


1. Introduction
Mass spectrometry is a powerful technique to determine the chemical composition of a substance (De Hoffmann and Stroobant, Reference De Hoffmann and Stroobant2007). However, the mass spectrum of a substance does not reveal its exact molecular structure, but merely the possible ratios of elements in the compound and its fragments. To identify a sample, researcher may use commercial databases (for common compounds), or software tools that can discover molecular structures from the partial information available. The latter leads to a combinatorial search problem that is a natural fit for answer set programming (ASP). Molecules can be modeled as undirected graphs, representing the different elements and atomic bonds as node and edge labels, respectively. ASP is well-suited to encode chemical domain knowledge (e.g., possible number of bonds for carbon) and extra information about the sample (e.g., that it has an
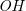 $\textit {OH}$
group), so that each answer set encodes a valid molecular graph.
$\textit {OH}$
group), so that each answer set encodes a valid molecular graph.
Unfortunately, this does not work: a direct ASP encoding yields exponentially many answer sets for each molecular graph due to the large number of symmetries (automorphisms) in such graphs. For example,
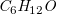 $\textit {C}_6\textit {H}_{12}\textit {O}$
admits 211 distinct molecule structures but leads to 111,870 answer sets. Removing redundant solutions and limiting the search to unique representations are common techniques used in the ASP community where they have motivated research on symmetry-breaking. Related approaches work by rewriting the ground program before solving, see (Drescher et al., Reference Drescher, Tifrea and Walsh2011, sbass) and (Devriendt et al., Reference Devriendt, Bogaerts, Bruynooghe and Denecker2016a; Devriendt and Bogaerts, Reference Devriendt and Bogaerts2016, BreakID), or augmenting ASP programs with additional constraints learned from generated instances (Tarzariol et al., Reference Tarzariol, Gebser, Schekotihin and Law2023, ilasp using sbass). Some methods also integrate symmetry-breaking with existing solvers (Devriendt et al., Reference Devriendt, Bogaerts, Bruynooghe and Denecker2016b, idp3), or provide dedicated solvers (Khaled and Benhamou, Reference Khaled and Benhamou2018, HC-asp). In addition to these general approaches, there are also methods that explicitly define symmetry-breaking constraints for undirected graphs (Codish et al., Reference Codish, Miller, Prosser and Stuckey2019). However, our experiments with some of these approaches still produced 10–10,000 times more answer sets than molecules even in simple cases.
$\textit {C}_6\textit {H}_{12}\textit {O}$
admits 211 distinct molecule structures but leads to 111,870 answer sets. Removing redundant solutions and limiting the search to unique representations are common techniques used in the ASP community where they have motivated research on symmetry-breaking. Related approaches work by rewriting the ground program before solving, see (Drescher et al., Reference Drescher, Tifrea and Walsh2011, sbass) and (Devriendt et al., Reference Devriendt, Bogaerts, Bruynooghe and Denecker2016a; Devriendt and Bogaerts, Reference Devriendt and Bogaerts2016, BreakID), or augmenting ASP programs with additional constraints learned from generated instances (Tarzariol et al., Reference Tarzariol, Gebser, Schekotihin and Law2023, ilasp using sbass). Some methods also integrate symmetry-breaking with existing solvers (Devriendt et al., Reference Devriendt, Bogaerts, Bruynooghe and Denecker2016b, idp3), or provide dedicated solvers (Khaled and Benhamou, Reference Khaled and Benhamou2018, HC-asp). In addition to these general approaches, there are also methods that explicitly define symmetry-breaking constraints for undirected graphs (Codish et al., Reference Codish, Miller, Prosser and Stuckey2019). However, our experiments with some of these approaches still produced 10–10,000 times more answer sets than molecules even in simple cases.
We therefore develop a new approach that prevents symmetries in graph representations already during grounding, and use it as the core of an ASP-based prototype implementation for enumerating molecular structures based on partial chemical information. In Section 2, we explain the problem and our prototype tool from a user perspective. We then define the problem formally in Section 3, using an abstract notion of tree representations of molecular graphs that takes inspiration from the chemical notation simplified molecular-input line-entry system (Weininger, Reference Weininger1988). We then derive a new canonical representation for molecular graphs (Section 4) to guide our ASP implementation (Section 5). In Section 6, we evaluate the correctness, symmetry-breaking capabilities, and performance of our tool in comparison to other ASP-based approaches and a leading commercial software for analytical chemistry (Gugisch et al., Reference Gugisch, Kerber, Kohnert, Laue, Meringer, Rücker and Wassermann2015). We achieve perfect symmetry-breaking, that is the removal of all redundant solutions, for acyclic graph structures and up to three orders of magnitude reduction in answer sets for cyclic cases in comparison to other ASP approaches. Overall, ASP therefore appears to be a promising basis for this use case, and possibly for other use cases concerned with undirected graph structures.
Our ASP source code, evaluation helpers, and data sets are available online at https://github.com/knowsys/eval-2024-asp-molecules. The sources of our prototype application are at https://gitlab.com/nkuechen/genmol/. This work is an extended version of a conference paper that was presented at LPNMR 2024 (Küchenmeister et al., Reference Küchenmeister, Ivliev and Krötzsch2024).
2. Analysis of mass spectra with genmol
Many mass spectrometers break up samples into smaller fragments and measure their relative abundance. The resulting mass spectrum forms a characteristic pattern, enabling inferences about the underlying sample. High-resolution spectra may contain information such as “the molecule has six carbon atoms” or “there is an
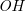 $\textit {OH}$
group,” but cannot reveal the samples’ full molecular structure. In chemical analysis, we are looking for molecular structures that are consistent with the measured mass spectrum.
$\textit {OH}$
group,” but cannot reveal the samples’ full molecular structure. In chemical analysis, we are looking for molecular structures that are consistent with the measured mass spectrum.
To address this task, we have developed Genmol, a prototype application for enumerating molecular structures for a given composition of fragments. It is available as a command-line tool and as a progressive web application (PWA), shown in Figure 1. Genmol is implemented in Rust, with the web front-end using the Yew framework on top of a JSON-API, whereas the search for molecular structures is implemented in Answer Set Programming (ASP) and solved using clingo (Gebser et al., Reference Gebser, Kaufmann, Kaminski, Ostrowski, Schaub and Schneider2011). An online demo of Genmol is available for review at https://tools.iccl.inf.tu-dresden.de/genmol/.
User interface of Genmol.

The screenshot shows the use of Genmol with a sum formula
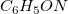 $C_6H_5ON$
and two fragments as input. Specifying detected fragments and restricting bond types helps to reduce the search space. Alternatively, users can provide a molecular mass or a complete mass spectrum, which will then be associated with possible chemical formulas using, for example information about the abundance of isotopes.
$C_6H_5ON$
and two fragments as input. Specifying detected fragments and restricting bond types helps to reduce the search space. Alternatively, users can provide a molecular mass or a complete mass spectrum, which will then be associated with possible chemical formulas using, for example information about the abundance of isotopes.
The core task of Genmol then is to find molecules that match the given input constraints. Molecules in this context are viewed as undirected graphs of atoms, linked by covalent bonds that result from sharing electrons.Footnote 1 Many chemical elements admit a fixed number of bonds, the so-called valence, according to the number of electrons available for binding (e.g., carbon has a valence of 4). Bonds may involve several electrons, leading to single, double, triple bonds, etc. The graph structure of molecules, the assignment of elements, and the possible types of bonds can lead to a large number of possible molecules for a single chemical formula, and this combinatorial search task is a natural match for Answer Set Programming.
3. Problem definition: Enumeration of molecules
We begin by formalizing the problem of molecule enumeration, and by introducing a chemistry-inspired representation of molecules. We consider a set
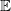 $\mathbb{E}$
of elements, with each
$\mathbb{E}$
of elements, with each
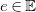 $e\in \mathbb{E}$
associated with a valence
$e\in \mathbb{E}$
associated with a valence
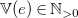 $\mathbb{V}(e)\in \mathbb{N}_{\gt 0}$
. We assume that
$\mathbb{V}(e)\in \mathbb{N}_{\gt 0}$
. We assume that
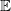 $\mathbb{E}$
contains a distinguished element
$\mathbb{E}$
contains a distinguished element
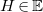 $H\in \mathbb{E}$
(hydrogen) with
$H\in \mathbb{E}$
(hydrogen) with
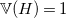 $\mathbb{V}(H)=1$
. Note that in reality, elements may have multiple valences. To simplify presentation, we will ignore this aspect in the formalization. The implementation, however, supports multi-valence elements by representing them as separate elements. We model molecules as undirected graphs with edges labeled by natural numbers to indicate the type of bond.
$\mathbb{V}(H)=1$
. Note that in reality, elements may have multiple valences. To simplify presentation, we will ignore this aspect in the formalization. The implementation, however, supports multi-valence elements by representing them as separate elements. We model molecules as undirected graphs with edges labeled by natural numbers to indicate the type of bond.
Definition 1.
A molecular graph
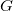 $G$
is a tuple
$G$
is a tuple
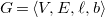 $G=\langle {V, E, \ell , b}\rangle$
with vertices
$G=\langle {V, E, \ell , b}\rangle$
with vertices
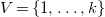 $V=\{1,\ldots ,k\}$
for some
$V=\{1,\ldots ,k\}$
for some
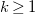 $k\geq 1$
, undirected edges
$k\geq 1$
, undirected edges
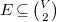 $E \subseteq \binom {V}{2}$
, where
$E \subseteq \binom {V}{2}$
, where
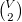 $\binom {V}{2}$
is the set of all 2-element subsets of
$\binom {V}{2}$
is the set of all 2-element subsets of
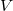 $V$
, and labeling functions
$V$
, and labeling functions
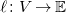 $\ell \colon V \to \mathbb{E}$
and
$\ell \colon V \to \mathbb{E}$
and
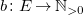 $b \colon E \to \mathbb{N}_{\gt 0}$
.
$b \colon E \to \mathbb{N}_{\gt 0}$
.
The degree
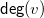 ${\textsf {deg}}(v)$
of a vertex
${\textsf {deg}}(v)$
of a vertex
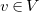 $v\in V$
is defined as
$v\in V$
is defined as
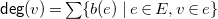 ${\textsf {deg}}(v)=\sum \{b(e)\mid e\in E, v\in e\}$
. A list of
${\textsf {deg}}(v)=\sum \{b(e)\mid e\in E, v\in e\}$
. A list of
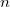 $n$
distinct vertices
$n$
distinct vertices
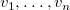 $v_1, \ldots , v_n$
is a simple path in
$v_1, \ldots , v_n$
is a simple path in
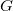 $G$
if
$G$
if
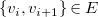 $\{v_i, v_{i+1}\} \in E$
for every
$\{v_i, v_{i+1}\} \in E$
for every
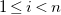 $1 \leq i \lt n$
. The graph
$1 \leq i \lt n$
. The graph
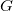 $G$
is connected if there is a simple path from
$G$
is connected if there is a simple path from
 $v$
to
$v$
to
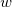 $w$
for every pair
$w$
for every pair
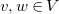 $v, w \in V$
.
$v, w \in V$
.
Since we assume that atoms in a molecule use all available bonds according to their valence, the (very frequent) hydrogen atoms do not need to be mentioned explicitly. Such hydrogen-suppressed molecular graphs are common in computational chemistry, and the basis for the next definition:
Definition 2.
A molecular formula is a function
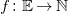 $f\colon \mathbb{E}\to \mathbb{N}$
. A (hydrogen-suppressed) molecular graph
$f\colon \mathbb{E}\to \mathbb{N}$
. A (hydrogen-suppressed) molecular graph
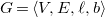 $G=\langle {V, E, \ell , b}\rangle$
is valid for
$G=\langle {V, E, \ell , b}\rangle$
is valid for
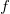 $f$
, if it satisfies the following properties:
$f$
, if it satisfies the following properties:
-
(1)
 $G$
is connected,
$G$
is connected,
-
(2) for every
$e \in \mathbb{E}$
with
$e \neq H$
,
$\#\{v \in V \mid \ell (v) = e \} = f(e)$
, -
(3) for every
$v \in V$
,
${\textsf {deg}}(v) \leq \mathbb{V}(\ell (v))$
, -
(4)
$\sum _{v \in V} \left (\mathbb{V}(\ell (v)) - {\textsf {deg}}(v)\right ) = f(H)$
.
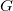
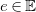
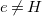
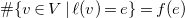

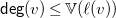
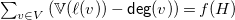
The enumeration problem can now be stated as follows: for a given molecular formula
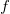 $f$
, enumerate, up to isomorphism, all valid molecular graphs for
$f$
, enumerate, up to isomorphism, all valid molecular graphs for
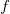 $f$
. In general, the number of distinct isomorphic molecular graphs is exponential in the number of atoms. It is therefore important to reduce the enumeration of redundant isomorphic graphs.
$f$
. In general, the number of distinct isomorphic molecular graphs is exponential in the number of atoms. It is therefore important to reduce the enumeration of redundant isomorphic graphs.
A first step towards this is the use of a more restricted representation of molecular graphs. Here, we take inspiration from the simplified molecular-input line-entry system (SMILES), a widely used serialization format for molecular graphs. SMILES strings start from an (arbitrary) spanning tree of the molecular graph, serialized in a depth-first order, with subtrees enclosed in parentheses. Edges not covered by the spanning tree (since they would complete a cycle) are indicated by pairs of numeric cycle markers.
Hydrogen-suppressed molecular graph of adenine (
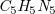 $\mathit{C}_5\mathit{H}_5\mathit{N}_5$
) and corresponding spanning tree with cycle edges (dotted); superscripts indicate correspondence of vertices.
$\mathit{C}_5\mathit{H}_5\mathit{N}_5$
) and corresponding spanning tree with cycle edges (dotted); superscripts indicate correspondence of vertices.

