


Background
The global rise in obesity amongst reproductive aged men is a substantial public health concern with potential intergenerational implications for offspring health. 1,Reference Billah, Khatiwada, Morris and Maloney2 Although the negative impact of maternal obesity on offspring health is well-established. Reference Hu, Yang and Zhang3 the intergenerational influence of paternal obesity is relatively underexplored and less well characterized.
The Paternal Origins of Health and Disease is a relatively new paradigm that emphasizes the importance of paternal pre-conception diet and metabolic health in directly and indirectly impacting the health of offspring. Reference Soubry4 Studies in rodent models have demonstrated that suboptimal diet and obesity in fathers can directly alter epigenetic markers in sperm, modifying early embryonic development and increasing the long-term risk of cardiometabolic disorders in offspring including obesity, reduced glycemic control, increased hepatic lipogenesis, and altered hypothalamic/feeding behaviors. Reference Ornellas, Carapeto, Mandarim-de-Lacerda and Aguila5–Reference Ornellas, Souza-Mello, Mandarim-de-Lacerda and Aguila9 In humans, paternal overweight at conception has been reported to double offspring obesity risk and adversely influence offspring metabolic health, independent of the mother’s weight. Reference Tomar, Gomez-Velazquez and Gerlini10,Reference Campbell and McPherson11 A large cohort study recently highlighted the impact of paternal obesity as a modifiable risk factor that negatively affects both maternal-neonatal outcomes and the health of adolescent offspring. Reference Lin, Chen and Qian12 Further, Zalbahar et al. Reference Zalbahar, Najman, McIntrye and Mamun13 reported a strong positive association between paternal (and maternal) BMI and offspring BMI and waist circumference, with the strongest effect observed in early adulthood (21 years of age), highlighting the need to identify interventions for paternal obesity in fathers during the preconception period.
From a metabolic programming perspective, our group has interest in intergenerational drivers of dyslipidemia and cardiovascular disease (CVD) risk, particularly changes in lipoprotein distribution parameters. Pregnancies complicated by obesity are characterized by an unfavorable lipoprotein profile, Reference Meyer, Stewart and Brown14 and multiple adverse pregnancy states have been associated with atherogenic lipoprotein subfractions in fetuses and offspring, including non-physiological hypercholesterolemia, Reference Mathew, Huang, Dumolt, Patel and Rideout15 gestational diabetes, Reference Algaba-Chueca, Maymo-Masip and Ballesteros16,Reference Meyer, Cortie, Dekker-Nitert, Barrett and Freeman17 and intrauterine growth restriction. Reference Miranda, Simoes and Paules18 Santos Ferreira et al. (2017) Reference Santos Ferreira, Williams and Kangas19 reported that increasing maternal and paternal BMI was associated with aberrant lipoprotein distribution patterns in children. However, beyond this, evidence linking parental obesity (particularly paternal) to lipoprotein particle parameters in children remains limited. This is a significant knowledge gap as assessment of lipoprotein distribution parameters in children of parents with obesity may provide mechanistic insight into the intergenerational transmission of cardiometabolic risk and reveal early alterations in lipid metabolism that are not captured by conventional lipid measures.
Thus, in the current study we used a diet-induced obese Sprague-Dawley rat model to investigate the independent effects of paternal obesity on offspring obesity and associated metabolic dysfunction across early life (newly weaned stage) and adulthood with particular emphasis on lipids and lipoprotein sub-particle distribution. We hypothesize that offspring from fathers with obesity would demonstrate obesity and metabolic dysfunction (i.e., reduced glycemic control and dyslipidemia) throughout the life course.
Methods
Twelve two-month-old male Sprague-Dawley rats (Charles River, obese prone, Crl: OP-CD) were single-housed in a controlled environment (12 h light:12 h dark, 20°C, and 50% humidity) in the Laboratory Animal Facility at the University at Buffalo. The animals had free access to food and water throughout the study. The rats were randomized using a random number table to two experimental diets for a 6-week pre-conception period (n = 6/group): (i) a low calorie control (CON) diet (Lean; total energy 3.8 kcal/g; % energy from fat, 10; protein, 20; and carbohydrate, 70) (Research Diets, D12450K) or (ii) a high calorie (HC) obese-inducing diet (Obese; total energy 4.7 kcal/g; % energy from fat, 45; protein, 20; and carbohydrate, 35) (Research Diets, D12451). Body weights and feed intake were recorded weekly.
