Genetic basis of LQTS subtypes

Modelling LQTS using patient-specific iPSCs
Before the advent of the iPSC techniques, researchers mainly relied on heterologous expression systems using transformed cell lines (e.g., HEK293, CHO cells) with transgenic expression of human ion channel genes. Because these transformed cells are not relevant to human cardiomyocytes, phenotypes in heterologous expression systems could be significantly different from cardiac phenotypes in clinical manifestations. In addition, genotype-phenotype correlation is challenged due to the splicing variance in human cardiomyocytes (Refs Reference Tsuji-Wakisaka90, Reference Gong91) and absence of genetic modifiers in the heterologous system (Refs Reference Watanabe92, Reference Schwartz, Crotti and George93). Human iPSC-derived cardiomyocytes (iPSC-CMs) can overcome these challenges as they are patient-specific and clinically relevant. Human iPSC-CMs can be used to customise medical treatment and quickly test hundreds of new therapeutic molecules for cardiovascular disease (Refs Reference Del Alamo94, Reference Sharma95). A large population of human iPSC-CMs were exploited to serve as a reliable electrophysiological model for comprehensive in vitro proarrhythmia assay (CiPA) (Ref. Reference Yim96). Moreover, patient-specific iPSCs could be employed to stratify subpopulations who may respond to treatment and receive the benefits of medicine while experiencing minimal side effects (Ref. Reference Klaren and Rusyn97).
As most LQTS is caused by the cell-autonomous pathology of cardiomyocytes, iPSC-CMs are able to recapitulate abnormal electrophysiological features of LQTS-associated ion channels in the dish and provide a versatile platform for cardiac toxicity testing and novel drug discovery. Combined with CRISPR/Cas9 genome editing tools, LQTS-causing genetic variants can be corrected in diseased iPSC-CMs to study phenotype-genotype interactions using patient-specific cardiomyocytes (Ref. Reference Schwartz98). Table 2 summarises the cellular phenotypes of major subtypes of LQTS that have been recapitulated using patient-specific cardiomyocytes.
Summary of studies on modelling LQTS using patient-specific iPSCs

Patient-specific iPSCs have been utilised to create a platform where pathogenesis of LQTS is investigated in vitro. One advantage of patient-specific iPSC-CMs is the ability to evaluate the impact of genetic abnormality on the phenotypic reflection of an individual. For instance, iPSC-CMs could recapitulate most of electrophysiological characteristics seen in LQTS patients. The opportunity to investigate functions of mutated proteins directly in LQTS patient-derived cardiomyocytes could provide novel insights into molecular basis of LQTS for novel drug discovery.
Moretti et al. established a patient iPSC-based platform for modelling LQTS1 (Ref. Reference Moretti35). In iPSC-CMs derived from LQTS1 patients carrying KCNQ1-R190Q mutations, ventricular-, atrial- and nodal-like cells were categorised by specific myocyte lineage markers MLC2v, MLC2a and HCN4, respectively. The KCNQ1-R190Q mutation significantly reduced IKs peak and tail currents, thus prolonging APD in iPSC-CMs. Mutations in Arginine 190 led to abnormal expression of Kv7.1 in the plasma membrane in the KCNQ1-R190Q carrier (Refs Reference Wilson99, Reference Pan100). In addition, KCNQ1 mutation altered the IKs amplitude by adjusting the sensitivity to adrenergic activation (Ref. Reference Moretti35). β-adrenergic agonist isoproterenol increased the IKs current in healthy iPSC-CMs, which was abrogated in LQTS1 cardiomyocytes. Early after-depolarisations (EADs) were elicited in LQTS1 cardiomyocytes in the presence of isoproterenol, which recapitulated clinical observations in LQTS1 patients who are vulnerable to emotional arousal or exercise (Ref. Reference Schwartz7). As β-blockers are predominantly prescribed to treat arrhythmic symptoms, LQTS-specific iPSC-CMs in the dish also phenocopy the beneficial effects of β-blockers in suppressing arrhythmias (Ref. Reference Shah and Carter101).