Example 3.
Adenine (
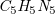 $\textit {C}_5\textit {H}_5\textit {N}_5$
) has the graph structure shown in Figure
2
(left), with a spanning tree on the right. Hydrogen atoms are omitted, as they take up any free binding place. For instance,
$\textit {C}_5\textit {H}_5\textit {N}_5$
) has the graph structure shown in Figure
2
(left), with a spanning tree on the right. Hydrogen atoms are omitted, as they take up any free binding place. For instance, ![]() has a double- and a single-bond and a valence of four, leaving one hydrogen atom. The SMILES is C1(N = CN = C2N)=C2N=CN1 where consecutive atoms are connected by bonds and double bonds are marked (=). The segment from vertex 2 to 6 is in parentheses to indicate a branch. Additionally, the two non-sequential (dotted) connections are denoted by matching pairs of numerical markers.
has a double- and a single-bond and a valence of four, leaving one hydrogen atom. The SMILES is C1(N = CN = C2N)=C2N=CN1 where consecutive atoms are connected by bonds and double bonds are marked (=). The segment from vertex 2 to 6 is in parentheses to indicate a branch. Additionally, the two non-sequential (dotted) connections are denoted by matching pairs of numerical markers.
Definition 4.
A molecular graph
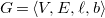 $G=\langle {V, E, \ell , b}\rangle$
is a molecular tree if it is free of cycles and the natural order of vertices
$G=\langle {V, E, \ell , b}\rangle$
is a molecular tree if it is free of cycles and the natural order of vertices
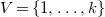 $V=\{1,\ldots ,k\}$
corresponds to a depth-first search of the tree (in particular, vertex
$V=\{1,\ldots ,k\}$
corresponds to a depth-first search of the tree (in particular, vertex
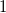 $1$
is the root).
$1$
is the root).
A tree representation of an arbitrary molecular graph
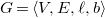 $G=\langle {V, E, \ell , b}\rangle$
is a set
$G=\langle {V, E, \ell , b}\rangle$
is a set
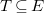 $T\subseteq E$
such that
$T\subseteq E$
such that
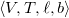 $\langle {V, T, \ell , b}\rangle$
is a molecular tree. In this case, we denote
$\langle {V, T, \ell , b}\rangle$
is a molecular tree. In this case, we denote
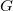 $G$
as
$G$
as
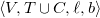 $\langle {V, T \cup C, \ell , b}\rangle$
where
$\langle {V, T \cup C, \ell , b}\rangle$
where
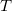 $T$
are the tree edges and
$T$
are the tree edges and
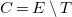 $C=E\setminus T$
are the cycle edges.
$C=E\setminus T$
are the cycle edges.
Note that the tree edges
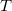 $T$
by definition visit every vertex of
$T$
by definition visit every vertex of
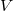 $V$
, and the cycle edges
$V$
, and the cycle edges
 $C$
merely make additional connections between vertices of the tree. In SMILES, the edges in
$C$
merely make additional connections between vertices of the tree. In SMILES, the edges in
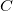 $C$
and their labels (bond types) are encoded with special markers, while the order of vertices is given by the order of appearance in the SMILES string.
$C$
and their labels (bond types) are encoded with special markers, while the order of vertices is given by the order of appearance in the SMILES string.
A tree representation for a given molecular graph is uniquely determined by the following choices: (a) a spanning tree (corresponding to a choice of tree edges and cycle edges), (b) a root vertex, and (c) for every vertex, an order of visiting its child vertices in depth-first search. For a given graph structure, the number of tree representations can be significantly lower than the number of isomorphic molecular graphs. For example, a graph that is a chain has only linearly many tree representations, but still admits exponentially many graphs. Nevertheless, the number of tree representations can still be exponential, and we will investigate below how the choices in (a)–(c) can be further constrained to reduce redundancy.
4. Canonical tree representations of molecular graphs
To eliminate redundant isomorphic solutions, we first define a canonical tree representation of any molecular graph. The defining conditions of this unique representation will then be used to constrain the search for possible graphs in our implementation. We first consider the simpler case of molecular trees.
4.1 Canonical molecular trees
We define a total order on molecular trees, which will allow us to define a largest tree among a set of candidates. To define this order inductively, we need to consider subtrees that may not have
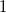 $1$
as their root. For a molecular tree
$1$
as their root. For a molecular tree
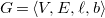 $G=\langle {V, E, \ell , b}\rangle$
with vertex
$G=\langle {V, E, \ell , b}\rangle$
with vertex
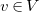 $v\in V$
, let
$v\in V$
, let
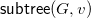 ${\textsf {subtree}}(G,v)$
be the tuple
${\textsf {subtree}}(G,v)$
be the tuple
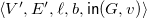 $\langle {V', E', \ell , b, {\textsf {in}}(G,v)}\rangle$
where
$\langle {V', E', \ell , b, {\textsf {in}}(G,v)}\rangle$
where
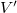 $V'$
and
$V'$
and
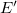 $E'$
are the restriction of
$E'$
are the restriction of
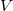 $V$
and
$V$
and
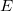 $E$
, respectively, to vertices that are part of the subtree with root
$E$
, respectively, to vertices that are part of the subtree with root
 $v$
in
$v$
in
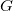 $G$
, and either
$G$
, and either
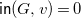 ${\textsf {in}}(G,v)=0$
if
${\textsf {in}}(G,v)=0$
if
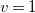 $v=1$
is the root of
$v=1$
is the root of
 $G$
, or
$G$
, or
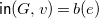 ${\textsf {in}}(G,v)=b(e)$
is the edge label
${\textsf {in}}(G,v)=b(e)$
is the edge label
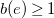 $b(e)\geq 1$
of the edge
$b(e)\geq 1$
of the edge
 $e$
between
$e$
between
 $v$
and its parent in
$v$
and its parent in
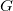 $G$
. Moreover, if
$G$
. Moreover, if
 $\langle {c_1,\ldots ,c_k}\rangle$
are the ordered children of
$\langle {c_1,\ldots ,c_k}\rangle$
are the ordered children of
 $v$
, then
$v$
, then
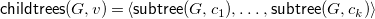 ${\textsf {childtrees}}(G,v)=\langle {{\textsf {subtree}}(G,c_1),\ldots ,{\textsf {subtree}}(G,c_k)}\rangle$
.
${\textsf {childtrees}}(G,v)=\langle {{\textsf {subtree}}(G,c_1),\ldots ,{\textsf {subtree}}(G,c_k)}\rangle$
.
Finally, let
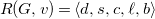 $R(G,v)=\langle {d,s,c,\ell ,b}\rangle$
be the tuple with
$R(G,v)=\langle {d,s,c,\ell ,b}\rangle$
be the tuple with
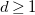 $d\geq 1$
the depth of
$d\geq 1$
the depth of
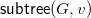 ${\textsf {subtree}}(G,v)$
;
${\textsf {subtree}}(G,v)$
;
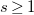 $s\geq 1$
the size (number of vertices) of
$s\geq 1$
the size (number of vertices) of
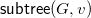 ${\textsf {subtree}}(G,v)$
;
${\textsf {subtree}}(G,v)$
;
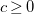 $c\geq 0$
the number of children of
$c\geq 0$
the number of children of
 $v$
;
$v$
;
 $\ell \in \mathbb{E}$
the element
$\ell \in \mathbb{E}$
the element
 $\ell (v)$
of
$\ell (v)$
of
 $v$
; and
$v$
; and
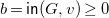 $b={\textsf {in}}(G,v)\geq 0$
. Intuitively, we use
$b={\textsf {in}}(G,v)\geq 0$
. Intuitively, we use
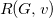 $R(G, v)$
to define an order between nodes, which we extend to an order between molecular trees. For the following definition, recall that the lexicographic extension of a strict order
$R(G, v)$
to define an order between nodes, which we extend to an order between molecular trees. For the following definition, recall that the lexicographic extension of a strict order
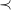 $\prec$
to tuples of the same size
$\prec$
to tuples of the same size
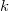 $k$
is defined by setting
$k$
is defined by setting
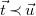 $\vec {t}\prec \vec {u}$
if there is
$\vec {t}\prec \vec {u}$
if there is
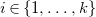 $i\in \{1,\ldots ,k\}$
such that
$i\in \{1,\ldots ,k\}$
such that
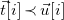 $\vec {t}[i]\prec \vec {u}[i]$
and
$\vec {t}[i]\prec \vec {u}[i]$
and
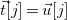 $\vec {t}[j]=\vec {u}[j]$
for all
$\vec {t}[j]=\vec {u}[j]$
for all
 $j\lt i$
.
$j\lt i$
.
Definition 5.
Let
 $\sqsubset$
be an arbitrary but fixed strict total order on
$\sqsubset$
be an arbitrary but fixed strict total order on
 $\mathbb{E}$
, overloaded to also denote the usual order
$\mathbb{E}$
, overloaded to also denote the usual order
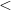 $\lt$
on natural numbers. We further extend
$\lt$
on natural numbers. We further extend
 $\sqsubset$
to 5-tuples of the form
$\sqsubset$
to 5-tuples of the form
 $R(G,v)$
lexicographically.
$R(G,v)$
lexicographically.
We define a strict order
 $\prec$
on subtrees as the smallest relation where, for each pair of subtrees
$\prec$
on subtrees as the smallest relation where, for each pair of subtrees
 $S_i={\textsf {subtree}}(G_i,v_i)$
of molecular trees
$S_i={\textsf {subtree}}(G_i,v_i)$
of molecular trees
 $G_i$
(
$G_i$
(
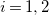 $i=1,2$
)
$i=1,2$
)
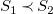 $S_1\prec S_2$
holds if
$S_1\prec S_2$
holds if
-
(1)
$R(G_1,v_1)\sqsubset R(G_2,v_2)$
, or
-
(2)
$R(G_1,v_1)=R(G_2,v_2)$
, that is
$S_1$
and
$S_2$
are locally indistinguishable, with (necessarily equal) number of children
$k$
, such that
${\textsf {childtrees}}(G_1,v_1)\prec {\textsf {childtrees}}(G_2,v_2)$
where
$\prec$
is the lexicographic extension of
$\prec$
to
$k$
-tuples of subtrees.
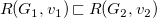
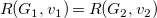
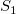

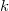

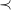

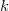
For molecular trees
 $G_1$
and
$G_1$
and
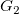 $G_2$
, we define
$G_2$
, we define
 $G_1\prec G_2$
if
$G_1\prec G_2$
if
 ${\textsf {subtree}}(G_1,1)\prec {\textsf {subtree}}(G_2,1)$
.
${\textsf {subtree}}(G_1,1)\prec {\textsf {subtree}}(G_2,1)$
.
Example 6.
A tree representation of threonine, annotated with the respective
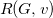 $R(G, v)$
tuple for all of its nodes, is shown in Figure
3
. In two instances, pairs of nodes share the same
$R(G, v)$
tuple for all of its nodes, is shown in Figure
3
. In two instances, pairs of nodes share the same
 $R(G, v)$
value. For example, nodes
$R(G, v)$
value. For example, nodes ![]() and
and ![]() are indistinguishable by Definition
5
(1), but Definition
5
(2) yields
are indistinguishable by Definition
5
(1), but Definition
5
(2) yields
 ${\textsf {subtree}}(G,2) \prec {\textsf {subtree}}(G,5)$
.
${\textsf {subtree}}(G,2) \prec {\textsf {subtree}}(G,5)$
.
Canonical molecular tree of threonine (
 $\mathit{C}_4\mathit{H}_9\mathit{N}\mathit{O}_3$
); central vertex is circled.
$\mathit{C}_4\mathit{H}_9\mathit{N}\mathit{O}_3$
); central vertex is circled.