Following the pre-conception period, male rats from each group were bred with lean, CON-fed female breeders (8-weeks-old) to establish a timed pregnancy, Reference Heyne, Plisch, Melberg, Sandgren, Peter and Lipinski20 confirmed by the presence of a vaginal plug. Mothers continued the CON diet throughout gestation and lactation (21 days) and weekly body weights and feed intake were recorded. Following birth on postnatal day 1, the pups were separated, individually weighed, and sexed based on anogenital distance. Litters were culled to 8 pups per dam (4 males and 4 females where possible), and litter weights were recorded weekly.
On postnatal day (PND) 21, six pups (three females and three males) from each mother were euthanized for blood and liver collection, pooled by sex from each mother for analysis. Blood was collected by cardiac puncture and allowed to clot for 30 min at room temperature and serum was separated and stored in aliquots at –80°C following centrifugation. The livers were rapidly excised, weighed, and frozen in liquid nitrogen, and stored at −80°C for further analysis. The remaining littermates (1 male and 1 female from each mother) were weaned onto the control diet until for fourteen weeks until adulthood (PND 120) for blood and liver collection. The animals were cared for in accordance with the University at Buffalo Institutional Animal Care and Use Committee’s guidelines (protocol #202100085). To ensure rigor and transparency, ARRIVE guidelines were followed in the reporting of this research. Reference du Sert, Hurst and Ahluwalia21
Serum biochemistry
Serum glucose was assessed by colorimetric detection (Invitrogen, Frederick, MD, USA; EIAGLUC) and insulin by ELISA (Millipore, Billerica, MA, USA; EZRMI-13K). Direct measures of serum cholesterol (total cholesterol (TC), HDL-C, and LDL/VLDL-C), were assessed enzymatically (Bioassay Systems, Hayward, CA, USA; EHDL-100) and triglycerides (TG) were assessed with a commercial enzymatic kit (Zenbio, Durham, NC, USA; STG- 1NC).
Direct measurement of lipoprotein subclass profiles, including particle concentration (nM) and particle size (nm) was performed using nuclear magnetic resonance (NMR) spectroscopy via automated signal acquisition followed by computational analysis using proprietary signal-processing algorithms (LabCorp Inc., Raleigh, NC). Lipoprotein subclass diameter ranges were defined as follows: Particle concentrations - triglyceride-rich lipoprotein (TRL) particles, 24–240 nm; total LDL particles, 19–23 nm; and total HDL particles, 7.4–13 nm; Mean lipoprotein sizes: TRL, 30–100 nm; LDL, 19–22.2 nm, and HDL, 7.4–13 nm. Apolipoprotein A–1 and B (ApoB) concentrations were quantified using immunologic assays (Liposcience, Raleigh, NC).
Liver lipids
Liver TG was extracted from frozen pulverized tissue and analyzed with a commercial kit (Zenbio, STG-1-NC). For liver cholesterol, pulverized liver tissue (∼50 mg) was spiked with α-cholestane (internal standard) and saponified in KOH–methanol at 100°C for 2 h. Petroleum diethyl ether was used to extract the non-saponifiable sterol fraction, which was then dried under N2 gas. A Shimadzu GC-17A gas chromatograph with a flame ionization detector using a SAC-5 capillary column (30 m × 0.25 mm × 0.25 mm, Supelco, Bellefonte, CA, USA) was used to analyze the sterol fractions.