Itzhaki et al. generated an iPSC model from dermal fibroblasts of a female LQTS2 patient with the KCNH2-A614 V mutation (Ref. Reference Itzhaki102). KCNH2-A614 V iPSC-CMs had prolonged repolarisation and higher susceptibility to the development of EADs. IKr amplitude was reduced in KCNH2-A614 V iPSC-CMs compared to healthy cardiomyocytes. Matsa et al. reported an iPSC model of a symptomatic and an asymptomatic carrier of the KCNH2-A561 T mutation (Ref. Reference Matsa103). A prolonged AP was found in atrial-like and ventricular-like cardiomyocytes but not in pacemaker cells differentiated from KCNH2-A561 T iPSCs. Lahti et al. generated iPSC-CMs from an asymptomatic carrier with R176W mutation in KCNH2 and ventricular-like cardiomyocytes showed prolonged APD (Ref. Reference Lahti104). Point mutations (A614 V, A561 T and R176W) in KCNH2 interfered with channel complex assembly and chaperone interaction, resulting in abnormal degradation or defective transport to the plasma membrane (Refs Reference Itzhaki102, Reference Matsa103, Reference Lahti104). Additionally, Chai et al. identified modifier genes responsible for differential severity in an LQTS2 family (Ref. Reference Chai105). Lower IKr and augmented ICa,L amplitude were observed in iPSC-CMs derived from symptomatic patients with a KCNH2-R752W mutation.
Fatima et al. reported the generation of iPSCs from two LQTS3 patients with V240 M or R535Q mutations in SCN5A (Ref. Reference Fatima46). APD was longer in LQTS3 iPSC-CMs compared to healthy controls. Electrophysiological profiling revealed that Nav1.5 took a longer time to reach inactivation in LQTS3 iPSC-CMs, which contributed to the prolonged APD. Terrenoire et al. derived iPSC-CMs from a proband with an F1473C mutation in SCN5A, which was located in the channel inactivation gate (Ref. Reference Terrenoire106). The F1473C mutation increased late sodium current and delayed repolarisation, leading to an extended QT interval and a higher susceptibility to arrhythmia. They also found that the sodium channel blocker mexiletine exhibited stronger inhibitory activities when applying a higher pacing rate to iPSC-CMs. Together, these findings support patient-specific iPSC models for studying pathophysiology of LQTS1-3 that are associated with mutations in potassium and sodium channels in cardiomyocytes, and for developing patient- and disease-specific drug treatment.
Yazawa et al. examined iPSC-CMs originally derived from two LQTS8 patients (Ref. Reference Yazawa107). LQTS8 iPSC-CMs showed an abnormal delay in ICa,L inactivation and prolonged Ca2 + transients. The APs were three times longer in LQTS8 ventricular-like iPSC-CMs than healthy controls. Roscovitine enhanced the inactivation of the voltage-dependent Ca2 + channel and restored the regular Ca2 + transients in LQTS8 iPSC-CMs (Ref. Reference Yazawa107). Dysfunction in CALM1-F142L is a major cause of LQTS14 which is characterised by relapsed cardiac arrests (Ref. Reference Rocchetti108). Rocchetti et al. explored effects of heterozygous CALM1-F142L mutations on repolarisation frequency, membrane currents and intracellular Ca2 + oscillations using patient-specific iPSC-CMs. The prolonged APD and altered repolarisation frequency were linked to a perturbation of Ca2 + -dependent inactivation of ICa,L. In addition, another group created iPSCs from an LQTS15 patient with CALM2-D130 G mutation (Ref. Reference Limpitikul109). Intracellular Ca2 + handling in LQTS15 iPSC-CMs were abrogated and Ca2 + -dependent inactivation of ICa,L was reduced. Furthermore, they employed CRISPR genome editing to selectively inhibit the mutant allele and rescued Ca2 + handing abnormalities in LQTS15 iPSC-CMs.