Proposition 7.
The relation
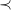 $\prec$
of Definition
5
is a strict total order on molecular trees.
$\prec$
of Definition
5
is a strict total order on molecular trees.
Proof.
The claim follows by showing that
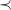 $\prec$
is a strict order on subtrees. The order
$\prec$
is a strict order on subtrees. The order
 $\sqsubset$
on tuples is a strict total order since it is the lexicographic extension of strict total orders. Hence, all subtrees
$\sqsubset$
on tuples is a strict total order since it is the lexicographic extension of strict total orders. Hence, all subtrees
 $S_i={\textsf {subtree}}(G_i,v_i)$
(
$S_i={\textsf {subtree}}(G_i,v_i)$
(
 $i=1,2$
) with
$i=1,2$
) with
 $R(G_1,v_1)\neq R(G_2,v_2)$
are
$R(G_1,v_1)\neq R(G_2,v_2)$
are
 $\prec$
-comparable by 1. Totality for case
$\prec$
-comparable by 1. Totality for case
 $R(G_1,v_1)= R(G_2,v_2)$
is shown by induction on the (equal) depth of the subtrees
$R(G_1,v_1)= R(G_2,v_2)$
is shown by induction on the (equal) depth of the subtrees
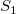 $S_1$
and
$S_1$
and
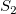 $S_2$
. For depth
$S_2$
. For depth
 $1$
,
$1$
,
 $S_1$
and
$S_1$
and
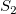 $S_2$
have no children, and
$S_2$
have no children, and
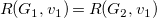 $R(G_1,v_1)=R(G_2,v_1)$
implies
$R(G_1,v_1)=R(G_2,v_1)$
implies
 $S_1=S_2$
. For depth
$S_1=S_2$
. For depth
 $i$
with
$i$
with
 $i\gt 1$
, we can assume all subtrees of depth
$i\gt 1$
, we can assume all subtrees of depth
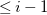 $\leq i-1$
to be
$\leq i-1$
to be
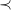 $\prec$
-comparable unless equal. If
$\prec$
-comparable unless equal. If
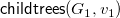 ${\textsf {childtrees}}(G_1,v_1)$
and
${\textsf {childtrees}}(G_1,v_1)$
and
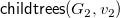 ${\textsf {childtrees}}(G_2,v_2)$
are comparable under the lexicographic extension of
${\textsf {childtrees}}(G_2,v_2)$
are comparable under the lexicographic extension of
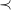 $\prec$
, then
$\prec$
, then
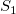 $S_1$
and
$S_1$
and
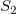 $S_2$
are
$S_2$
are
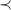 $\prec$
-comparable by 2. Otherwise,
$\prec$
-comparable by 2. Otherwise,
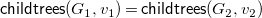 ${\textsf {childtrees}}(G_1,v_1)={\textsf {childtrees}}(G_2,v_2)$
, and therefore
${\textsf {childtrees}}(G_1,v_1)={\textsf {childtrees}}(G_2,v_2)$
, and therefore
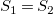 $S_1=S_2$
.
$S_1=S_2$
.
By Proposition7, we could define the canonical molecular tree to be the
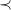 $\prec$
-largest tree among a set of isomorphic trees. However, this would force us to select a root that is the start (or end) of a longest path in the graph to maximize the depth of the tree. In general, many such nodes might exist. It is therefore more efficient to compare a smaller set of potential roots that are closer together:
$\prec$
-largest tree among a set of isomorphic trees. However, this would force us to select a root that is the start (or end) of a longest path in the graph to maximize the depth of the tree. In general, many such nodes might exist. It is therefore more efficient to compare a smaller set of potential roots that are closer together:
Definition 8.
Let
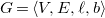 $G=\langle {V, E, \ell , b}\rangle$
be a molecular tree. A vertex
$G=\langle {V, E, \ell , b}\rangle$
be a molecular tree. A vertex
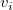 $v_i$
is central in a simple path
$v_i$
is central in a simple path
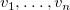 $v_1,\ldots ,v_n$
in
$v_1,\ldots ,v_n$
in
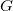 $G$
if
$G$
if
 $i\in \{\lceil (n+1)/2\rceil ,\lfloor (n+1)/2\rfloor \}$
(a singleton set if
$i\in \{\lceil (n+1)/2\rceil ,\lfloor (n+1)/2\rfloor \}$
(a singleton set if
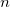 $n$
is odd). A vertex is central in
$n$
is odd). A vertex is central in
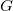 $G$
if it is central in any longest simple path in
$G$
if it is central in any longest simple path in
 $G$
.
$G$
.
The canonical molecular tree
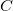 $C$
of
$C$
of
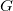 $G$
is the
$G$
is the
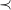 $\prec$
-largest molecular tree that is obtained by permutation of vertices in
$\prec$
-largest molecular tree that is obtained by permutation of vertices in
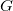 $G$
such that the root of
$G$
such that the root of
 $C$
is central in
$C$
is central in
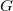 $G$
.
$G$
.
In every tree, the central vertices of all longest simple paths are the same, and hence there are at most two. Indeed, two distinct longest paths always share at least one vertex in a tree. So if two such paths
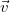 $\vec {v}$
and
$\vec {v}$
and
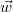 $\vec {w}$
would have different central vertices
$\vec {w}$
would have different central vertices
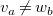 $v_a\neq w_b$
, and a shared vertex
$v_a\neq w_b$
, and a shared vertex
 $v_i=w_j$
with (w.l.o.g.)
$v_i=w_j$
with (w.l.o.g.)
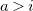 $a\gt i$
and
$a\gt i$
and
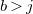 $b\gt j$
, then the path
$b\gt j$
, then the path
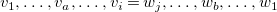 $v_1,\ldots ,v_a,\ldots ,v_i=w_j,\ldots ,w_b,\ldots ,w_1$
would be longer than
$v_1,\ldots ,v_a,\ldots ,v_i=w_j,\ldots ,w_b,\ldots ,w_1$
would be longer than
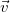 $\vec {v}$
and
$\vec {v}$
and
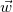 $\vec {w}$
, contradicting their assumed maximal length. Using this insight, our implementation can find the canonical molecular tree by considering at most two possible roots.
$\vec {w}$
, contradicting their assumed maximal length. Using this insight, our implementation can find the canonical molecular tree by considering at most two possible roots.
Example 9.
Two alternative tree representations for glycine are depicted in Figure
4. The red circles indicate central vertices, which are considered as candidates for the root of the spanning tree. Comparing the roots of both trees, we find that the tree representation on the right has more children, while the corresponding subtrees are identical in depth and number of vertices. Therefore,
 $R(G,1) \sqsubset R(G',1)$
, which implies that the right-hand tree is
$R(G,1) \sqsubset R(G',1)$
, which implies that the right-hand tree is
 $\prec$
-larger and thus the canonical molecular tree by Definition
8
.
$\prec$
-larger and thus the canonical molecular tree by Definition
8
.
Molecular trees of glycine (
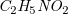 $\mathit{C}_2\mathit{H}_5\mathit{N}\mathit{O}_2$
); central vertices are circled.
$\mathit{C}_2\mathit{H}_5\mathit{N}\mathit{O}_2$
); central vertices are circled.

4.2 Canonical molecular graphs
Next, we define a canonical tree representation for arbitrary molecular graphs, allowing us to extend the above order on trees to an order on graphs. Let
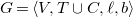 $G=\langle {V, T \cup C, \ell , b}\rangle$
be a tree representation with
$G=\langle {V, T \cup C, \ell , b}\rangle$
be a tree representation with
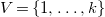 $V=\{1,\ldots ,k\}$
. We construct a molecular tree
$V=\{1,\ldots ,k\}$
. We construct a molecular tree
 $G'={\textsf {tr}}(G)$
by replacing each cycle edge
$G'={\textsf {tr}}(G)$
by replacing each cycle edge
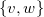 $\{v, w\}$
in
$\{v, w\}$
in
 $C$
with two new edges, one from
$C$
with two new edges, one from
 $v$
and one from
$v$
and one from
 $w$
leading to fresh vertices. Hence, let
$w$
leading to fresh vertices. Hence, let
 $V' = \{k + 1, \dots , k + 2 \cdot |C|\}$
be the set of fresh vertices and assume that the cycle edges
$V' = \{k + 1, \dots , k + 2 \cdot |C|\}$
be the set of fresh vertices and assume that the cycle edges
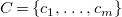 $C = \{c_1, \dots , c_m\}$
are numbered in an arbitrary way. The edges in
$C = \{c_1, \dots , c_m\}$
are numbered in an arbitrary way. The edges in
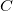 $C$
are replaced by new tree edges
$C$
are replaced by new tree edges
 \begin{equation*} T' = \{ \{\min c_i, k + 2i - 1\}, \{\max c_i, k + 2i\} \mid c_i \in C \}. \end{equation*}
\begin{equation*} T' = \{ \{\min c_i, k + 2i - 1\}, \{\max c_i, k + 2i\} \mid c_i \in C \}. \end{equation*}
Each new edge inherits the label of its corresponding cycle edge, and each new vertex receives the label of the node that was connected to its parent in the cycle edge. Formally, we obtain the labeling functions
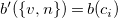 $b'(\{v, n\}) = b(c_i)$
and
$b'(\{v, n\}) = b(c_i)$
and
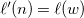 $\ell '(n) = \ell (w)$
for
$\ell '(n) = \ell (w)$
for
 $c_i = \{v, w\} \in C$
,
$c_i = \{v, w\} \in C$
,
 $\{v, n\} \in T'$
, with
$\{v, n\} \in T'$
, with
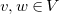 $v, w \in V$
and
$v, w \in V$
and
 $n \in \{k + 2i - 1, k + 2i\} \subseteq V'$
. Thus, the transformed molecular tree is
$n \in \{k + 2i - 1, k + 2i\} \subseteq V'$
. Thus, the transformed molecular tree is
 $G' = \langle {V \cup V', T \cup T', \ell \cup \ell ', b \cup b'}\rangle$
.
$G' = \langle {V \cup V', T \cup T', \ell \cup \ell ', b \cup b'}\rangle$
.
Tree representation of adenine and molecular tree with replaced cycle edges.