Data analyses
All statistical analyses were conducted using SPSS 16 (SPSS Inc., Chicago, IL, USA). Litters (not individual pups) from each paternal-maternal pair were considered as a single observation. Based on previously published work, Reference Raghavendhira, Srinivasan and Bhaskaran22 an a priori power analysis conducted using G*Power indicated that a total sample size of 12 fathers (n = 6 per group) would be required to detect a difference in blood cholesterol concentration between groups using a two-tailed test (d = 1.5, α = 0.05, and 80% power).
Lab members were blinded to the treatment groups while conducting biochemical assays and data analysis. Repeated measures analysis was used to assess the trajectory of maternal growth throughout pregnancy and gestation along with offspring growth during the postweaning period. Data were checked for normality using the Shapiro-Wilk test. The effects of paternal obesity status on offspring health outcomes were evaluated using two-way analysis of variance (ANOVA), with paternal obesity status and offspring sex included as fixed factors, allowing assessment of main effects and their interaction. Data are presented as means ± SE for PND21 metabolic outcomes and box and whisker plots (min to max, where the center line represents the median) for adult offspring (PND 120) outcomes. Data are presented as means ± SE. Differences were considered significant at p ≤ 0.05.
Results
Growth parameters
Growth parameters are presented in Fig. 1. At the end of the pre-conception period prior to breeding, body weights in male breeder rats fed the high calorie diet (‘obese’ fathers) were higher (p ≤ 0.05) compared with the control-fed counterparts (‘lean’ fathers) (573.25 ± 12.94 vs. 487.0 ±12.80 g) (Fig. 1a). As expected, no body weight differences (p > 0.05) were observed in mothers mated with lean versus obese males throughout gestation and lactation (Fig. 1b). Similarly, litter weights throughout the lactation period (21 days) were not different (p > 0.05) between offspring from lean vs. obese fathers (Fig. 1c). Repeated measures analysis of body weight in male and female offspring from birth to adulthood (PND 120) were similar (p > 0.05) between lean and obese groups.
Growth trajectories during the experimental period. (a) Paternal body weight of lean or obese male breeders prior to breeding during the obesity-induction phase, (b) maternal (lean) body weights throughout gestation and lactation following mating with lean or obese male breeders, (c) litter body weights throughout the lactation period, and (d) male and female offspring body weights from lean or obese fathers in the postnatal period. Data represents mean ± se, n = 6 (paternal/maternal pairs) per group.

Newly-weaned offspring
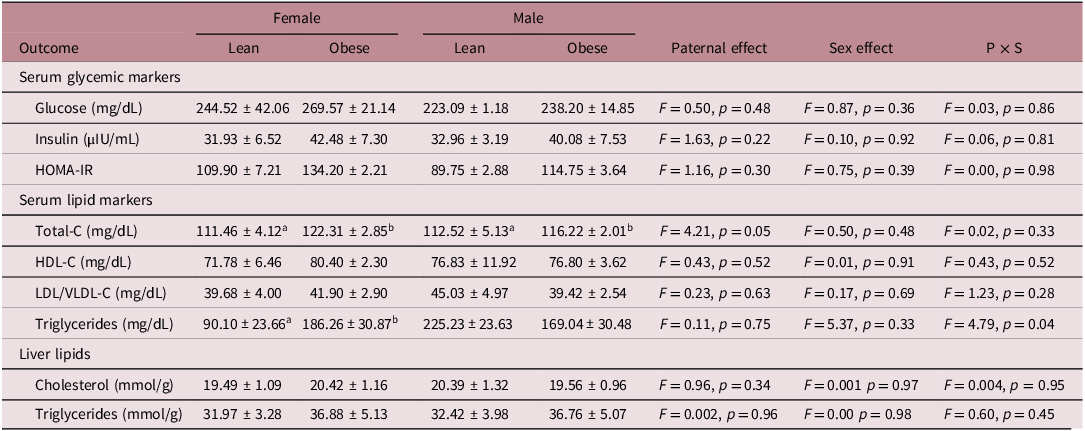
Metabolic health markers in newly-weaned male and female offspring at PND 21 are presented in Table 1. No significant main effects of paternal obesity (lean vs. obese) or offspring sex, nor any significant interaction effect, were noted for serum glucose, insulin, and HOMA-IR. Serum total-C concentration was higher in newly-weaned male and female offspring from obese vs. lean fathers [main paternal effect, F(1,21) = 4.21, p = 0.05]. No significant effects of paternal obesity status, offspring sex, or their interaction were detected for serum HDL-C or LDL/VLDL-C in newly-weaned offspring. An interaction effect was observed for serum TG [F(1,21) = 4.79, p = 0.04], with newly-weaned female (but not male) offspring from obese fathers demonstrating higher serum TG vs. female offspring from lean fathers. Offspring liver lipids (cholesterol and TG) were not significantly influenced by paternal obesity status, offspring sex, or their interaction.
