


Current monoamine-targeting antidepressants show only partial success in achieving remission from depressive symptoms; only half of patients with non-psychotic MDD respond to a single antidepressant treatment and only 20–35% achieved remission. Reference Rush, Fava, Wisniewski, Lavori, Trivedi and Sackeim1 Furthermore, treatment failure (failure to respond to at least two different antidepressants) occurs in approximately a third of patients. Reference Gaynes, Lux, Gartlehner, Asher, Forman‐Hoffman and Green2 MDD patient adherence to antidepressant therapy also may be impacted due to medication side-effects. Reference Clayton3 Various documented pathways in addition to monoamine depletion are involved in the pathogenesis of MDD, implying that other pharmacological mechanisms should be explored.
Alterations of the HPA axis and hypercortisolism are biochemical features consistently associated with MDD; Reference Kling, Coleman and Schulkin4–Reference Gregory, Hill, Grey, Ketelbey, Miller and Muniz-Terrera7 increased cortisol levels are observed in approximately 40–60% of people with severe depression. Reference Kling, Coleman and Schulkin4 Elevated cortisol levels have also been associated with treatment failure, Reference Juruena, Pariante, Papadopoulos, Poon, Lightman and Cleare8 depression severity, melancholic symptomology and cognitive impairment. Reference Nandam, Brazel, Zhou and Jhaveri9,Reference Mikulska, Juszczyk, Gawrońska-Grzywacz and Herbet6 There is, therefore, interest in development of therapeutics that target the HPA axis and/or cortisol regulation/action. Reference Nandam, Brazel, Zhou and Jhaveri9,Reference Ding, Wei, Yan and Guo10 Inhibition of cortisol production with mifepristone has been reported to improve psychotic depression and bipolar disorder; Reference Block, Kushner, Kalin, Nelson, Belanoff and Schatzberg11,Reference Young, Gallagher, Watson, Del-Estal, Owen and Nicol Ferrier12 one recent meta-analysis of cortisol-modifying therapies in depression confirmed the potential efficacy of such an approach. Reference Ding, Wei, Yan and Guo10 However, compounds targeting systemic cortisol production in the adrenal gland are unsuitable for long-term therapy due to significant adverse peripheral hormonal safety effects. An agent that could inhibit brain cortisol production merits study in MDD.
Emestedastat (Xanamem) is a brain-penetrant and highly selective small molecule inhibitor of 11β-HSD1, which catalyses the reduction of inert cortisone within cells to the active glucocorticoid hormone cortisol. 11β-HSD1 is found predominantly in the liver, but it is also expressed in adipose tissue, blood vessels, brain and immune cells. Emestedastat’s hypothesised targeted mechanism of action for cognition and neuropsychiatric symptoms is primarily the selective reduction of intracellular cortisol levels in brain cells without impacting adrenal cortisol production, thus leaving systemic cortisol levels unaffected. Reference Seckl and Walker13
Emestedastat has been investigated in 8 clinical trials, showing adequate target engagement by positron emission tomography imaging at low doses (5–10 mg/day) in the brain and an acceptable safety profile. Reference Villemagne, Doré, Chong, Kassiou, Mulligan and Feizpour14 Clinically meaningful improvements in cognitive performance in an attention composite (Att. Comp.; including working memory) of a cognitive test battery (CTB; Cogstate) were seen at doses of 5, 10 and 20 mg emestedastat for 6–12 weeks in older, cognitively normal volunteers in two Phase 1 studies. Reference Rolan, Taylor, Seckl, Harrison, Chen and Farrell15
Cognitive symptoms associated with MDD occur in approximately two-thirds of those affected and can include memory impairment, difficulty in making decisions and loss of cognitive flexibility. Reference Millan, Agid, Brüne, Bullmore, Carter and Clayton16 These symptoms are associated with poorer response to conventional antidepressant therapy, disability and limited functional recovery. Reference Trivedi and Greer17,Reference Hack, Tozzi, Zenteno, Olmsted, Hilton and Jubeir18 However, there are few examples of clinical drug trials specifically designed to evaluate improvements in cognition in MDD patients. The previous positive results for emestedastat enhancing cognition in healthy, older participants, and the potential for cerebral cortisol synthesis inhibition to improve mood, provided the rationale for this trial. This randomised, placebo-controlled, double-blind trial was designed as a proof-of-concept evaluation of efficacy on cognition, depression and safety in adult participants with MDD with ongoing depression and cognitive impairment.
Method
Trial design
This Phase 2a, randomised, placebo-controlled, parallel-group, double-blind, proof-of-concept trial was conducted at 16 clinical trial centres in Australia and the UK.