Example 10
(Continuation of Example 3). Figure
5
shows the tree representation
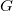 $G$
of Adenine on the left, which has two cycle edges (depicted in orange). Modifying it by adding two fresh nodes each, the molecular tree
$G$
of Adenine on the left, which has two cycle edges (depicted in orange). Modifying it by adding two fresh nodes each, the molecular tree
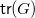 ${\textsf {tr}}(G)$
shown on the right emerges.
${\textsf {tr}}(G)$
shown on the right emerges.
Given two tree representations
 $G_1$
and
$G_1$
and
 $G_2$
, we define
$G_2$
, we define
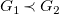 $G_1\prec G_2$
if
$G_1\prec G_2$
if
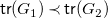 ${\textsf {tr}}(G_1)\prec {\textsf {tr}}(G_2)$
. This does not define a total order, since
${\textsf {tr}}(G_1)\prec {\textsf {tr}}(G_2)$
. This does not define a total order, since
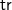 $\textsf {tr}$
is not injective. However, on any set of tree representations with the same number of vertices (and especially on any set of isomorphic tree representations),
$\textsf {tr}$
is not injective. However, on any set of tree representations with the same number of vertices (and especially on any set of isomorphic tree representations),
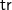 $\textsf {tr}$
is injective and
$\textsf {tr}$
is injective and
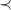 $\prec$
is total.
$\prec$
is total.
Though
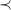 $\prec$
defines a largest tree representation of any molecular graph, it is impractical to consider every possible such representation in search of this optimum. We therefore restrict to tree representations where the tree edges are identified by iterative addition of longest simple paths that do not create cycles.
$\prec$
defines a largest tree representation of any molecular graph, it is impractical to consider every possible such representation in search of this optimum. We therefore restrict to tree representations where the tree edges are identified by iterative addition of longest simple paths that do not create cycles.
Definition 11.
A pre-tree representation is a molecular graph
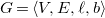 $G=\langle {V, E, \ell , b}\rangle$
where
$G=\langle {V, E, \ell , b}\rangle$
where
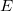 $E$
is a disjoint union
$E$
is a disjoint union
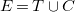 $E=T\cup C$
such that the edges of
$E=T\cup C$
such that the edges of
 $T$
define a tree (possibly not a spanning tree for
$T$
define a tree (possibly not a spanning tree for
 $G$
).
$G$
).
An extension of
 $G$
is a simple path
$G$
is a simple path
 $v_1,\ldots ,v_n$
such that
$v_1,\ldots ,v_n$
such that
 $v_1\in T$
and
$v_1\in T$
and
 $v_2,\ldots ,v_n\in C$
or
$v_2,\ldots ,v_n\in C$
or
 $v_1,\ldots ,v_n \in C$
if
$v_1,\ldots ,v_n \in C$
if
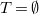 $T = \emptyset$
. A longest extension is one of maximal length among all extensions of
$T = \emptyset$
. A longest extension is one of maximal length among all extensions of
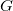 $G$
. A refinement of
$G$
. A refinement of
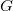 $G$
is a pre-tree representation
$G$
is a pre-tree representation
 $G'=\langle {V, T'\cup C', \ell , b}\rangle$
, where
$G'=\langle {V, T'\cup C', \ell , b}\rangle$
, where
 $T'=T\cup P$
and
$T'=T\cup P$
and
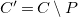 $C'=C\setminus P$
for a set of edges
$C'=C\setminus P$
for a set of edges
 $P$
of some longest extension of
$P$
of some longest extension of
 $G$
.
$G$
.
We can view any molecular graph as a pre-tree representation with
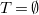 $T=\emptyset$
and refine it iteratively. Refinements exist whenever there is a vertex that is not reached in
$T=\emptyset$
and refine it iteratively. Refinements exist whenever there is a vertex that is not reached in
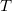 $T$
. Hence, a pre-tree representation admits no further refinement exactly if it is a tree representation.
$T$
. Hence, a pre-tree representation admits no further refinement exactly if it is a tree representation.
Refinement steps on a molecule with dotted edges in
 $C$
and solid edges in
$C$
and solid edges in
 $T$
.
$T$
.

Example 12.
Figure
6
visualizes how a pre-tree representation is refined along the lines of Definition
11
, starting with
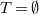 $T = \emptyset$
depicted on the left and arriving at a spanning tree in the rightmost depiction.
$T = \emptyset$
depicted on the left and arriving at a spanning tree in the rightmost depiction.
Definition 13.
A maximal refinement of a molecular graph
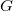 $G$
is a tree representation that is obtained from
$G$
is a tree representation that is obtained from
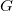 $G$
by a finite sequence of refinements. A centralized maximal refinement is a tree representation obtained from a maximal refinement by a permutation of vertices such that the root is central in its spanning tree (analogous to Definition
8
). The canonical tree representation of a molecular graph
$G$
by a finite sequence of refinements. A centralized maximal refinement is a tree representation obtained from a maximal refinement by a permutation of vertices such that the root is central in its spanning tree (analogous to Definition
8
). The canonical tree representation of a molecular graph
 $G$
is its
$G$
is its
 $\prec$
-largest centralized maximal refinement.
$\prec$
-largest centralized maximal refinement.
In particular, the canonical tree representation coincides with the canonical molecular tree if
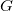 $G$
is free of cycles.
$G$
is free of cycles.
Centralized maximal refinement (right) of the maximal refinement (left).

5. ASP implementation
We compute the
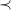 $\prec$
-largest molecular graphs using Answer Set Programming (ASP), a declarative logic programming language that is well-suited for combinatorial search problems. Compared to a direct implementation, ASP offers two main advantages: the solver efficiently handles traversal of the search space, and ASP can serve as a domain-specific language to encode additional constraints on the molecules.
$\prec$
-largest molecular graphs using Answer Set Programming (ASP), a declarative logic programming language that is well-suited for combinatorial search problems. Compared to a direct implementation, ASP offers two main advantages: the solver efficiently handles traversal of the search space, and ASP can serve as a domain-specific language to encode additional constraints on the molecules.
5.1 Answer set programming
We give a brief introduction to Answer Set Programming in this section, and direct interested readers to Eiter et al. (Reference Eiter, Ianni and Krennwallner2009) and Faber (Reference Faber2020) for a comprehensive treatment. ASP programs consist of facts, rules, and constraints that encode a problem, with stable models corresponding to solutions. To illustrate, consider the following simple ASP program:

Line 2 of the above program declares three available ions as facts. Line 4 contains a choice rule, selecting between one and two ![]() facts. The rule on line 6 is read from right to left, specifying that
facts. The rule on line 6 is read from right to left, specifying that ![]() is derived if sodium and chloride were selected. The constraint on line 8 filters out solutions that contain both potassium and chloride.
is derived if sodium and chloride were selected. The constraint on line 8 filters out solutions that contain both potassium and chloride.
Evaluation of ASP programs follows the guess-and-check paradigm: choice rules or negations are used to “guess” candidate solutions, which are then tested by rules and constraints to determine whether they constitude valid solutions. For our example program, we obtain the following solutions:

Although the order of rule appearance in ASP programs is of no consequence semantically, their logical interdependence prescribes an intuitive sequence of consequences. For ease of presentation, we therefore may use vocabulary such as “first” and “then” in description of such programs where suitable.
Modern solvers like clingo (Gebser et al., Reference Gebser, Kaufmann, Kaminski, Ostrowski, Schaub and Schneider2011) in addition allow for more advanced features, of which we make occasional use in our encoding. This includes aggregates like ![]() , which compute for example the minimum in a set of values, syntactic sugar like range notation
, which compute for example the minimum in a set of values, syntactic sugar like range notation ![]() , which expands to
, which expands to ![]() , and Python script blocks that allow us to define custom functions.
, and Python script blocks that allow us to define custom functions.
5.2 Application to molecular graph generation
Our implementation incorporates many of the conditions on canonical tree representations in the rules that infer these structures, rather than relying on constraints to filter redundant representations later. This is in contrast to other known approaches (Gebser et al., Reference Gebser, Janhunen and Rintanen2020) for tree generation, which first guess an edge relation and then prune inadequate graphs, thereby producing many isomorphic results. For molecular trees, our implementation achieves full symmetry-breaking along the lines of Definition8. For graphs with cycles, we merely approximate the conditions from Definition13, since the required
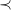 $\prec$
-maximality in this case seems to require a computationally prohibitive search in ASP.Footnote
2
It proceeds in the following steps:
$\prec$
-maximality in this case seems to require a computationally prohibitive search in ASP.Footnote
2
It proceeds in the following steps:
-
1. Choosing main-chain length, number of multi-bonds, and cycle edges.
-
2. Distributing the element symbols, multi-bonds, and cycle edges on the vertices.
-
3. Building the spanning tree.
-
4. Comparing sibling subtrees and ensure they are
$\prec$
-decreasing from left to right. -
5. Pruning representations that cannot be
$\prec$
-maximal due to bad choice of cycle edges.

As in Definition4, our implementation identifies vertices with integers, with ![]() being the root. Input molecular formulas are encoded in facts
being the root. Input molecular formulas are encoded in facts ![]() and
and ![]() , for elements
, for elements
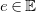 $e\in \mathbb{E}$
with atomic number
$e\in \mathbb{E}$
with atomic number
 $n_e$
(for usual ordering) and valence
$n_e$
(for usual ordering) and valence
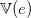 $\mathbb{V}(e)$
. We encode tree representations
$\mathbb{V}(e)$
. We encode tree representations
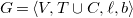 $G = \langle {V, T \cup C, \ell , b}\rangle$
by facts
$G = \langle {V, T \cup C, \ell , b}\rangle$
by facts ![]() and
and ![]() for
for
 $v \in V$
;
$v \in V$
; ![]() for
for
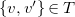 $\{v, v'\}\in T$
where
$\{v, v'\}\in T$
where
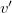 $v'$
is the
$v'$
is the
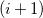 $(i+1)$
th child of
$(i+1)$
th child of
 $v$
; and
$v$
; and ![]() for
for
 $\{ v, v' \} \in C$
with
$\{ v, v' \} \in C$
with
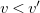 $v \lt v'$
, where
$v \lt v'$
, where
 $c$
is a unique integer id for this edge. Multiplicities of bonds
$c$
is a unique integer id for this edge. Multiplicities of bonds
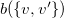 $b(\{ v, v' \})$
are encoded only if
$b(\{ v, v' \})$
are encoded only if
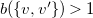 $b(\{ v, v' \})\gt 1$
: for
$b(\{ v, v' \})\gt 1$
: for
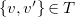 $\{ v, v' \} \in T$
, we associate bonds with the child
$\{ v, v' \} \in T$
, we associate bonds with the child
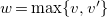 $w=\max \{ v, v' \}$
using
$w=\max \{ v, v' \}$
using ![]() , whereas for
, whereas for
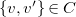 $\{ v, v' \} \in C$
, we encode
$\{ v, v' \} \in C$
, we encode
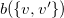 $b(\{ v, v' \})$
single-bond cycles for the same
$b(\{ v, v' \})$
single-bond cycles for the same
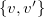 $\{ v, v' \}$
, which showed better performance in this case. See A for a tabular overview of all predicates used in our encoding.
$\{ v, v' \}$
, which showed better performance in this case. See A for a tabular overview of all predicates used in our encoding.
Example 15
(Continuation of Example 3). The encoding for the sum formula
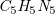 $\textit {C}_5\textit {H}_5\textit {N}_5$
would look like:
$\textit {C}_5\textit {H}_5\textit {N}_5$
would look like:
![]()
It necessitates the following valence information:
![]()
Matching this sum formula, Adenine (see also Figure 2 ) would be encoded like so:

Given the input, we guess facts for ![]() , as well as
, as well as ![]() . For efficiency, we avoid aggregates and instead proceed iteratively, updating counters as we make guesses. We constrain possible guesses based on Definition2. For example, given a molecular formula
. For efficiency, we avoid aggregates and instead proceed iteratively, updating counters as we make guesses. We constrain possible guesses based on Definition2. For example, given a molecular formula
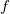 $f$
, the number of “additional” bonds used in cycles and multi-bonds
$f$
, the number of “additional” bonds used in cycles and multi-bonds
 \begin{equation*} |C| + \sum _{\{v, v'\} \in T \cup C} \big ( b(\{v, v'\})-1 \big ) \end{equation*}
\begin{equation*} |C| + \sum _{\{v, v'\} \in T \cup C} \big ( b(\{v, v'\})-1 \big ) \end{equation*}
known as the degree of unsaturation in chemistry, can be computed as
 \begin{equation*} 1 + \frac {1}{2} \sum _{e \in \mathbb{E}} f(e) \cdot (\mathbb{V}(e) - 2). \end{equation*}
\begin{equation*} 1 + \frac {1}{2} \sum _{e \in \mathbb{E}} f(e) \cdot (\mathbb{V}(e) - 2). \end{equation*}
When guessing multi-bonds and cycles, we therefore ensure that this number is met. Multi-bonds ![]() or
or ![]() are guessed for non-root vertices
are guessed for non-root vertices
 $v$
.Footnote
3
For cycle markers, first we guess the number of
$v$
.Footnote
3
For cycle markers, first we guess the number of ![]() at each vertex and thereafter generate these facts with their unique cycle ids. Second, we guess the number of
at each vertex and thereafter generate these facts with their unique cycle ids. Second, we guess the number of ![]() at each vertex, making sure to not exceed the total count of
at each vertex, making sure to not exceed the total count of ![]() at smaller vertices. Using additional constraints, we ensure that each cycle has a single end, start vertices are always smaller than end vertices, cycles never span a single edge (which should be represented as a multi-bond instead), and two cycle edges with the same start are indexed according to their end vertex.
at smaller vertices. Using additional constraints, we ensure that each cycle has a single end, start vertices are always smaller than end vertices, cycles never span a single edge (which should be represented as a multi-bond instead), and two cycle edges with the same start are indexed according to their end vertex.
This completes the initial guessing phase for
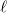 $\ell$
,
$\ell$
,
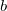 $b$
, and
$b$
, and
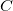 $C$
. Facts that have the form
$C$
. Facts that have the form ![]() store the number of bonding places
store the number of bonding places
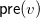 ${\textsf {pre}}(v)$
that have been used up in the process for vertex
${\textsf {pre}}(v)$
that have been used up in the process for vertex
 $v$
. In the next phase, the program specifies possible spanning trees to establish Definition2 (1). Choices are limited since we aim at
$v$
. In the next phase, the program specifies possible spanning trees to establish Definition2 (1). Choices are limited since we aim at
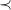 $\prec$
-maximal tree representations, for example, the subtree depth cannot increase from left to right.
$\prec$
-maximal tree representations, for example, the subtree depth cannot increase from left to right.
We first guess the length of the longest path in the tree representation, whose central elements are the only possible roots by Definition13. This length is encoded as ![]() . It ranges from
. It ranges from
 $1$
to
$1$
to
 $|V|$
, but performance is gained by a better lower bound estimate:
$|V|$
, but performance is gained by a better lower bound estimate:
![]()
Proposition 16.
Let
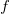 $f$
be a molecular formula with
$f$
be a molecular formula with
 $N := \sum _{e \in \mathbb{E} \setminus \{H\}} f(e)$
non-hydrogen atoms, and let
$N := \sum _{e \in \mathbb{E} \setminus \{H\}} f(e)$
non-hydrogen atoms, and let
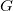 $G$
be a valid molecular graph for
$G$
be a valid molecular graph for
 $f$
. Given that the maximal valence is
$f$
. Given that the maximal valence is
 $X=\max _{e\in \mathbb{E}}{\mathbb{V}(e)}$
,
$X=\max _{e\in \mathbb{E}}{\mathbb{V}(e)}$
,
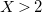 $X \gt 2$
, a longest simple path in
$X \gt 2$
, a longest simple path in
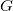 $G$
is at least of length
$G$
is at least of length
 \begin{equation} \min \left \{2 \cdot \left \lceil \log _{X-1}\left ((X-2)\cdot \frac {N - 1}{X}+1\right )\right \rceil + 1, 2 \cdot \left \lceil \log _{X-1}\left ((X-2)\cdot \frac {N}{2} + 1\right )\right \rceil \right \} \end{equation}
\begin{equation} \min \left \{2 \cdot \left \lceil \log _{X-1}\left ((X-2)\cdot \frac {N - 1}{X}+1\right )\right \rceil + 1, 2 \cdot \left \lceil \log _{X-1}\left ((X-2)\cdot \frac {N}{2} + 1\right )\right \rceil \right \} \end{equation}
Proof.
Consider a tree
 $G'$
with
$G'$
with
 $N$
vertices where all nodes except the leaves have
$N$
vertices where all nodes except the leaves have
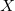 $X$
neighbors and let
$X$
neighbors and let
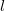 $l$
be the length of the longest simple paths in
$l$
be the length of the longest simple paths in
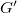 $G'$
. A longest simple path in
$G'$
. A longest simple path in
 $G$
is at least of length
$G$
is at least of length
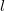 $l$
, as it is only increased by cycles, multi-bonds and lower-valence atoms. The two expressions in the
$l$
, as it is only increased by cycles, multi-bonds and lower-valence atoms. The two expressions in the
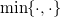 $\min \{\cdot ,\cdot \}$
estimate the minimal lengths in the case that
$\min \{\cdot ,\cdot \}$
estimate the minimal lengths in the case that
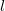 $l$
is odd or even, respectively.
$l$
is odd or even, respectively.
In case
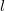 $l$
is odd,
$l$
is odd,
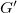 $G'$
is a complete tree where the root has
$G'$
is a complete tree where the root has
 $X$
children and inner non-root vertices have
$X$
children and inner non-root vertices have
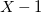 $X-1$
children. Let
$X-1$
children. Let
 $s_n^{odd}$
be the number of vertices in
$s_n^{odd}$
be the number of vertices in
 $G'$
if it has depth
$G'$
if it has depth
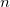 $n$
:
$n$
:
 \begin{align} s_n^{odd} = 1 + X \cdot \frac {(X-1)^{n-1}-1}{(X-2)} \end{align}
\begin{align} s_n^{odd} = 1 + X \cdot \frac {(X-1)^{n-1}-1}{(X-2)} \end{align}
There is one root vertex which has
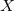 $X$
subtrees of depth
$X$
subtrees of depth
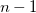 $n-1$
. The longest path in this tree traverses first
$n-1$
. The longest path in this tree traverses first
 $n-1$
vertices in one of these
$n-1$
vertices in one of these
 $X$
subtrees, then the root vertex, and finally another
$X$
subtrees, then the root vertex, and finally another
 $n-1$
vertices in another child tree, totaling
$n-1$
vertices in another child tree, totaling
 $2 \cdot ( n-1 ) + 1$
nodes.
$2 \cdot ( n-1 ) + 1$
nodes.
Rearranging Equation (2) to
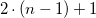 $2 \cdot ( n-1 ) +1$
, we get
$2 \cdot ( n-1 ) +1$
, we get
 $2 \cdot \left ( \log _{X-1}\left ((X-2)\cdot \frac {s_n^{odd} - 1}{X}+1\right )\right ) + 1$
.
$2 \cdot \left ( \log _{X-1}\left ((X-2)\cdot \frac {s_n^{odd} - 1}{X}+1\right )\right ) + 1$
.
In case
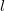 $l$
is even,
$l$
is even,
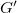 $G'$
is combined of two equal trees where inner vertices have
$G'$
is combined of two equal trees where inner vertices have
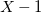 $X-1$
children. Let
$X-1$
children. Let
 $s_n^{even}$
be the number of vertices in
$s_n^{even}$
be the number of vertices in
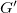 $G'$
if both trees have depth
$G'$
if both trees have depth
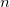 $n$
:
$n$
:
 \begin{align} s_n^{even} = 2 \cdot \frac {(X-1)^{n}-1}{(X-2)} \end{align}
\begin{align} s_n^{even} = 2 \cdot \frac {(X-1)^{n}-1}{(X-2)} \end{align}
The longest path in this tree traverses first
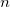 $n$
vertices in one and then further
$n$
vertices in one and then further
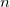 $n$
vertices in the other tree. Rearranging Equation (3) to
$n$
vertices in the other tree. Rearranging Equation (3) to
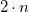 $2 \cdot n$
yields
$2 \cdot n$
yields
 $2 \cdot \left ( \log _{X-1}\left ((X-2)\cdot \frac {s_n^{even}}{2} + 1\right )\right )$
.
$2 \cdot \left ( \log _{X-1}\left ((X-2)\cdot \frac {s_n^{even}}{2} + 1\right )\right )$
.
As the logarithm may give fractional values, both terms have to be rounded up: the minimal length of a longest path in
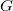 $G$
is the next integer.
$G$
is the next integer.
We use a Python #script-block during grounding to compute this lower bound procedurally (as this is not natively feasible in ASP).
Next, we iteratively guess ![]() , and
, and ![]() for
for
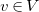 $v\in V$
, where
$v\in V$
, where
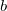 $b$
is the number of children of
$b$
is the number of children of
 $v$
, and
$v$
, and
 $d$
and
$d$
and
 $s$
are the depth and size of the subtree with root
$s$
are the depth and size of the subtree with root
 $v$
. We require
$v$
. We require
 $d\leq s$
and
$d\leq s$
and
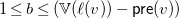 $1\leq b\leq (\mathbb{V}(\ell (v))-{\textsf {pre}}(v))$
. Moreover, if
$1\leq b\leq (\mathbb{V}(\ell (v))-{\textsf {pre}}(v))$
. Moreover, if
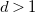 $d\gt 1$
then
$d\gt 1$
then
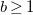 $b\geq 1$
, and
$b\geq 1$
, and
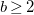 $b\geq 2$
for the root
$b\geq 2$
for the root ![]() unless
unless
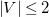 $\left |V\right |\leq 2$
. The rules for
$\left |V\right |\leq 2$
. The rules for ![]() are:
are:


At this point, the used-up binding places due to ![]() at child vertices are captured in a fact
at child vertices are captured in a fact ![]() . Definition2 3 is equivalent to a check of
. Definition2 3 is equivalent to a check of
 ${\textsf {pre}}(v) + {\textsf {post}}(v) \leq \mathbb{V}(\ell (v))$
for each
${\textsf {pre}}(v) + {\textsf {post}}(v) \leq \mathbb{V}(\ell (v))$
for each
 $v \in V$
.
$v \in V$
.
Next, we split the main chain evenly between the first two children of root ![]() , which have indices
, which have indices
 $2$
and
$2$
and
 $2+{\textsf {size}}(2)$
. If the length is odd, the first child’s depth is greater by
$2+{\textsf {size}}(2)$
. If the length is odd, the first child’s depth is greater by
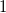 $1$
:
$1$
:

In general, the depth of a first child is always set to its parent’s depth minus
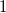 $1$
. Depths for further children are chosen iteratively to be non-increasing.
$1$
. Depths for further children are chosen iteratively to be non-increasing.
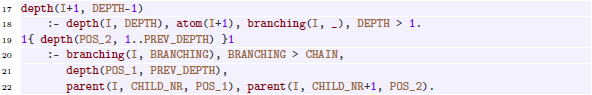
The first child of a non-final vertex
 $v$
is always
$v$
is always
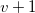 $v+1$
(line 23 below). Vertex ids for further children are chosen iteratively such that their left neighbor can reach its depth and the parent’s size is not exceeded (lines 24–25). These choices also determine the
$v+1$
(line 23 below). Vertex ids for further children are chosen iteratively such that their left neighbor can reach its depth and the parent’s size is not exceeded (lines 24–25). These choices also determine the ![]() of each child (not shown).
of each child (not shown).
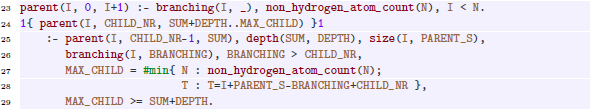
Next, we materialize the total order
 $\prec$
from Section 4.1 in a predicate
$\prec$
from Section 4.1 in a predicate ![]() . For graphs with cycles, we use the number of cycle markers per vertex as an additional ordering criterion instead of the (more costly) tree transformation of Section 4.2. The following constraints exclude cases that cannot be
. For graphs with cycles, we use the number of cycle markers per vertex as an additional ordering criterion instead of the (more costly) tree transformation of Section 4.2. The following constraints exclude cases that cannot be
 $\prec$
-maximal, due to children traversed in
$\prec$
-maximal, due to children traversed in
 $\prec$
-increasing order (line 30) or choice of a non-optimal central vertex as root (line 31).
$\prec$
-increasing order (line 30) or choice of a non-optimal central vertex as root (line 31).
![]()
At this point, perfect symmetry-breaking for acyclic graphs has been achieved. Cyclic graphs, however, can still have isomorphic representations, since the implementation (a) does not compare all possible choices of main chain, and (b) does not ensure that tree edges are obtained from longest extensions as in Definition11. For (b), repeated longest path computations are impractical, but we can heuristically eliminate many non-optimal choices by excluding obvious violations.
Definition 17.
Let
 $G=\langle {V,T\cup C,\ell ,b}\rangle$
be a tree representation with cycle edge
$G=\langle {V,T\cup C,\ell ,b}\rangle$
be a tree representation with cycle edge
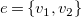 $e=\{v_1,v_2\}$
. Let
$e=\{v_1,v_2\}$
. Let
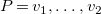 $P=v_1, \ldots , v_2$
be the unique path in
$P=v_1, \ldots , v_2$
be the unique path in
 $G$
that consists only of tree edges. We say that
$G$
that consists only of tree edges. We say that
 $e$
is shortening, if an
$e$
is shortening, if an
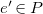 $e' \in P$
exists, s.t.
$e' \in P$
exists, s.t.
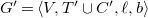 $G'=\langle {V,T'\cup C',\ell ,b}\rangle$
with
$G'=\langle {V,T'\cup C',\ell ,b}\rangle$
with
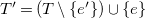 $T'=\left (T \setminus \{e'\}\right ) \cup \{e\}$
and
$T'=\left (T \setminus \{e'\}\right ) \cup \{e\}$
and
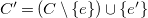 $C'=\left (C \setminus \{e\}\right ) \cup \{e'\}$
is deeper.
$C'=\left (C \setminus \{e\}\right ) \cup \{e'\}$
is deeper.
Patterns for detecting shortening cycles.