Metabolic markers in newly weaned (postnatal day 21) male and female offspring from lean and obese fathers

Data are means ± SE. n = 6 (paternal/maternal pairs) per group. Groups (within sex) not sharing a superscript are significantly different (p < 0.05). The ab’s represent the post-hoc tests based on an overall p-value that is shown in the ‘paternal effect’ or ‘P × S’ column.
Adult offspring
Paternal obesity status did not affect (p≤0.05) offspring glucose, insulin, or HOMA-IR, either independently or in interaction with offspring sex (Fig. 2a–c). Male and female offspring from obese fathers demonstrated higher serum total-cholesterol [F(1,21) = 20.74, p < 0.001] and LDL-VLDL-C [F(1,21) = 11.4, p = 0.003] compared to offspring from lean fathers (Fig. 3a, b). Male, but not female, adult offspring from obese fathers exhibited higher HDL-C compared to the lean group [P×S, F(1,21) = 5.83, p = 0.03] (Fig. 3c). No difference (p > 0.05) was observed in serum TG between offspring from lean vs. obese fathers (Fig. 3d). Serum apo-A1 concentrations were similar between groups (Fig. 3e), however, male offspring exhibited higher serum apoB concentrations [P × S, F(1,21) = 0.94, p = 0.05] compared to male offspring from lean fathers (Fig. 3f). No main effects of paternal obesity status or offspring sex, nor any interaction between these variables, were observed for lipoprotein subclass profiles (i.e., particle number or size) for triglyceride-rich (TRL) (Fig. 4a, d) or HDL lipoprotein particles (Fig. 4b, e). Alternatively, male offspring from obese fathers demonstrated higher LDL particle concentration [F(1,11) = 44.92, p < 0.001] (Fig. 4c) and size [F(1,11) = 11.1, p < 0.009] (Fig. 4f) compared to males from lean fathers. Paternal obesity did not influence LDL subclass distribution in female offspring (Fig. 4c, f). Liver lipid concentration, including cholesterol (Fig. 5a) and triglyceride (Fig. 5b) were not influenced by paternal obesity, offspring sex, or their interaction.
Serum markers of glycemic control in adult (postnatal day 120) male and female offspring from lean or obese fathers including (a) glucose (mg/dL), (b) insulin (uIU/mL), and (c) homeostatic model assessment of insulin resistance (HOMA-IR). Data are means ± SE. n = 6 (paternal/maternal pairs) per group. Groups (within sex) not sharing a superscript are significantly different (p < 0.05).

Serum markers of lipid control in adult (postnatal day 120) male and female offspring from lean or obese fathers including (a) total cholesterol (mg/dL), (b) HDL cholesterol (mg/dL), (c) LDL/VLDL cholesterol (mg/dL), (d) triglycerides (mg/dL), (e), Apo-A1 (mg/dL), and (f) ApoB (mg/dL). Data are means ± SE. n = 6 (paternal/maternal pairs) per group. Groups (within sex) not sharing a superscript are significantly different (p < 0.05).

Serum lipoprotein distribution in adult (postnatal day 120) male and female offspring from lean or obese fathers including (a) triglyceride rich (TRL) lipoprotein # (nmol/L), (b) LDL particle # (nmol/L) (c) HDL particle # (umol/L), (d) TRL particle size (nm), (e), LDL particle size (nm), and (f) HDL particle size (nm). Data are means ± SE. n = 6 (paternal/maternal pairs) per group. Groups (within sex) not sharing a superscript are significantly different (p < 0.05).