The trial was conducted in accordance with the World Medical Association Declaration of Helsinki, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Guideline for Good Clinical Practice, the Australian National Health & Medical Research Council (NHMRC) National Statement on Ethical Conduct in Human Research (2007), incorporating all updates, and the Medicines for Human Use (Clinical Trials) Regulations 2004 (applicable to clinical studies run in the UK). The trial was registered with ClinicalTrials.gov, NCT05657691, and is complete. It was conducted with the approval of The Alfred Human Research Ethics Committee and Bellberry Human Research Ethics Committee (Australia; respective approval nos HREC/90113/Alfred-2022 and 2022-08-834) and Wales Research Ethics Committee 1 (UK; approval no. 23/WA/0069).
Participants
Enrolled participants had a confirmed diagnosis of MDD with ongoing cognitive impairment and residual depressive symptoms. Individuals aged ≥18 and ≤75 years, diagnosed according to DSM-5 criteria and with a current MDD episode (using the Mini-International Neuropsychiatric Interview at Screening) were eligible for inclusion in the trial. Participants were required either to have been on a stable dose of an approved treatment for depression (but not a tricyclic antidepressant, monoamine oxidase inhibitor, esketamine, vortioxetine or bupropion) for at least 6 weeks, or to have had a history of prior antidepressant use, have persistent symptoms of depression (Hamilton Depression Rating Scale (HAM-D) ≥17) and cognitive symptoms as determined by both subjective self-report of cognitive impairment and objective cognitive impairment (as measured by 0.5 s.d. deficit on a symbol-coding task compared with education and age norms). This cut-off was chosen as the operational definition of impairment, and is based on the typical cognitive deficit observed in patients living with depression. Reference Rhee, Shim, Manning, Tennen, Kaster and d’Andrea19
Participants were recruited by investigators using either site databases or other methods including clinician referral, community outreach and social media. Local research ethics committees approved the trial design, and all participants provided written informed consent.
Randomisation and masking
Eligible participants were randomly assigned 1:1 to receive either 10 mg emestedastat or matching placebo (with no active ingredient) by a computer-generated sequence using interactive web response systems; the randomisation block size was four. The participants, site staff, end-point assessors, investigators, statisticians and sponsor personnel were blinded to treatment assignment throughout the 10-week trial, and until final database lock.
Procedures
Following a 4-week screening period, eligible participants entered a 6-week blinded treatment and a 4-week blinded follow-up period. Participants took a single dose of investigational product (10 mg emestedastat or placebo) orally each morning with or without food. Concomitant antidepressant medications remained stable throughout the trial. Following the baseline visit, four on-site visits were conducted at weeks 2, 4, 6 and 10 (4 weeks of blinded follow-up post-trial drug treatment), for safety and efficacy assessments.
Outcome measures
Efficacy end-points included measures of cognition and depression. The primary end-point was change in the Att. Comp. score from baseline to week 6. Att. Comp. comprised three tests of attention and working memory (detection, identification and one-back – speed of performance) of the Cogstate computerised CTB (Cogstate Ltd., Melbourne). Att. Comp. was chosen as the primary end-point because of high construct and criterion validity, Reference White, Schembri, Prenn-Gologranc, Ondrus, Katina and Novak20 its sensitivity in assessing cognitive deficits in depression Reference Davis, DellaGioia, Matuskey, Harel, Maruff, Pietrzak and Esterlis21 and also because it has shown reproducible results with minimal placebo response in previous emestedastat trials in cognitively normal, older volunteers. Reference Rolan, Taylor, Seckl, Harrison, Chen and Farrell15 The derivation of Att. Comp. is detailed in the statistical analysis plan (supplementary materials).
Prespecified secondary outcomes included changes from baseline to week 6 and durability to week 10 on the Montgomery–Åsberg Depression Rating Scale (MADRS); Executive Function Composite (one-back test, category fluency test, letter fluency test and international digit symbol substitution test – symbols); Episodic Memory Composite (one-card learning and Hopkins verbal learning test – revised, immediate recall only); individual cognition tests used in the cognitive composites; and Patient Global Impression of Severity (PGI-S). Secondary outcomes relating to responder rates were also examined, including participants achieving 30 and 50% reduction in MADRS scores and those achieving clinical remission (MADRS score <10).
Exploratory outcomes prespecified included change from baseline to week 6 in trail-making A and B tests, the Pittsburgh Sleep Quality Index and Brief Pain Inventory.
Statistical methodology
With allowance for 10% drop-out, the estimated sample size for this trial was approximately 160 participants. Based on a two-sample, t-test statistic, a sample size of 72 participants per treatment group has approximately 80% power to detect an approximate 0.25-point difference in mean change from baseline to week 6 in Att. Comp. z-score between emestedastat and placebo, assuming a common standard deviation of 0.6 and one-sided alpha of 5%. A one-sided test was considered reasonable for the detection of potential emestedastat benefit on cognitive impairment and MADRS at week 6 in this first, proof-of-concept, randomised trial conducted in this population. Two-sided tests were used for all other evaluations. There was no correction for multiple comparisons, and thus p-values reported for secondary end-points are considered ‘nominal’.