In summary, LQTS patient-specific iPSCs can be used for testing drug efficacy and toxicity in a disease-specific context. Numerous regimens with the potential to rectify the disease phenotype have been investigated using LQTS2-specific iPSC-CMs. Sala et al. exploited patient-specific iPSC-CMs to examine the effects of KCNH2 selective activator on both acquired and congenital LQTS. The small chemical LUF7236 restored IKr by delaying the KCNH2 channel deactivation (Ref. Reference Sala110). In some instances, nicorandil, PD118057, and ICA-105574 treatment in LQTS2 iPSC-CMs resulted in APD shortening and EAD elimination (Refs Reference Matsa111, Reference Meng112), which is consistent with clinical regimens. In addition, LQTS-specific iPSC-CMs have tremendous potential in evaluating the likelihood of regimens on restoring cardiac abnormalities, allowing for quick identification of unanticipated toxicity and laying the foundation for risk prediction. LQTS iPSC-CMs from an individual carrying a KCNH2-R165W mutation exhibited more sensitive to arrhythmogenic medications compared to healthy controls (Ref. Reference Lahti104). Significant APD elongation and decreased IKs amplitude were observed in LQTS1 iPSC-CMs exposed to E-4031, which was manifested by the formation of new EADs (Ref. Reference Moretti35). These findings suggest greater sensitivity to malignant arrhythmias in response to culprit drugs in LQTS, which might substantially suppress the remaining IKr level (Ref. Reference Itzhaki102). Therefore, modelling LQTS and testing new drugs using patient-specific iPSC-CMs could be a big step forward in the realm of personalised health care.
Treatment strategies for LQTS
Beta-blockers are the mainstream drugs of choice for LQTS. A study conducted on LQTS1 and LQTS2 patients showed that not all beta-blockers are equally effective (Ref. Reference Chockalingam113). One possible explanation is that ion channels exhibit varying degrees of pharmacological response to beta-blocker therapies. Propranolol is widely considered as the most effective drug, owing to sodium channel inhibitory activity with a meagre impact on potassium channels (Ref. Reference Chockalingam113). Additionally, the precise nature of the genetic mutation in ion channel genes may be a determinant of the efficacy of beta-blocker therapy for LQTS (Ref. Reference Bhuiyan114). For example, LQTS1 patients who have a missense mutation appear to benefit more from beta-blocker medication than those who have other genetic variants. Besides beta-blockers, the sodium channel blocker mexiletine may also lessen the recurrence of cardiac events in LQTS2 patients (Ref. Reference Bos115). Combination treatment with mexiletine resulted in a significant reduction in polymorphic ventricular tachycardia and no syncope episodes occurred after the treatment. LQTS3 patients rely on pharmacological treatment (e.g. beta-blockers, sodium channel blockers), ICD and pacemaker treatment (Ref. Reference Wilde and Remme116). Specifically, beta-blockers are routine regimens, and ICD is suggested for those at a higher risk. Pacemaker treatment is for LQTS3 patients with symptomatic bradycardia as well as QT lengthening who don't respond to regular therapeutics.
In patients with Andersen-Tawil Syndrome, beta-blockers are commonly prescribed even without reducing the frequency of ventricular ectopy. Flecainide can mitigate the arrhythmic symptoms of certain Andersen-Tawil Syndrome patients by activating IK1 (Refs Reference Miyamoto117, Reference Caballero118). However, amiodarone and acetazolamide are more effective in Andersen-Tawil Syndrome patients with a heterozygous R218W mutation in KNCJ2 (Ref. Reference Junker119). Verapamil, a Cav1.2 blocker, is shown to effectively restrain the bidirectional ventricular tachycardia in Andersen-Tawil Syndrome (Ref. Reference Tristani-Firouzi and Etheridge120). Increasing the potassium concentration in the blood is beneficial in controlling ventricular arrhythmias in Andersen-Tawil Syndrome (Ref. Reference Sansone and Tawil121).
ICD implantation is a standard approach for CACNA1C-LQTS patients. Numerous medications with the potential to prevent CACNA1C-LQTS arrhythmias have been explored, including nifedipine, pinacidil and ranolazine (Ref. Reference Casini, Verkerk and Remme122). Owing to Ca2 + influx through L-type Ca2 + channels, suppression of ICa,L by nifedipine was postulated to be anti-arrhythmic. One alternative strategy to alleviate arrhythmia is to raise the repolarisation currents with the KATP channel opener pinacidil. Both therapies reduced AP and eliminated the proclivity to EAD. Ranolazine, a sodium channel blocker, suppressed EADs without affecting the APD. Inhibition of the activity of L-type Ca2 + channels by verapamil can attenuate ventricular arrhythmia in LQTS8 through shortening the repolarisation period (Ref. Reference Jacobs123).