Liver lipids, including (a) cholesterol (mmol/g) and triglycerides (mmol/g) in adult (postnatal day 120) male and female offspring from lean or obese fathers. Data are means ± SE. n = 6 (paternal/maternal pairs) per group. Groups (within sex) not sharing a superscript are significantly different (p < 0.05).

Discussion
In this study, paternal obesity was associated with adverse metabolic programming of lipid and lipoprotein profiles in male and female offspring, independent of abnormal early-life growth patterns or adulthood obesity. This dyslipidemic phenotype was detectable in early life, with newly weaned pups demonstrating significantly higher TC (male and female) and serum triglycerides (female only). Adult offspring further exhibited increased total and LDL/VLDL cholesterol and a male-specific increase in apoB and shift toward a more atherogenic lipoprotein subclass distribution profile, including higher concentrations of small LDL particles. Collectively, this data may suggest that paternal obesity, even in the absence of postnatal overnutrition or obesity, may represent an early determinant of elevated cardiometabolic disease risk in progeny by programming an advanced dyslipidemia profile.
Several human cohort studies have reported positive associations between paternal pre-conception obesity and childhood BMI. Reference Zhang, Clayton, Overvad, Olsen, Lawlor and Dahm23 These findings have been mechanistically linked to epigenetic modifications in sperm Reference Akhatova, Jones, Coward and Yeste24 and to differentially methylated CpG sites in targeted gene regions of the offspring epigenome, detected from cord and peripheral blood samples. Reference Lin, Chen and Qian12,Reference Davey Smith, Steer, Leary and Ness25–Reference Sharp, Alfano and Ghantous28 However, a recent assessment of the overall strength of the evidence supporting an impact of paternal factors on epigenetic programming in offspring concluded a lack of consistent associations due to unclear exposure measures and numerous confounding factors. Reference Sum, Burton, Alcazar, Teh, Godfrey and Huang29 By allowing rigorous control of key confounding factors – such as nutrient/caloric intake and duration of obesity exposure, animal model studies may help elucidate direct mechanisms underlying the paternal transmission of obesity risk to offspring. Although several animal studies have reported higher body weights in offspring born to fathers with obesity, Reference Fullston, Ohlsson Teague and Palmer7,Reference Haberman, Menashe and Cohen30,Reference Masuyama, Mitsui, Eguchi, Tamada and Hiramatsu31 we did not observe a difference in body weight between offspring from obese and lean fathers. Consistent with our findings, several other animal studies have likewise reported no such association, Reference Yang, Shrestha and Ramalingam32,Reference Tahiri, Llana and Fos-Domenech33 while others have observed effects restricted to male offspring only. Reference Sanchez-Garrido, Ruiz-Pino and Velasco34 Similarly, we did not observe detectable impairments in glycemic control in offspring from obese fathers, a phenotype that has been reported in several previous animal reports, Reference Sanchez-Garrido, Ruiz-Pino and Velasco34–Reference Larque, Lugo-Martinez and Mendoza37 though not all. Reference Schellong, Melchior, Ziska, Rancourt, Henrich and Plagemann38 These discrepant findings may stem from experimental design factors, particularly the length of the pre-conception period which was limited to 6 weeks in our study versus substantially longer durations of 8–14 weeks in other work. However, previous studies with longer exposure windows of 10 weeks Reference Yang, Shrestha and Ramalingam32 and 6 months, Reference Tahiri, Llana and Fos-Domenech33 have also reported no influence of paternal obesity on offspring obesity risk. Thus, it’s conceivable that additional design factors, including diet composition, caloric intake, animal model/strain, and offspring age and sex, likely interact with exposure duration to shape programming outcomes.