Att. Comp. z-score was analysed using a restricted maximum likelihood-based mixed model for repeated measures (MMRM) with fixed effects for treatment, week and treatment-by-week interaction; and with baseline score as a covariate. The outcome variable was change from baseline scores. Trial week was included in the model as a categorical variable (weeks 2, 4 and 6), along with the treatment-by-week interaction. Least squares means were used to compare treatments. Pairwise treatment comparisons were performed each week; however, the primary comparison was the contrast between emestedastat and placebo at week 6. The primary comparison was tested at the 0.05 level using a one-sided test. The secondary end-points were summarised descriptively and analysed using a MMRM similar to that employed to evaluate the primary efficacy end-point, but also included week 10. Results for the modified intention-to-treat population, defined as all randomised participants with at least one post-baseline efficacy data point, are presented for both the primary and secondary end-points. Analyses of responder rates used chi-squared tests. Treatment groups were merged for the purposes of exploring correlations between cognitive improvements and depression.
Prespecified subgroup analyses were conducted for the primary end-point and secondary end-points, including MADRS and PGI-S. Subgroups included baseline Att. Comp. score (< median or ≥ median), baseline MADRS score (< median or ≥ median), concurrent use of an antidepressant at baseline (yes or no) and country of participation (Australia or UK). Further delineation of the subtype of concurrent antidepressant use (selective serotonin reuptake inhibitors (SSRIs), serotonin-noradrenaline reuptake inhibitors (SNRIs) or other) was examined in a post hoc subgroup analysis.
Effect size measurements were derived using Cohen’s d statistic. This was defined as the least squares mean difference in change from baseline divided by the pooled baseline standard deviation. An effect size of ≥0.2 between emestedastat and placebo in this initial trial in MDD was defined as indicating potential clinical effect(s).
Results
Of 720 participants screened, a total of 167 were enrolled in this trial between 8 December 2022 and 22 April 2024 (Supplementary Fig. 1). Participants were randomly assigned (1:1) to either active 10 mg emestedastat (n = 84) or placebo (n = 83). Of the 167 participants enrolled and randomised, 2 withdrew prior to dosing, 147 (88%) completed the trial as per protocol and 20 (12%) discontinued prior to completion.
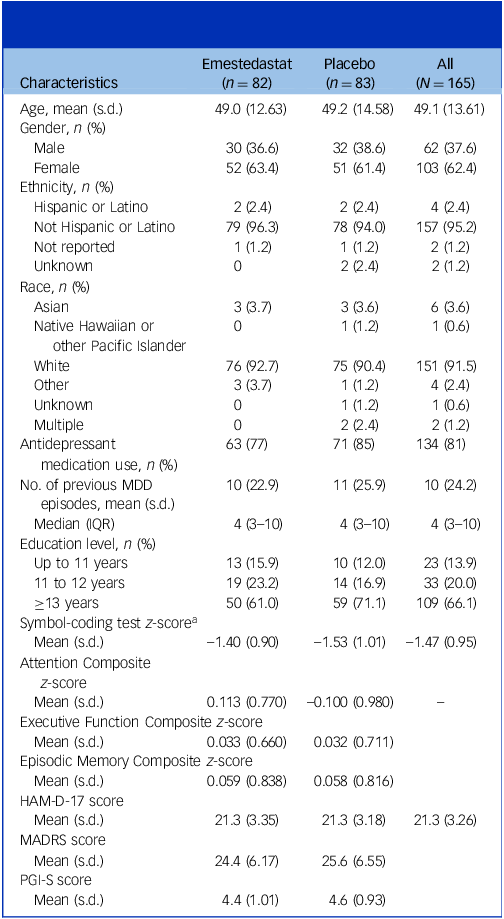
Baseline characteristics are summarised by treatment group and combined population (Table 1). Demographic and baseline parameters were broadly similar across treatment groups. The mean age of participants was 49.1 years (ranging 19–75 years), and there were 103 (62%) females and 62 (38%) males. Overall, the population was characterised as having moderate MDD (baseline mean MADRS score of 25) and recurrent illness, with a mean and median number of previous depressive episodes of 10 and 4, respectively. Concurrent antidepressant treatment was common, with 77% of the emestedastat group and 85% of the placebo group taking background antidepressant therapy.
Baseline demographics and clinical characteristics summarised by treatment

Table 1 Long description
The table presents baseline characteristics of participants in a study, divided by treatment group (Emestedastat and Placebo) and combined population. It includes data on age, gender, ethnicity, race, antidepressant medication use, number of previous major depressive disorder (MDD) episodes, education level, and various cognitive and depression scores. The table has 17 rows and 5 columns, with headers for Characteristics, Emestedastat (n = 82), Placebo (n = 83), and All (N = 165). Key trends include a mean age of 49.1 years, 62% females, and 38% males. The population is characterized by moderate MDD with a mean MADRS score of 25 and a mean of 10 previous depressive episodes. Concurrent antidepressant treatment was common, with 77% in the Emestedastat group and 85% in the Placebo group.