Note that for any graph
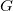 $G$
, its canonical tree representation
$G$
, its canonical tree representation
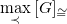 $\max \limits _\prec {[G]_{\cong }}$
cannot contain shortening cycles. Otherwise, one could construct a representation with greater depth, which would therefore be
$\max \limits _\prec {[G]_{\cong }}$
cannot contain shortening cycles. Otherwise, one could construct a representation with greater depth, which would therefore be
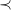 $\prec$
-larger.
$\prec$
-larger.
Our implementation detects shortening cycles in a tree representation by matching it against the patterns depicted in Figure 8. Note that this listing is not necessarily exhaustive. In green, we mark paths that use a cycle edge connecting
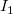 $I_1$
and
$I_1$
and
 $I_2$
, which would increase the depth of the node
$I_2$
, which would increase the depth of the node
 $I$
in the cases (1) – (4), or would increase the length of the main chain in the cases (5) – (9). For instance, in case (1) this is detected by computing the lengths
$I$
in the cases (1) – (4), or would increase the length of the main chain in the cases (5) – (9). For instance, in case (1) this is detected by computing the lengths
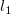 $l_1$
(between node
$l_1$
(between node
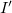 $I'$
and
$I'$
and
 $I_1$
) and
$I_1$
) and
 $l_2$
(between
$l_2$
(between
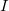 $I$
and
$I$
and
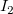 $I_2$
), and comparing them with the length between
$I_2$
), and comparing them with the length between
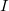 $I$
and
$I$
and
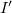 $I'$
. This computation is immediate since the respective nodes lie on the same branch, whose nodes are numbered consecutively according to the depth-first labeling required by Definition 4. The extended path created by the cycle edge is represented in blue, and the nodes that are no longer part of the path are colored red.
$I'$
. This computation is immediate since the respective nodes lie on the same branch, whose nodes are numbered consecutively according to the depth-first labeling required by Definition 4. The extended path created by the cycle edge is represented in blue, and the nodes that are no longer part of the path are colored red.
5.3 Generalization to graph problems
While our encoding is optimized for the generation of molecules, many insights of our implementation also apply in the general case for generating any undirected graph, optionally with degree-constraints. Hence, we also provide ASP programs that
-
1. Enumerate all graphs that satisfy prescribed degree specifications for a given set of nodes.Footnote 4
-
2. Enumerate all undirected graphs for a given number of nodes.Footnote 5
The ASP program in item 4 takes predicates like ![]() as input instead of
as input instead of ![]() and
and ![]() . We guess
. We guess ![]() facts for each node, rather than assigning elements through
facts for each node, rather than assigning elements through ![]() , which previously determined node degrees. Since multi-edges are no longer considered, there is no need to guess
, which previously determined node degrees. Since multi-edges are no longer considered, there is no need to guess ![]() facts. The degree of unsaturation is still used to infer the number of cycle edges that must be distributed. In addition, the definition of the
facts. The degree of unsaturation is still used to infer the number of cycle edges that must be distributed. In addition, the definition of the ![]() predicate was adapted to use
predicate was adapted to use ![]() instead of
instead of ![]() , and to omit checks related to multi-bonds.
, and to omit checks related to multi-bonds.
To generate undirected graph structures in item 5, we first guess the degree of each node and then proceed analogously to the previous case.
6. Experimental evaluation
We evaluate our ASP implementation (“Genmol”) for correctness, avoidance of redundant solutions, and runtime. All of our experiments were conducted on a mid-end server (2
 $\times$
QuadCore Intel Xeon 3.5 GHz, 768GiB RAM, Linux NixOS 23.11) using clingo v5.7.1 for ASP reasoning. Evaluation data, scripts, and results are available online at https://github.com/knowsys/eval-2024-asp-molecules.
$\times$
QuadCore Intel Xeon 3.5 GHz, 768GiB RAM, Linux NixOS 23.11) using clingo v5.7.1 for ASP reasoning. Evaluation data, scripts, and results are available online at https://github.com/knowsys/eval-2024-asp-molecules.
Evaluated systems The ASP-based core of our system Genmol consists of 174 rules (including 44 constraints).Footnote 6 As a gold standard, we use the existing commercial tool Molgen (https://molgen.de), which produces molecular graphs using a proprietary canonicalization approach. Moreover, we compare our approach to four ASP-based solutions, labeled Naive, Graph, sbass, and BreakID. Naive is a direct ASP encoding of Definition2, which serves as a baseline:

The above listing skips the construction of the ![]() and
and ![]() , which can easily be obtained from the
, which can easily be obtained from the ![]() .
.
Graph refines Naive with symmetry-breaking constraints based on Definition 12 of (Codish et al., Reference Codish, Miller, Prosser and Stuckey2019), which apply to partitioned simple graphs
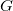 $G$
represented by their adjacency matrix
$G$
represented by their adjacency matrix
 $\mathcal{A}_G$
:
$\mathcal{A}_G$
:
 \begin{align} \text{sb}(G) = \bigwedge\nolimits _{e \in \mathbb{E}}\; \bigwedge\nolimits _{\substack {\ell (i) = \ell (j) = e, \\i \lt j,\, j - i \neq 2}} \mathcal{A}_G[i] \preceq _{\{i, j\}} \mathcal{A}_G[j]. \end{align}
\begin{align} \text{sb}(G) = \bigwedge\nolimits _{e \in \mathbb{E}}\; \bigwedge\nolimits _{\substack {\ell (i) = \ell (j) = e, \\i \lt j,\, j - i \neq 2}} \mathcal{A}_G[i] \preceq _{\{i, j\}} \mathcal{A}_G[j]. \end{align}
Here,
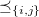 $\preceq _{\{i, j\}}$
denotes the lexicographic order comparing the
$\preceq _{\{i, j\}}$
denotes the lexicographic order comparing the
 $i$
th and
$i$
th and
 $j$
th row of the adjacency matrix
$j$
th row of the adjacency matrix
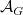 $\mathcal{A}_G$
of a molecular graph
$\mathcal{A}_G$
of a molecular graph
 $G$
, ignoring columns
$G$
, ignoring columns
 $i$
and
$i$
and
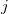 $j$
. Graph representations that do not satisfy (4) are pruned. These constraints can be succinctly represented in ASP, and are appended to the naive implementation:Footnote
7
A graph
$j$
. Graph representations that do not satisfy (4) are pruned. These constraints can be succinctly represented in ASP, and are appended to the naive implementation:Footnote
7
A graph
 $G$
satisfies
$G$
satisfies
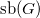 $\text{sb}(G)$
if the rows of its adjacency matrix are lexicographically ordered. This means that every nonzero entry in the matrix must be followed by rows that either:
$\text{sb}(G)$
if the rows of its adjacency matrix are lexicographically ordered. This means that every nonzero entry in the matrix must be followed by rows that either:
-
1. Contain a nonzero entry further to the left (see lines 1–3), or
-
2. Have a larger entry in the same column (see lines 4–6).
This is captured by the auxiliary predicate ![]() , which specifies whether rows
, which specifies whether rows ![]() and
and ![]() are in lexicographic order up to column
are in lexicographic order up to column ![]() , as illustrated in Figure 9. Note that we only compare rows, whose nodes are assigned to the same element.
, as illustrated in Figure 9. Note that we only compare rows, whose nodes are assigned to the same element.
Visualization of graph symmetry-breaking encoding.


sbass (Drescher et al., Reference Drescher, Tifrea and Walsh2011) and BreakID (Devriendt and Bogaerts, Reference Devriendt and Bogaerts2016) append instance-specific symmetry-breaking constraints to the ground program by considering the automorphisms of its graph representation. BreakID is run on the Naive implementation, while we used sbass with an equivalent aggregate-free version.Footnote 8
All evaluated ASP encodings were passed through the heuristic non-ground optimizer ngo (https://potassco.org/ngo/), but no performance improvements were observed, suggesting that our encodings are reasonably efficient.
Data set For evaluation, we have extracted a dataset of molecules with molecular formulas and graph structures using the Wikidata SPARQL service (Malyshev et al., Reference Malyshev, Krötzsch, González, Gonsior and Bielefeldt2018). We selected 8980 chemical compounds of up to 17 atoms (due to performance constraints) with SMILES and an article on English Wikipedia as a proxy for practical relevance. Compounds with unconnected molecular graphs, atoms of non-standard valence, and subgroup elements were excluded, resulting in a dataset of 5625 entries, of which we found 152 to have non-parsable SMILES.
Evaluation of correctness Given the complexity of the implementation, we also assess its correctness empirically. To this end, we augment our program with ASP rules that take an additional direct encoding of a molecular graph as input, and that check if the molecular graph found by Genmol is isomorphic to it. This allows us to determine if the given structures of molecules in our data set can be found in our tool. The validation graph structure is encoded in facts ![]() that were extracted from the SMILES representation in Wikidata.
that were extracted from the SMILES representation in Wikidata.
Correctness experiments were measured with a timeout of 7 min. Out of 5473 compounds, a matching molecular structure was found for 5338, whereas 132 could not be processed within the timeout. For three compounds, Sandalore (Wikidata ID Q21099635), and Eythrohydrobupropion (Q113691142) as well as Threodihydrobupropion (Q72518680), the given structures could not be reproduced, which we traced back to errors in Wikidata that we have subsequently corrected.
The evaluation therefore suggests that Genmol can find the correct molecular structures across a wide range of actual compounds. Timeouts occurred primarily for highly unsaturated, larger compounds (over 16 atoms), where millions of solutions exist.
Evaluation of symmetry-breaking To assess to what extent our approximated implementation of canonical tree representations succeeds in avoiding redundant isomorphic solutions, we consider the smallest 1750 distinct molecular formulas from our data set. We then computed molecular graph representations for all 1750 cases for each of our evaluated systems, using Molgen as a gold standard to determine the actual number of distinct molecular graphs. The timeout for these experiments was 60 s. The number of returned solutions are shown in Figure 10 (left), with samples sorted by their number of distinct graphs according to Molgen. As expected, Molgen is a lower bound, and in particular no implementation finds fewer representations (which would be a concern for correctness), while Naive is an upper bound. As expected, no ASP tool achieves perfect canonization of results, but the difference between the number of solutions and the optimum vary significantly. In particular, BreakID rarely improves over Naive (just 24 such cases exist), though it does cause one third more timeouts.
Number of models for each compound in the data set (left) and ratio of compounds with model counts within a factor of the gold standard (right).

For Naive, some samples led to over 20,000 times more models than Molgen, whereas the largest such factor was just above
 $39$
for Genmol (for
$39$
for Genmol (for
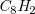 $\mathit{C}_8\mathit{H}_2$
). Figure 10 (right) shows the ratio of samples with model counts within a certain factor of the gold standard. For example, the values at
$\mathit{C}_8\mathit{H}_2$
). Figure 10 (right) shows the ratio of samples with model counts within a certain factor of the gold standard. For example, the values at
 $10$
show the ratios of samples for which at most ten times as many models were computed than in Molgen: this is
$10$
show the ratios of samples for which at most ten times as many models were computed than in Molgen: this is
 $99\%$
for Genmol,
$99\%$
for Genmol,
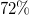 $72\%$
for Graph, and
$72\%$
for Graph, and
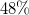 $48\%$
for BreakID, sbass, and Naive. All ratios refer to the same total, so the curves converge to the ratio of cases solved within the timeout. Their starting point marks the ratio with exact model counts:
$48\%$
for BreakID, sbass, and Naive. All ratios refer to the same total, so the curves converge to the ratio of cases solved within the timeout. Their starting point marks the ratio with exact model counts:
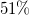 $51\%$
for Genmol and
$51\%$
for Genmol and
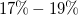 $17\%-19\%$
for the others.
$17\%-19\%$
for the others.
We conclude that symmetry-breaking in Genmol, while not perfect, performs very well in comparison with generic approaches. In absolute terms, the results are often close enough to the optimum that any remaining redundancies could be removed in a post-processing step using conventional graph isomorphism tools. Although applying such checks to the entire solution space would be computationally prohibitive, users are typically interested only in a relatively small subset of the generated molecules that are to be sorted according to some chosen criterion.
Performance and scalability To assess the runtime of our approach, we conduct experiments with series of uniformly created molecular formulas of increasing size. We consider two patterns: formulas of the form
 $\mathit{C}_n\mathit{H}_{2n+2}O$
belong to tree-shaped molecules (such as ethanol with SMILES OCC), whereas formulas of the form
$\mathit{C}_n\mathit{H}_{2n+2}O$
belong to tree-shaped molecules (such as ethanol with SMILES OCC), whereas formulas of the form
 $\mathit{C}_n\mathit{H}_{2n}O$
require one cycle (like oxetane, C1COC1) or double bond (like acetone, CC(=O)C). We use a timeout of 10 min for all tools except Molgen, whose free version is limited to 1 min runtime. All runs are repeated five times and the median is reported. Moreover, clingo allows to specify the search strategy via the — configuration parameter. For the ASP-based approaches, we tested all available options on the second pattern (with a 1 min timeout) and found no significant improvements compared to the default auto setting.
$\mathit{C}_n\mathit{H}_{2n}O$
require one cycle (like oxetane, C1COC1) or double bond (like acetone, CC(=O)C). We use a timeout of 10 min for all tools except Molgen, whose free version is limited to 1 min runtime. All runs are repeated five times and the median is reported. Moreover, clingo allows to specify the search strategy via the — configuration parameter. For the ASP-based approaches, we tested all available options on the second pattern (with a 1 min timeout) and found no significant improvements compared to the default auto setting.
The results are shown in Figure 11. As before, Molgen serves as a gold standard. As seen in the graphs on the top, the number of distinct molecular structures grows exponentially, and this optimum is closely tracked by Genmol (perfectly for the tree-shaped case). Graph reduces model counts too, albeit less effectively, whereas BreakID and sbass do not achieve any improvements over Naive in these structures.
Model numbers (top) and runtimes (bottom) for molecules of increasing size.