LCSD is an effective, minimally invasive anti- fibrillatory treatment for individuals with LQTS (Ref. Reference Niaz124). LCSD is advised for individuals with LQTS who are suffering breakthrough cardiac episodes. Patients who are unable to tolerate the adverse effects of beta blockers may benefit from supplementary treatment with LCSD. Recent studies on LCSD revealed that LCSD monotherapy exhibited superior efficacy, which is supposed to push it forward into a guideline-directed therapy (Ref. Reference Schwartz and Ackerman125)
Novel drug discovery using the patient-specific iPSC platform would drive the next-generation precision treatment regime for LQTS. Voltage-sensitive dyes can be applied to 2D iPSC-CMs monolayer to assess APs under certain drug treatments. To evaluate whether a new drug could cause potential ventricular arrhythmia, CiPA using patient-specific iPSC-CMs has been used for testing drug-induced arrhythmias (Refs Reference Hortigon-Vinagre126, Reference Blinova127). A non-invasive platform called multi-electrode array (MEA) is also available for extensive electrophysiological profiling of human iPSC-CMs under different drug conditions (Refs Reference Millard128, Reference Qu and Vargas129). Taking advantage of patient-specific iPSC-CMs and cutting-edge techniques would facilitate novel drug screening and discovery process for treating LQTS.
Precision medicine in LQTS
Precision medicine is a multidisciplinary approach to the prevention and treatment of cardiovascular disease while integrating various individuals' genetic profiles, environmental cues and lifestyles that are factors influencing disease phenotypes (Ref. Reference Leopold and Loscalzo130). This approach addresses the constraints of reductionism in medicine, which assumes that all patients with the same symptoms have the identical phenotype. Precision medicine utilises omics approaches (proteomics, transcriptomics, metabolomics, etc.) to interpret clinical data and further characterise the phenotype (Refs Reference Lindsey131, Reference Shah and Newgard132). These phenotypic data can subsequently be evaluated using molecular interaction frameworks to identify previously unknown clinical phenotypes and disease connections and to choose the most effective therapeutics.
Precision medicine is involved in the accurate diagnosis approach of LQTS. The recently developed AI-ECG model was able to differentiate individuals with electrocardiographically concealed LQTS from the healthy cohorts. Several limitations have been considered in applying AI-enabled tools for a better interpretation of clinical data, such as ECG. The strongest illustration of how errors may occur in an AI-driven approach is overfitting, which occurs when there are excessive features compared to the quantity of data in the training set (Ref. Reference Krittanawong133). The AI-driven approach performs extremely well on the training set but badly on the test set with low generalisability because of overfitting. There are additional clinical and technical obstacles to the implementation of AI–ECG in patient care. AI analysis depends on standardised digital ECG sampling, which is not ubiquitously accessible even in high-resource clinical situations (Ref. Reference Siontis134). AI essentially relies on the integrity of data. Poor-quality data will disable appropriate training and validation on AI algorithms, precluding the development of AI-enabled techniques (Ref. Reference Goldstein, Navar and Carter135).
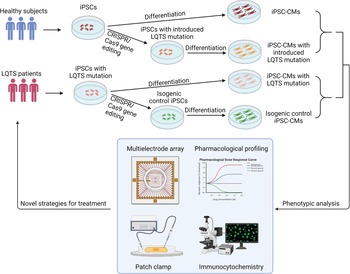
To implement precision medicine in cardiovascular disease field, it is necessary to establish a modelling platform to test various strategies. Human iPSC-CMs act as a disease model in a dish by keeping the patient's genetic traits and recapitulating the disease phenotypes in vitro. Human iPSCs are capable of being differentiated into cardiomyocytes (Refs Reference Zwi136, Reference Lian137). Current protocols provide simplified roadmaps for generating a variety of cardiomyocytes to characterise the channelopathies (Refs Reference Gnecchi, Stefanello and Mura138, Reference van den Berg139). Consequently, translational approaches using iPSC technology have implemented these cellular models for elucidating novel mechanisms of disease (Refs Reference Itzhaki102, Reference Matsa111, Reference Gnecchi and Schwartz140, Reference Bellin141, Reference Zhang142, Reference Mura143). Emerging CRISPR/Cas9 genome editing techniques enable researchers to distinguish the impact of variants for monogenic disorders, such as cardiac channelopathies (Ref. Reference Garg144). Two strategies were developed to resolve the confounding factors behind the mutation: First, insert the variant allele into a wild-type cell line to validate whether the disease phenotype is attributed to the variant; Second, correct the sequence of the variant in a patient-specific iPSC line to determine whether a corrected genetic variant reverses the diseased phenotype (Fig. 3). In these complementary approaches, each genome-edited cell line serves as the isogenic control compared to the unedited parental line to identify the causative variant (Ref. Reference Sala, Bellin and Mummery145). Additionally, isogenic controls are beneficial in eliminating the intrinsic variability displayed by iPSC-CMs in various healthy and diseased individuals (Ref. Reference Sala, Bellin and Mummery145). iPSC-CMs provide an exceptional patient-specific model to study the underlying mechanisms of multiple diseases. However, several disadvantages have yet to be overcome. Human iPSC-CMs exhibit similar properties to neonatal stage cardiomyocytes in terms of morphology and protein expression profiles (Refs Reference Karbassi146, Reference Veerman147). Although iPSC-CMs sufficiently recapitulate the phenotype of many inherited heart defects, they may not mature enough to understand the fundamental mechanisms underpinning inherited LQTS. In addition, epigenetic modifications are altered in the iPSC reprogramming process (Ref. Reference Kim148) regardless of the rudimentary epigenetic memory of the original somatic cells, which eliminate the contribution of personal lifestyles or medication usage vital to reconstruct the manifestation of disease phenotypes.