MDD, major depressive disorder; IQR, interquartile range; HAM-D, Hamilton Depression Rating scale; MADRS, Montgomery–Åsberg Depression Rating Scale; PGI-S, Patient Global Impression of Severity scale.
a. Z-scores calculated with respect to age and educational norms.
As shown in Table 1, there were generally similar baseline clinical characteristics across the two treatment groups, including symbol-coding scores.
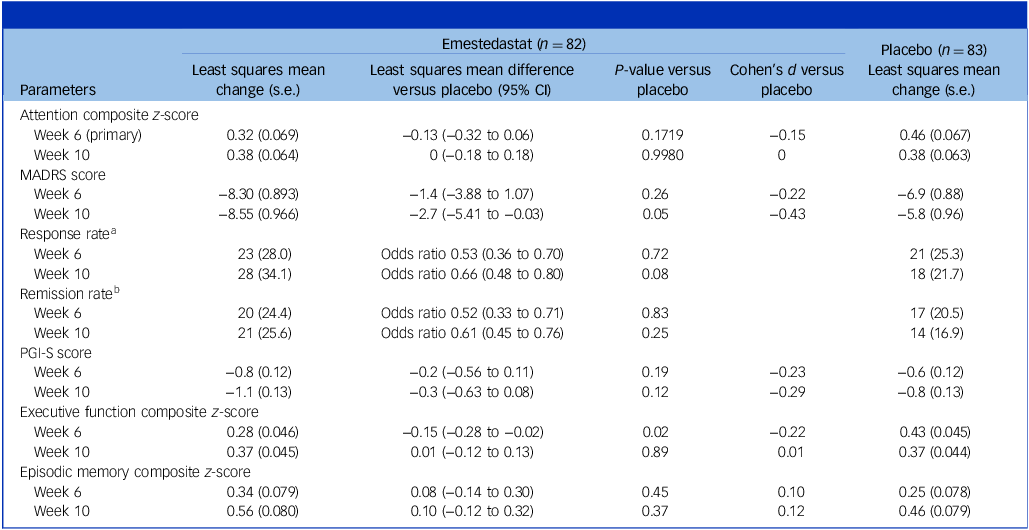
There was no observed difference in performance on Att. Comp. during the treatment period between the two groups, with a least squares mean difference of −0.13 (95% CI −0.32 to 0.06; p = 0.17) in favour of placebo at week 6, end of treatment (Fig. 1(a)). Both groups demonstrated improved performance on Att. Comp. during the treatment period from baseline to week 6 (least squares mean change values at week 6 were 0.32 (s.e. 0.07) for emestedastat and 0.46 (s.e. 0.07) for placebo; Fig. 1(a) and Table 2).
Change from baseline in cognitive (primary) and depression end-points. (a) Emestedastat (n = 82) and placebo (n = 83) group performance on the attention composite (Att. Comp., comprising identification, detection and one-back tests from the Cogstate test battery). (b) Montgomery–Åsberg Depression Rating Scale (MADRS) scores. (c) Participant-reported depression severity (Patient Global Impression of Severity (PGI-S)) scores over the blinded 6-week treatment period and 4-week follow up (week 10)). (d) Correlation of Att. Comp. change from baseline to week 6 and MADRS change from baseline to week 6, adjusted for baseline scores and concurrent antidepressant use. Error bars represent standard error. Data shown from the modified intention-to-treat population. Mixed model for repeated measures. *Nominal p = 0.05.

Fig. 1 Long description
The image contains four graphs depicting changes in cognitive and depression metrics over a 10-week period for two groups: Emestedastat and Placebo. The first graph shows the least squares mean change from baseline in the attention composite scores, with the Emestedastat group (n = 82) and the Placebo group (n = 83) tracked over weeks 2, 4, 6, and 10. The second graph illustrates the Montgomery-Asberg Depression Rating Scale (MADRS) scores, indicating a more significant reduction in depression severity for the Emestedastat group compared to the Placebo group. The third graph presents participant-reported depression severity scores (PGI-S), showing a similar trend of reduction over time for both groups. The fourth graph is a scatter plot correlating the change in attention composite scores with the change in MADRS scores from baseline to week 6, adjusted for baseline scores and concurrent antidepressant use. Error bars represent standard error, and data is shown from the modified intention-to-treat population. A nominal p-value of 0.05 is indicated for statistical significance.