Regardless, despite consumption of a postnatal control diet and no differences in offspring obesity status, adult offspring from fathers with obesity demonstrated a clear dyslipidemic phenotype, characterized by increased serum total and LDL/VLDL-cholesterol concentrations. Further, elevated apoB levels and a greater abundance of small LDL particles indicated a more pronounced dyslipidemic effect of paternal obesity in male versus female offspring. By comparison, although data on LDL-C seems limited, the majority of rodent model studies that have assessed serum biochemistry in offspring from high fat-fed and/or obese fathers have reported no change in serum total cholesterol. Reference Fullston, Ohlsson Teague and Palmer7,Reference Ornellas, Souza-Mello, Mandarim-de-Lacerda and Aguila9,Reference Aizawa, Tochihara and Yamamuro39,Reference Chambers, Morgan, Heger, Sharpe and Drake40 Interestingly, Guo et al. Reference Guo, Hu and Bao41 reported no change in serum lipid concentration in F1offspring from obese fathers but observed overt dyslipidemia (i.e., increased TC, HDL-C, and LDL-C) in subsequent generations (male offspring), demonstrating that sustained paternal high fat diet exposure has long-term intergenerational consequences on lipid metabolism Similarly, paternal obesity has been reported to have minimal effects on serum triglyceride levels, Reference Fullston, Ohlsson Teague and Palmer7,Reference Ornellas, Souza-Mello, Mandarim-de-Lacerda and Aguila9,Reference Aizawa, Tochihara and Yamamuro39,Reference Chambers, Morgan, Heger, Sharpe and Drake40 although some studies have documented increases. Reference Masuyama, Mitsui, Eguchi, Tamada and Hiramatsu31,Reference Cropley, Eaton and Aiken42 In line with this, human studies that have assessed associations between paternal obesity and blood lipids in children have largely reported no effect on total and LDL-C. Reference Santos Ferreira, Williams and Kangas19,Reference Zhang, Clayton, Overvad, Olsen, Lawlor and Dahm23,Reference McCarthy, Ye, Yuan and He43,Reference Gaillard, Steegers and Duijts44 Notably, there appears to be a substantial gap in studies examining lipoprotein subclass distribution in offspring of fathers with obesity, with only a single human study identified and no rodent model studies available for direct comparison with our findings. In an analysis of the serum metabolome in children (16, 17, and 31 years of age) from 3 European birth cohorts, Santos Ferreira et al. Reference Santos Ferreira, Williams and Kangas19 reported that increasing paternal BMI (and maternal) was positively associated very-low-density cholesterol and triglyceride and VLDL subclass distribution (i.e., particle number and diameter). Negative associations were noted for HDL-C and HDL particle number and diameter; however no clear associates were observed for LDL lipid concentration and particle subclass. Information on lipoprotein subclass distribution may provide a more nuanced assessment of CVD risk than standard lipid panels. For example, advanced profiling of LDL and VLDL subclasses suggests that atherogenic risk persists in people with low LDL-C, with small VLDL and elevated LDL particle number capturing residual risk not reflected by LDL-C alone. Reference Lawler, Akinkuolie and Chu45,Reference Morze, Melloni and Wittenbecher46 Further, information on HDL particle heterogeneity (subclass number and size) may provide functional insight (i.e., efflux capacity, antioxidant/anti-inflammatory actions) and strong surrogate CVD risk assessment. Reference Costacou, Vaisar and Miller47,Reference Schaefer, Asztalos and Vaisar48 The increased abundance of small LDL particles in male offspring of fathers with obesity in our study may reflect elevated CVD risk, given the enhanced susceptibility of small LDL particles to oxidation and their role in arterial lesion growth. Reference Vekic, Stromsnes, Mazzalai, Zeljkovic, Rizzo and Gambini49 The underlying reasons for the observed male-specific effect on LDL particle number are not known, however sex-specific responses to paternal obesity have been reported previously in other metabolic outcomes. Reference Jazwiec, Patterson and Ribeiro50,Reference Shi, Li and Yu51 Epigenetic alterations in sperm associated with paternal obesity – such as changes in DNA methylation or small non-coding RNAs – may differentially affect metabolic pathways in male versus female offspring, potentially due to interactions with sex chromosomes or postnatal sex hormone environments. Androgen-driven regulation of hepatic lipid handling and LDL