Patient-specific iPSCs take the lead for precision medicine in LQTS. Blood samples are collected for generation of patient-specific iPSCs. Pathogenic genetic variants are identified through whole-genome sequencing. CRISPR/Cas9 mediated genome editing can either introduce or correct a mutation in patient-specific iPSCs to study genotype-phenotype interactions. Deep phenotyping of LQTS iPSC-derived cardiomyocytes include multielectrode arrays, pharmacological profiling, electrophysiological characterisation and immunocytochemistry. Preclinical testing in LQTS patient-specific cardiomyocytes will guide clinicians for precise stratification and treatment of LQTS.

Precision medicine aims to administer the most appropriate treatment strategy for an individual based on their unique personal profiles. To incorporate complex data sets (i.e., next-generation sequencing, omics, clinical tests) into precision medicine, advanced biological systems are essential to interpret data and further provide guidance for identifying physiological and pathological factors. Precision medicine approaches to treating LQTS patients should consider various factors, including age, gender, electrolytes, QT-prolonging drugs, etc. Among LQTS1 patients, male adolescents are prone to develop initial cardiac issues earlier than female adolescents (Ref. Reference Zareba149). However, female adults are at higher risk than male adults in encountering the first cardiac event. In LQTS2 pathogenic subjects, the mortality rate remains higher in women than men (Ref. Reference Priori24). Extracellular K + concentration contributes to the regulation of QT duration in healthy individuals. Low serum K + levels serve as a highly susceptible factor in predisposing arrhythmic death (Ref. Reference Etheridge, Asaki and Niu150). Increased serum K + concentration by oral uptake and intravenous injection is beneficial in shortening QT interval in LQTS patients (Refs Reference Etheridge151, Reference Compton152). The QT interval measures the duration of cardiac repolarisation, which is primarily determined by equilibration of influx of sodium and calcium ions as well as efflux of potassium ions. QT prolongation is caused by an increase in inward current or a decrease in outward current. The suppression of the outward IKr, which prolongs individual cardiac ventricular APDs and ECG, is thought to be the mechanism by which medications prolong the QT interval (Ref. Reference Kannankeril, Roden and Darbar153). Ongoing pharmacogenetics (Ref. Reference Niemeijer154) and pharmacogenomics (Ref. Reference Strauss155) studies on drug-induced LQTS will probably lead to a stratified and customised strategy for treating acquired LQTS.
RNA interference (RNAi) and CRISPR genome editing have been used for treating LQTS in vitro. To circumvent the dominant-negative effect of KCNQ1 mutations in LQTS1, researchers developed a dual-component RNAi method that allows to knock down the mutant KCNQ1 transcript while leaving the wild-type KCNQ1 transcript intact (Ref. Reference Dotzler156). They further examined this RNAi-mediated strategy in rescuing KCNQ1 function in both TSA201 cell line and human iPSC-CMs to pave the way for completely repairing dysfunctional mutants in LQTS1. Lu et al. demonstrated that siRNA knockdown of KCNH2-E637 K mutation restored normal KCNH2 localisation to the membrane in cells co-expressing the KCNH2-E637 K mutant and recovered the normal amplitudes of maximum current and tailed current (Ref. Reference Lu157). In another study, impaired channel trafficking to plasma membrane was found in LQTS2 iPSC-CMs with a KCNH2-A561 T mutation, which could be rescued by a mutant allele-specific siRNA knockdown (Ref. Reference Matsa103). In addition, CRISPR/Cas9-based genome editing was employed to treat LQTS. Limpitikul et al. first generated iPSC-CMs from an LQTS patient carrying CALM2-D130 G mutation and subsequently characterised the electrophysiological properties of these iPSC-CMs (Ref. Reference Limpitikul109). Moreover, they utilised CRISPR interference (CRISPRi) to specifically knockdown the mutant CALM2-D130 G transcript and achieved a functional rescue in patient-specific iPSC-CMs. In summary, both RNAi and CRISPRi hold great promise for precisely treating LQTS patients.