Efficacy results: change from baseline to weeks 6 and 10 mITT population

Table 2 Long description
The table presents a comparison of efficacy results between emestedastat and placebo over weeks 6 and 10 in the mITT population. It includes parameters such as Attention Composite z-score, MADRS score, response rate, remission rate, PGI-S score, executive function composite z-score, and episodic memory composite z-score. The table has 82 rows for emestedastat and 83 rows for placebo, with columns for least squares mean change, least squares mean difference versus placebo, P-value versus placebo, Cohen’s d versus placebo, and least squares mean change. Notable trends include improvements in both groups for Attention Composite z-score and executive function composite z-score. The response rate and remission rate are also compared, with odds ratios provided. The table highlights the statistical differences and similarities between the two treatments over the specified weeks.
a. Response was defined as a reduction of ≥50% in MADRS score, and is presented as number of participants (%).
b. Remission was defined as achieving a MADRS score of <10, and is presented as number of participants (%). Odds ratios are accompanied by 95% CI.
mITT, modified intention to treat; MADRS, Montgomery−Åsberg Depression Rating Scale; PGI-S, Patient Global Impression of Severity scale.
During the blinded 4-week follow-up period, the emestedastat group remained stable and the placebo group decreased slightly in performance to the same level as the emestedastat group (least squares mean change values at week 10 were 0.38 (s.e. 0.06) for both emestedastat and placebo). Similar results were observed in the prespecified subgroups.
The least squares mean change from baseline in MADRS score at week 6 was −8.30 points (s.e. 0.89) for emestedastat and −6.89 (s.e. 0.88) for placebo (least squares mean difference −1.40; 95% CI −3.88 to 1.07; p = 0.26; Cohen’s d 0.22; Fig. 1(b)). Group separation favouring emestedastat increased at week 10, when the least squares mean difference between groups was −2.72 (95% CI −5.41 to −0.03; p = 0.048; Cohen’s d 0.43; Fig. 1(b)).
In general, the prespecified subgroups analysed displayed reductions in MADRS scores similar to those for the full population. In the subgroup not taking a concurrent antidepressant (n = 31), a reduction in MADRS was observed with emestedastat treatment at week 6, with least squares mean changes of 8.19 (s.e. 1.730) for emestedastat and −3.93 (s.e. 2.098) for placebo (p = 0.13; Cohen’s d 0.64). There were similar results in the subgroup taking concurrent antidepressant therapy, with least squares mean reductions in MADRS at week 6 of 8.37 (s.e. 1.03) for emestedastat and 7.4 (s.e. 0.97) for placebo (p = 0.49; Cohen’s d 0.15). Further subgroup data are presented in the Supplementary material.
Response and remission rates are shown in Table 2. Consistent with greater separation in MADRS score at week 10, the emestedastat-treated group displayed higher response and remission rates in the same time frame (response rate odds ratio 0.66, 95% CI 0.48 to 0.71, p = 0.08; remission rate odds ratio 0.61, 95% CI 0.45 to 0.76, p = 0.25).
On the patient-reported outcome measure, PGI-S, there was reduction in severity for both groups over the 10-week trial period, which separated earlier than MADRS and was more pronounced for emestedastat at all time points (Fig. 1(c)). At week 6, least squares mean change from baseline was −0.83 (s.e. 0.12) for emestedastat and −0.60 (s.e. 0.12) for placebo (least squares mean difference favouring emestedastat −0.23 (95% CI −0.56 to 0.11; p = 0.18; Cohen’s d 0.23)). There was a similar least squares mean difference of −0.29 (95% CI −0.63 to 0.08; p = 0.12; Cohen’s d 0.29) favouring emestedastat at week 10.
There were no consistent treatment effect differences observed in the trail-making A and B tests, Pittsburgh Sleep Quality Index and Brief Pain Inventory exploratory outcomes, with both groups improving modestly over the treatment period.
In an exploratory post hoc subgroup analysis, emestedastat showed a potentially clinically meaningful improvement in depressive symptoms on MADRS in participants on concurrent SSRI (n = 76) treatment compared with placebo at week 6 (least squares mean difference −2.56 (95% CI −6.04 to 0.91); p = 0.15; Cohen’s d 0.38; Supplementary Fig. 2(a)), and further improvement at week 10 (least squares mean difference −4.17 (95% CI −7.88 to −0.44); p = 0.03; Cohen’s d 0.61). This was supported by observed improvements for emestedastat on PGI-S compared with placebo at week 6 (least squares mean difference −0.41 (95% CI −0.88 to 0.06); p = 0.08; Cohen’s d 0.43; Supplementary Fig. 2(b)) and further improvement at week 10 (least squares mean difference −0.59 (95% CI −1.12 to −0.06); p = 0.03; Cohen’s d 0.62). Given the relatively small group size for the subgroups of SNRI (n = 36) and other antidepressants (n = 21), improvements on both MADRS and PGI-S were variable but generally favoured emestedastat (supplementary data).