As expected, the runtimes indicate similarly exponential behavior as inputs grow, but the point at which computation times exceed the timeout is different for each tool. Molgen achieves the best scalability overall, whereas Genmol is most scalable among the ASP-based approaches. BreakID and sbass are even slower than Naive, largely due to longer solving times, whereas the preprocessing of the grounding had no notable impact.
Learning symmetry-breaking constraints We also explored an approach proposed by (Tarzariol et al. (Reference Tarzariol, Gebser, Schekotihin and Law2023)) that aims to automatically learn symmetry-breaking constraints, which can be appended to the input program as ASP rules. This method relies on sbass to derive instance-specific constraints that are used to classify instances as positive or negative examples depending on whether they satisfy the derived constraints. These examples are provided as input to ilasp (Law et al., Reference Law, Russo and Broda2015), which attempts to learn ASP rules that eliminate negative examples while preseriving positive ones.
Since sbass did not yield any significant improvement over the Naive implementation in our case, we instead tested whether ilasp could automatically derive symmetry-breaking constraints using explicitly constructed examples. As a representative set of examples, we used instances of the form
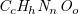 $\mathit{C}_c\mathit{H}_h\mathit{N}_n\mathit{O}_o$
with
$\mathit{C}_c\mathit{H}_h\mathit{N}_n\mathit{O}_o$
with
 $c\in \{3,4\}$
,
$c\in \{3,4\}$
,
 $n,o\in \{0,1\}$
,
$n,o\in \{0,1\}$
,
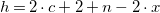 $h=2\cdot c +2+n-2\cdot x$
, and
$h=2\cdot c +2+n-2\cdot x$
, and
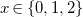 $x\in \{0,1,2\}$
cycles. For each class of isomorphic molecular graphs, we selected the one with the lexicographically smallest adjacency matrix as a positive example and randomly sampled a negative example from the others, yielding a total of 1228 examples.
$x\in \{0,1,2\}$
cycles. For each class of isomorphic molecular graphs, we selected the one with the lexicographically smallest adjacency matrix as a positive example and randomly sampled a negative example from the others, yielding a total of 1228 examples.
The hypothesis space for ilasp, that is the set of possible rules, was configured in such a way as to allow the construction of the Graph encoding, resulting in 388,098 candidate rules. We found that even with 768 GiB of RAM, the process exceeded available memory after approximately 9 h, making it infeasible to complete the computation. A considerably easier version of the problem where the definition of the ![]() predicate (lines 1–6) is given as background knowledge to ilasp successfully derives the missing constraint (lines 8–9) in 7.5 min.
predicate (lines 1–6) is given as background knowledge to ilasp successfully derives the missing constraint (lines 8–9) in 7.5 min.
We conclude that useful symmetry-breaking constraints cannot be learned in our setting without providing a significant amount of background knowledge, which is unrealistic in practice.
7. Conclusion
Motivated by the practical problem of interpreting results in mass spectrometry, we developed an ASP-based approach for enumerating molecular structures based on partial information about their chemical composition. We focused on molecular formulas as inputs, but our prototype Genmol can also use additional signals, such as known molecular fragments. Indeed, one of the main strengths of an ASP-based solution is that it is very simple to refine the search space with additional constraints. Our extensive evaluation showed that our approach improves upon direct ASP-based solutions and existing optimizations by several orders of magnitude, regarding both performance and conciseness, bringing it into the range of optimized commercial implementations that (presumably) lack the flexibility of ASP.
In general, we believe that our conceptual work towards canonical graph representations and their efficient realization in ASP is relevant beyond our motivating application scenario. Most of our ideas can be readily generalized to other graph-related search problems, and may therefore be of interest to ASP practitioners. Moreover, our real-world evaluation data can be a valuable benchmark for further research on automated symmetry-breaking in ASP.
Acknowledgements
This work has partly been supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) in project number 389792660 (TRR 248, Center for Perspicuous Systems) and 390696704 (CeTI Cluster of Excellence) and by the Bundesministerium für Forschung, Technologie und Raumfahrt (BMFTR, Federal Ministry of Research, Technology and Space) in the Center for Scalable Data Analytics and Artificial Intelligence (ScaDS.AI), in European ITEA project 01IS21084 (InnoSale), and in DAAD project 57616814 (SECAI, School of Embedded Composite AI) as part of the program Konrad Zuse Schools of Excellence in Artificial Intelligence.
Disclosure of interests
The authors have no further competing interests to declare.
Appendix A List of predicates

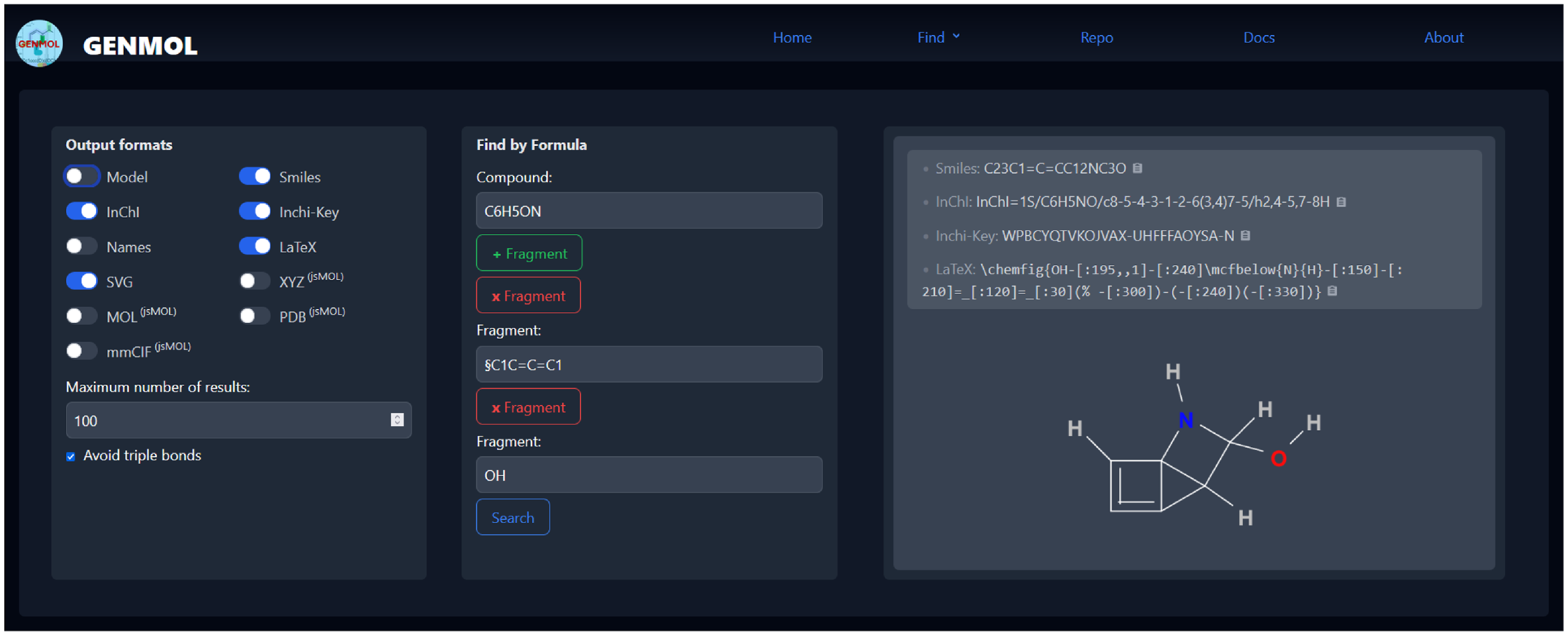
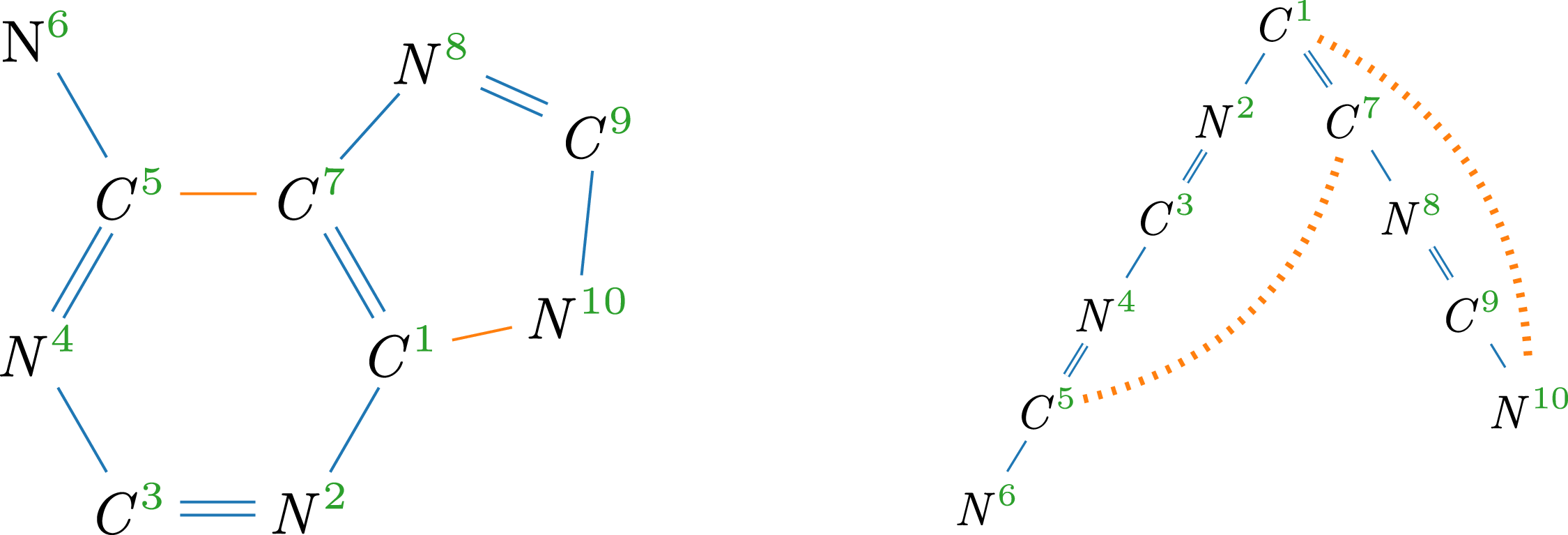