The combination of patient-specific iPSC-CMs and genome editing has advanced the identification of purported modifier genes involved in shaping the phenotype of LQTS1 (Ref. Reference Lee158) and LQTS2 (Ref. Reference Chai105). However, numerous intrinsic restrictions exist when searching for a single causative gene in certain LQTS cases. The primary challenge is to establish the genotype-phenotype link. For instance, mutations in SCN5A were previously identified as a single causative gene for congenital LQTS (Ref. Reference Wang159). However, additional abnormalities associated with SCN5A mutations have been identified in Brugada syndrome (Ref. Reference Chen160) and dilated cardiomyopathy (Ref. Reference McNair161). The second challenge is the low penetrance in pedigrees with familial LQTS (Ref. Reference Priori, Napolitano and Schwartz4). When determining penetrance, mutation screening in the family members of the proband is the most effective method. Low penetrance could implicate other factors including genetic variants or environmental cues on LQTS pathogenesis. Therefore, an increasing number of LQTS-associated variants pose potential challenges to developing precision medicine for each LQTS subtype. Enhanced understanding of LQTS pathophysiology has led to the development and production of genotype-specific therapies (Ref. Reference Dotzler156). RNAi and CRISPRi mediated approaches have been shown to be effective in vitro to shorten the prolonged APD (Refs Reference Limpitikul109, Reference Dotzler156, Reference Lu157). Although development of genotype-specific pharmacotherapies may overcome underlying molecular defects of LQTS, the complexities of gene-specific therapeutic approaches may exceed the initial expectations and may not become available to all LQTS patients. With enough scientific input, a balance between ‘one therapy fits all’ and ‘personalised treatment for each individual patient’ is likely to be accomplished using patient-specific iPSC-CMs. This ‘personalised treatment’ strategy contrasts with the ‘one therapy fits all’ approach, in which diseases treatment and preventive measures are established based on the outcomes of randomised trials without taking individual differences into account.
Future perspectives
Patient-specific iPSCs and next-generation genome sequencing have significantly advanced the implementation of precision medicine in LQTS, which strives to improve the stratification of the patient population and to identify patient-specific interventions through examining large population datasets and genetic traits of LQTS patients. To implement the precision treatment in LQTS, researchers and doctors need to work together to complete this unprecedented mission. In the future, the genome of individual LQTS patients will be sequenced to identify potential genetic variants associated with QT prolongation (Fig. 3). In parallel, their blood samples will be collected for generation of patient-specific iPSC lines and cardiomyocyte differentiation. Together with genomic information and CRISPR genome editing, deep phenotyping using patient-specific iPSC-CMs will be utilised to stratify LQTS patients into multiple subgroups based on their phenotypes in the petri dish. In addition, potential candidate drugs will be administered to patient-specific iPSC-CMs and identify the best candidate for each group of LQTS patients to restore normal QT prolongation. Finally, ML of deep phenotyping of patient-specific cardiomyocyte data, large and diverse population-based clinical database and genetic traits of LQTS will eventually assign the optimal treatment regime to each LQTS patient for the best outcomes. The future precision medicine will be on the horizon which would be heralded by the forward clinical trial and novel drug testing in a petri dish using LQTS patient-specific iPSCs.
Acknowledgements
We would like to thank Dr Dennis Lewandowski for critically reading and editing this manuscript. Research in Dr Ming-Tao Zhao's lab was supported by the American Heart Association (AHA) Career Development Award 18CDA34110293, NIH/NHLBI R01HL155282, R21HL165406, Additional Ventures Innovation Fund (AVIF) and Single Ventricle Research Fund (SVRF).
Conflict of interest
None.




 Open access
Open access