There was no correlation between change in Att. Comp. with that in depressive symptoms at week 6 as measured by either MADRS (Fig. 1(d); mean estimate −0.005; 95% CI −0.017 to 0.007; p = 0.42; R 2 = 0.022) (Fig. 1(d)) or PGI-S (Supplementary Fig. 3; mean estimate −0.067; 95% CI −0.156 to 0.022; p = 0.14; R 2 = 0.022); nor was there any correlation between change in Att. Comp. with baseline characteristics including age, cognition and depression severity (R 2 ≤ 0.03).
There were no differences between Australian and UK sites in any of the above analyses, and no outlier sites impacting Att. Comp. or MADRS data to any extent.
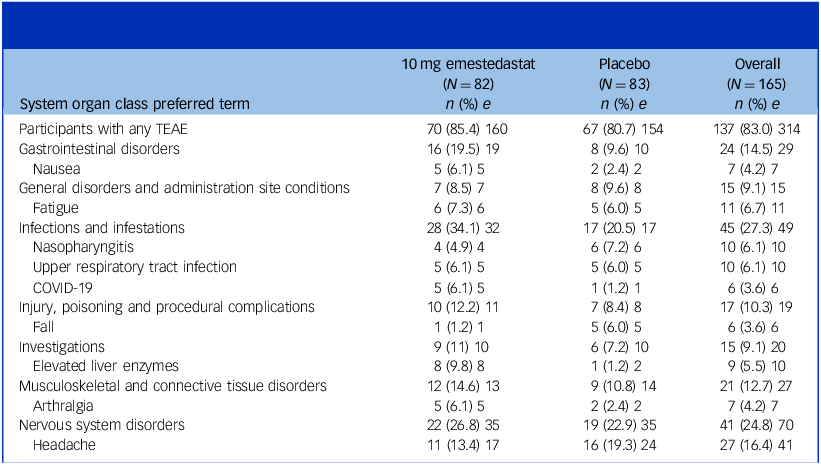
Table 3 summarises treatment-emergent adverse events by preferred-term ≥5% incidence. There were no treatment-related serious adverse events (SAEs), and emestedastat was generally well tolerated over the 6-week treatment period. Eight (9.8%) emestedastat-treated participants versus one (1.0%) placebo-treated had mild–moderate, asymptomatic elevation in liver function tests (LFTs) not associated with hyperbilirubinaemia. Of these, 7 had elevations ≤2 times the upper limit of normal (ULN) and 1 had LFTs 3.3 times ULN, which returned to normal in 5 days following discontinuation of emestedastat.
Summary of treatment-emergent adverse events (TEAEs) occurring in 5% or more participants by system organ class preferred term and treatment group

Table 3 Long description
The table presents data on treatment-emergent adverse events (TEAEs) occurring in five percent or more participants, categorized by system organ class preferred term and treatment group. It includes columns for 10 milligrams emestedastat, placebo, and overall, with rows listing various adverse events and their incidence. Key events include gastrointestinal disorders, infections and infestations, and nervous system disorders. Notable trends show higher incidences of certain events in the emestedastat group compared to the placebo group.
Data shown are from the safety population. N, group size; n, number of participants; e, number of events.
Discussion
To our knowledge, this is the first randomised, placebo-controlled trial assessing the ability of a drug to improve cognitive impairment in patients with MDD and a current depressive episode where measurable cognitive impairment was an inclusion criterion. Previously, vortioxetine trials had included patients on the basis of self-reported cognitive impairment, which may have been less reliable. It is also the first trial in this population assessing the efficacy and safety of a brain-penetrant 11β-HSD1 inhibitor designed to decrease brain cortisol levels. The population studied is best described as at least ‘moderately severe’ and ‘difficult to treat’, with a mean and median number of prior MDD episodes of 10 and 4, respectively, and a baseline HAM-D score of 21. More than 80% of participants were taking background antidepressant therapy.
The main cognitive findings were unexpected, in that large improvements in cognition in both treatment groups were observed without correlation with improvement in depressive symptoms or baseline characteristics. Previously reported placebo group cognitive improvements in MDD trials have been much smaller. For example, the cognition placebo response in an 8-week trial of vortioxetine in MDD patients, using the same cognitive tests, was less than half of that observed in this trial. Reference Mahableshwarkar, Zajecka, Jacobson, Chen and Keefe22 The placebo response was also approximately three times larger than that observed in the previous XanaMIA dose-ranging trial of emestedastat (NCT03830762) in cognitively normal, healthy, older adults. Reference Rolan, Taylor, Seckl, Harrison, Chen and Farrell15
Possible causes of a larger-than-expected placebo response in this trial include (a) focus on cognition as the primary end-point during participant consent, leading to participant expectations of improvement; (b) regression to the mean due to selection of an objectively cognitively impaired group versus the vortioxetine trials, where cognitive screening was by self-reported symptoms; and (c) a pseudo-specific effect on cognition due to the overall improvement in depression in both groups.