receptor expression may render male offspring more susceptible to early-life perturbations, whereas estrogen-mediated protective effects in females may help to buffer adverse lipid profiles in adulthood. Reference Seidemann, Lippold and Rohm52,Reference Palmisano, Zhu, Eckel and Stafford53 In addition, differences in developmental timing may contribute, as male and female offspring exhibit distinct trajectories of metabolic maturation, which could influence the timing and persistence of programmed effects. Reference Dearden, Bouret and Ozanne54 Sex-specific regulation of hepatic lipid metabolism, including differences in lipogenesis, fatty acid oxidation, and lipoprotein secretion, may further predispose males to dyslipidemia under conditions of metabolic challenge. Reference Palmisano, Zhu, Eckel and Stafford53 Finally, sex-specific differences in growth trajectories, and the gut microbiome may further contribute to divergent long-term lipid outcomes between male and female offspring. Reference Zhu, Qi and Shen55
We did not observe any group differences in liver cholesterol or triglyceride concentration in newly-weaned or adult animals from fathers with obesity. Alternatively, using a similar diet-induced obesity model (i.e., species, diet, exposure period) we have previously shown that maternal obesity is associated with liver steatosis in male offspring. Reference Andreani, Mahmood, Kua, Patel and Rideout56,Reference Rideout, Andreani and Pembroke57 This suggests that the in utero exposure to a maternal obesogenic environment may more strongly program offspring susceptibility to liver lipid accumulation. However, several previous rodent model studies have observed hepatic lipid accumulation in livers of offspring from fathers with obesity that was accompanied by enhanced lipogenic gene expression including acetyl-coA carboxylase alpha and fatty acid synthase. Reference Ornellas, Souza-Mello, Mandarim-de-Lacerda and Aguila9,Reference Yang, Shrestha and Ramalingam32,Reference Aizawa, Tochihara and Yamamuro39,Reference Guo, Hu and Bao41 Thus, despite limited characterization of the paternal influence, the available evidence suggests that both maternal and paternal obesity contribute to liver steatosis in offspring, with combined maternal and paternal obesity further exacerbating this phenotype. Reference Ornellas, Souza-Mello, Mandarim-de-Lacerda and Aguila9
While a key strength of this study is the comprehensive assessment of metabolic parameters, including lipoprotein subclass distribution, in both male and female offspring across early and adult life stages, the specific epigenomic alterations in spermatozoa that may influence the expression of lipid-regulatory genes in offspring were not examined. Despite these limitations, study findings suggest that paternal obesity programs dyslipidemia in male and female offspring, independent of early-life growth patterns or the development of obesity in adulthood. Alterations in circulating lipids were evident as early as weaning, with elevated TC and increased serum triglycerides in females. In adulthood, offspring exhibited higher LDL/VLDL-cholesterol, alongside male-specific increases in apoB and a shift toward a more atherogenic lipoprotein subclass profile characterized by greater abundance of small LDL particles. Together, these findings indicate that paternal obesity alone can predispose offspring to an adverse dyslipidemic profile, potentially increasing cardiometabolic disease risk even in the absence of postnatal overnutrition or obesity.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgements
We wish to thank the staff of the Laboratory Animal Facilities at the University at Buffalo.
Author contributions
TL assisted in sample and data analysis and contributed to the writing of the initial draft manuscript; GA and SM assisted in conducting the animal study and sample analysis; LR assisted in data interpretation and manuscript preparation; TR was involved in funding acquisition, data analysis, and manuscript preparation and revision. All authors read and approved the final manuscript.
Funding statement
This research was funded by the United States Department of Agriculture, USDA Pulse Crop Health Initiative (58-3060-0-049, to TCR).
Competing interests
The authors declare that they have no competing interests.
Ethical statement
The animals were cared for in accordance with the University at Buffalo Institutional Animal Care and Use Committee’s guidelines (protocol #202100085).
Consent for publication
Not applicable.






 Open access
Open access