Given the lack of correlation between improvements in MADRS score and Att. Comp., it appears unlikely that improvement in depressive symptoms led to improvement in cognition. Reference LaMonica, Biddle, Naismith, Hickie, Maruff and Glozier23 In the past, improvements in cognition in patients treated for depression have been potentially attributed to a pseudo-specific effect, i.e. as depression improves so does the patient’s ability to perform on cognition testing. 24 The fact that cognitive performance and depressive symptoms independently changed in this trial supports the hypothesis that these two symptom domains in MDD are independent. It is also unlikely that the observation was due to regression to the mean, because there was no correlation with baseline cognitive impairment. Thus, it appears likely that therapeutic expectations led to the unexpectedly high placebo response observed in cognitive performance.
Our findings raise questions about how to control for a large placebo response in future MDD trials where cognition is the primary end-point. One potential strategy would be to mask the primary end-point selection to both participants and investigators, as has been done in MDD trials with depression measures as the primary end-point, but intention masking carries ethical challenges. Another strategy could involve selection of cognitive end-points less subject to the placebo phenomenon. Site and participant placebo training, and potentially a double-blinded (not single-blind), placebo run-in trial design, could also be utilised. Reference Nasereddin, Alnajjar, Bashar, Abuarab, Al-Adwan and Chellappan25,Reference Schmidt, Kezic, Popova, Melkote, Van Der Ark and Pemberton26
The temporal pattern of emestedastat benefit appearing late in the 6-week treatment period, and increasing during blinded follow-up through to week 10, is potentially consistent with the biology of cortisol and its control in the brain leading to a slower onset of activity. This is demonstrated by the adverse effects of therapeutic glucocorticoids, which often require many weeks to emerge, and reversal of these has a similar time course. Reference Scott, Sharpe, Quinn and Colagiuri27 Cortisol has long been associated with the pathology of MDD, and signalling mechanisms comprise both fast alterations to receptor activation and slower alterations to gene and protein expression. Reference Ding, Wei, Yan and Guo10,Reference Liston, Cichon, Jeanneteau, Jia, Chao and Gan28,Reference Schouten, Bielefeld, Garcia-Corzo, Passchier, Gradari and Jungenitz29 Inhibiting CNS production of cortisol represents a distinctly different mechanism from most currently prescribed antidepressant medications. Available antidepressants are designed to work primarily by modifying monoaminergic mechanisms that increase the availability of monoaminergic neurotransmitters in the brain, particularly serotonin and norepinephrine, by inhibiting their reuptake. Antidepressant effects with this mechanism typically occur within 3–4 weeks of therapy initiation. Potential emestedastat-mediated changes in brain function are probably different mechanistically compared with alteration of neurotransmitter levels, and may be due to the ‘slower’ or ‘neural plasticity’ effects of inhibiting brain cortisol production.
The trend towards emestedastat benefit on MADRS scores was modest at the end of treatment, and increased at 4 weeks post-treatment to a potentially clinically meaningful level overall, with similar effects seen in participants on background SSRI therapy. Definitive demonstration of antidepressant effects with new agents can be problematic due to the large improvements in placebo-treated patients, and to the relatively smaller additional benefit from active drug. For example, a meta-analysis of 11 randomised controlled trials for vortioxetine estimated a mean MADRS benefit of two points compared with placebo (Cohen’s d 0.22) over 6–12 weeks of treatment. Reference Pae, Wang, Han, Lee, Patkar and Masand30 These effect sizes are comparable to other approved MDD therapies, with agomelatine displaying Cohen’s d effect size ranging from 0.18 to 0.26. Reference Taylor, Sparshatt, Varma and Olofinjana31,Reference Singh, Singh and Kar32 The results presented here are also comparable to those for agents approved for adjunctive treatment of difficult-to-treat depression. In two large, controlled trials, brexpiprazole combined with SSRI/SNRI treatment demonstrated benefits on MADRS of 1.31–3.21 points compared with placebo and SSRI/SNRI in patients with inadequate response to a standard trial of antidepressant therapy. Reference Thase, Youakim, Skuban, Hobart, Augustine and Zhang33 Considering these effect sizes, the findings in the current trial of ‘difficult-to-treat’ patients with moderately severe MDD could be regarded as a signal of potential clinical relevance. However, given the proof-of-concept nature of the trial design, the findings will require replication and expansion in larger and longer trials.
Emestedastat was generally found to be safe and well tolerated, with no SAEs related to treatment and only a slight excess of asymptomatic transaminase elevations. Given the prevalence of more serious adverse side-effects experienced by patients taking the currently available monoamine antidepressant agents, emestedastat’s adverse effect profile appears clinically acceptable. In particular, adverse effects such as sexual dysfunction, insomnia or drowsiness and increased anxiety were not observed as emestedastat adverse effects in this trial. The incidence of generally mild liver enzyme elevations in the emestedastat-treated group in this trial was higher than that observed in previous trials in patients clinically diagnosed with Alzheimer’s disease (NCT02727699), and in healthy older adults (NCT03830762) at doses of ≤10 mg daily (overall incidence approximately 6%; data on file, Actinogen Medical Ltd). None of the observed elevations in LFTs were associated with hyperbilirubinaemia or other liver function abnormalities, and most (7 of 8) were ≤2 times ULN, with only one trial participant discontinuing due to LFT changes.
The limitations of this first emestedastat trial in MDD include the following: (a) the trial may have been too short in duration given the cortisol control mechanism of action; and (b) the trial population may not be fully generalisable in that it was primarily Caucasian, heavily co/pre-treated with many prior depressive episodes and was conducted in participants with measurable cognitive impairment.
In conclusion, emestedastat had no measurable benefit on cognitive impairment in the population studied in this trial, but did show trends towards potential benefit on depression symptoms – particularly 4 weeks after the end of the 6-week treatment period. A pronounced placebo effect on cognitive impairment is instructive for those seeking to investigate cognitive impairment in an MDD population. Effects on depression support the hypothesis that decreasing CNS cortisol may be beneficial to patients with MDD, even in those with chronic recurrent depression on standard therapy. Emestedastat represents a novel pharmacological approach to the treatment of depression and merits further study, either as an add-on therapy or a stand-alone treatment in trials of longer duration.
Supplementary material
The supplementary material is available online at https://doi.org/10.1192/bjp.2026.10689
Data availability
Requests for trial data can be submitted to Actinogen Medical Ltd. Requests for the clinical study report, analytical code and anonymised individual participant data will be considered for researchers who provide a methodologically sound proposal for use of this information. Data will be available immediately following publication of this manuscript, with no anticipated end date.
Acknowledgements
This trial was funded by Actinogen Medical Ltd. The authors thank the participants and their families involved in this trial. The authors also thank the trial site staff and investigators involved in the conduct of the trial (see Supplementary Appendix for a list of principal investigators from enrolling trial sites. Lee Bergman (Actinogen Medical Ltd) provided editorial assistance with the manuscript.
Author contribution
All authors were involved in trial conceptualisation or design. S.H.R., with the trial investigator group, conducted the investigation. J.D.T., D.C.H., P.E.R. and M.J.J. carried out the data analysis. J.D.T., D.C.H., P.E.R. and M.J.J. verified the raw data. All authors contributed to data interpretation. J.D.T. wrote the first draft of the manuscript, with input from D.C.H. All authors provided critical revision of the manuscript for intellectual content. All authors had full access to the data in the trial and had final responsibility for the decision to submit for publication.
Declaration of interest
J.D.T. and D.C.H. are employees of Actinogen Medical Ltd. P.E.R. and M.J.J. report consulting fees from Actinogen Medical Ltd. J.E.H. reports consulting fees from ADvantage Ther., Actinogen Medical Ltd, AlzeCure, Altoida, Boehringer Ingelheim, Cambridge Cognition, Cognition Thera., Curasen, EQT Life Sciences, GfHEU, Julius Clinical, Kynexis, Manus Neuro, Novartis, Pearson, Scottish Brain Sciences, Shionogi, Signant, Teitur Ther., Therini Bio, Transposon, Virogenics, Vivoryon Thera. and Winterlight Labs; holds stock or stock options in Scottish Brain Sciences; and reports royalties or licences from Blackwell Publishers and Oxford University Press. S.H.R. reports consultancy fees from GW Pharma, Gruenenthal, GSK and Takeda; and clinical research grants from Noema, COMPASS, Pathways, AbbVie, Leo, Vertex, Eli Lilly, GSK, BMS and Vertanical. M.B. reports consulting fees from Lundbeck, St Bio Pharma, Milken Baszucki Fund, Minnesota University Wolters Kluwer, China Shanghai Health Promotion Assoc., Global Congress of Biological Psychiatry India, Medical Tribune Polska, Precision Psych Fondamental, ISBD and Janssen MDD; holds patents for the modulation of physiological processes and agents useful the same, and for modulation of diseases of the central nervous system and related disorders; reports royalties or licences from Cambridge University Press and Allen and Unwin, and support from a NHMRC Leadership 3 Investigator grant (no. GNT2017131), NHMRC, Australia Principal Research Fellowship, MRFF, PCORI Patient Centred Outcomes Research Institute, AEDRTC Australian Eating Disorders Research and Translation Centre and USA Dept Defense, office of the congressionally directed medical research program (CDMRP).




 Open access
Open access
eLetters
No eLetters have been published for this article.