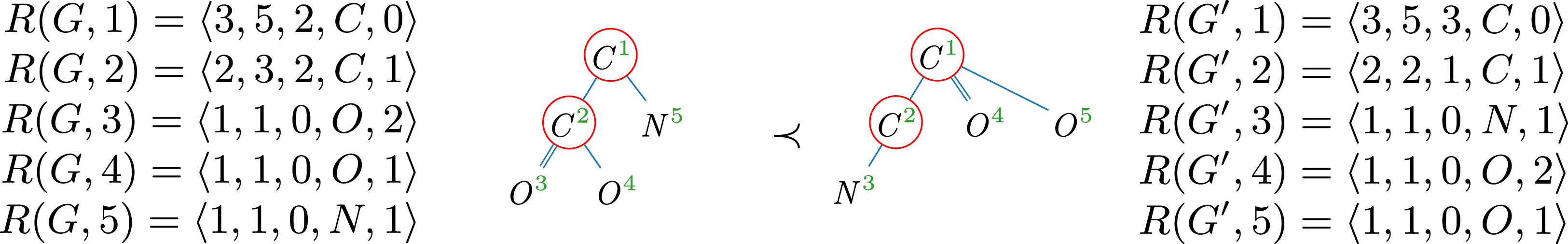

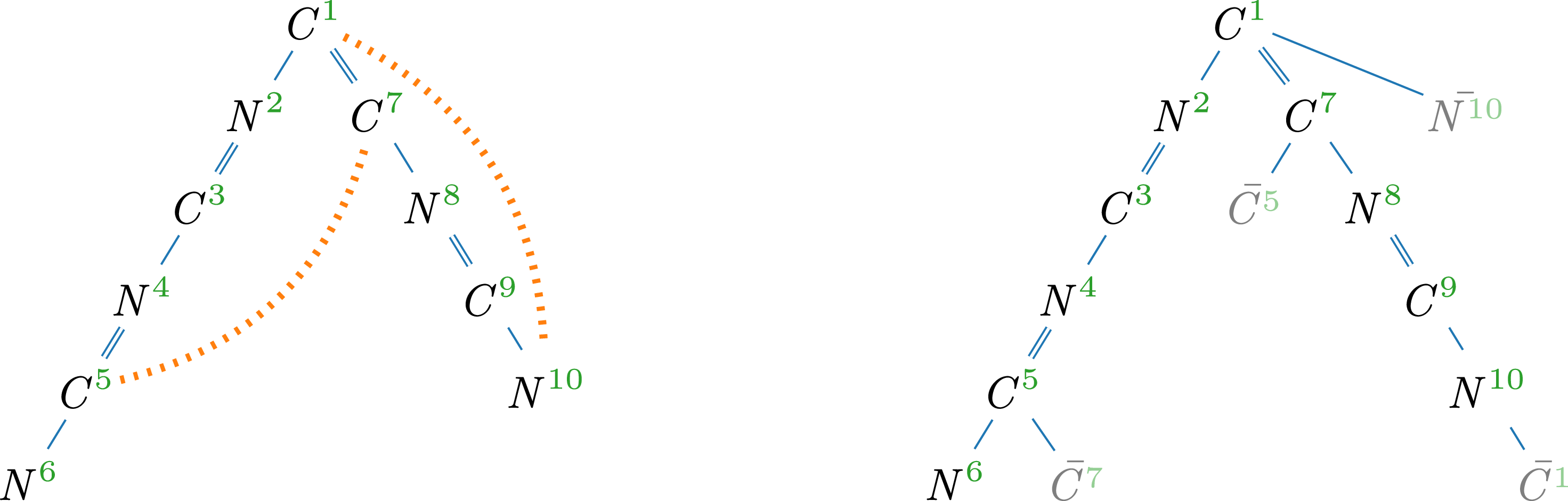
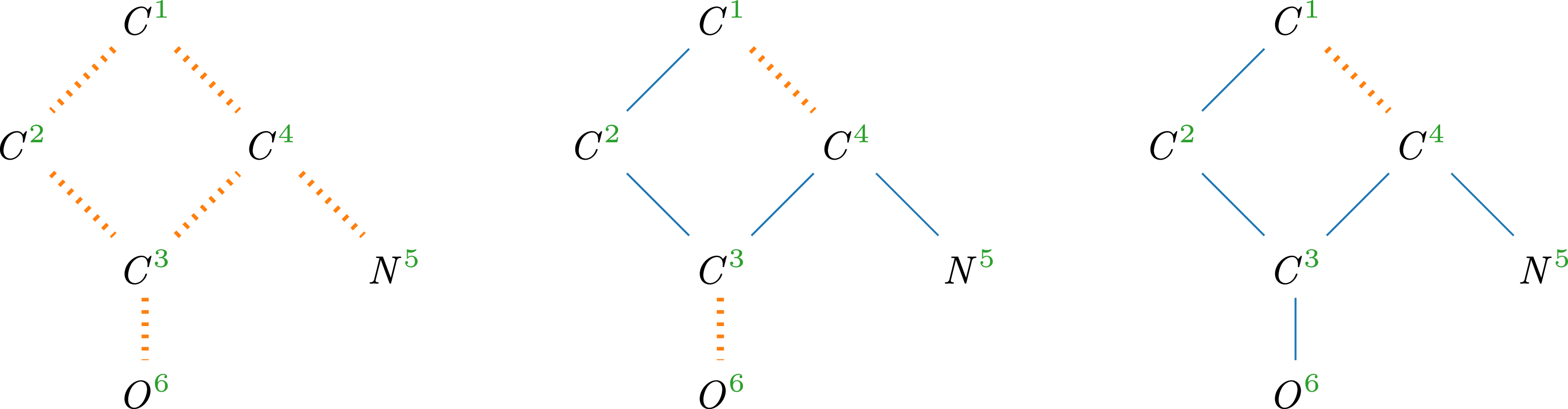


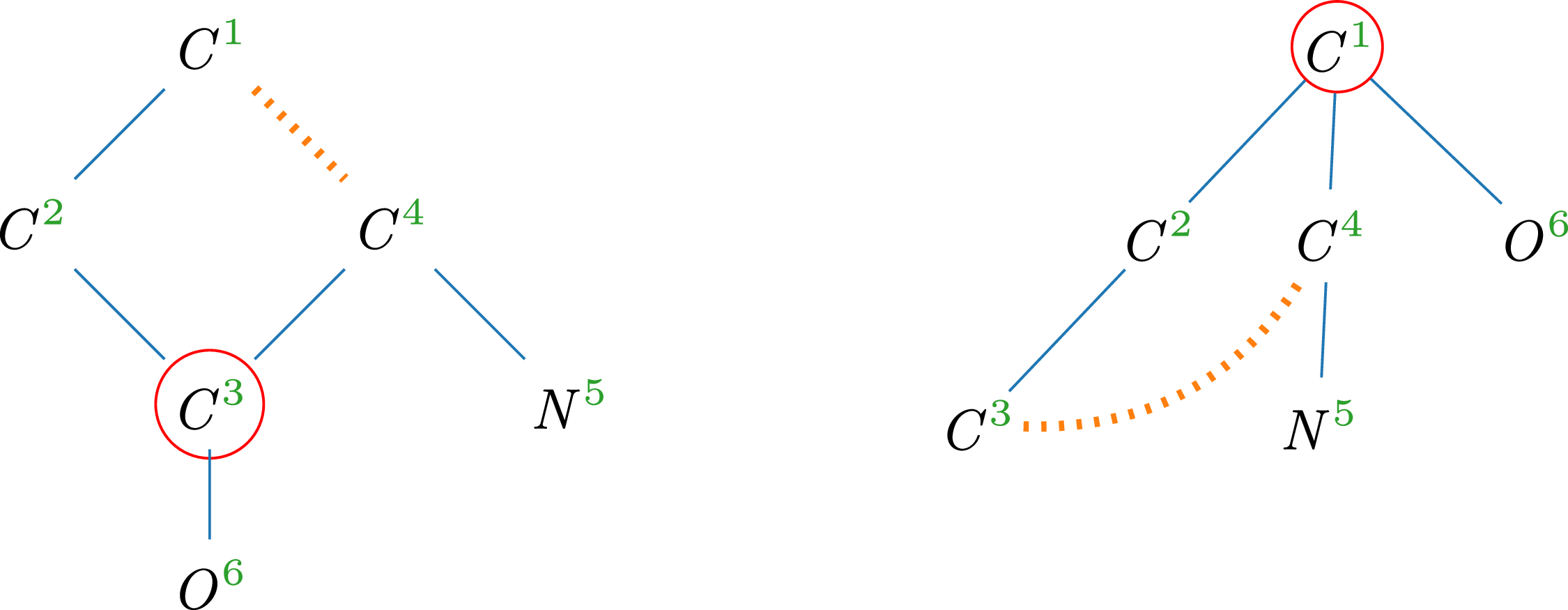
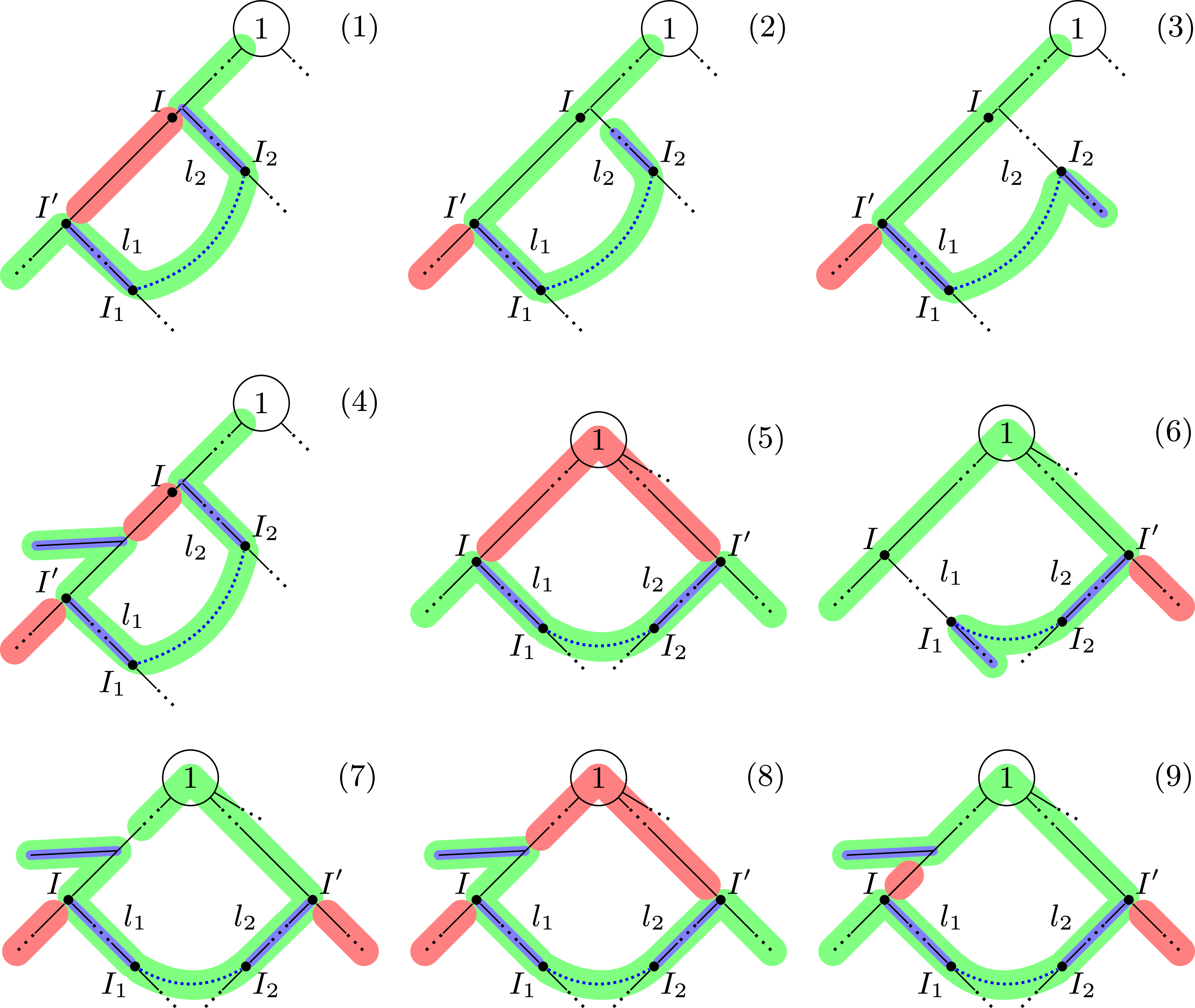
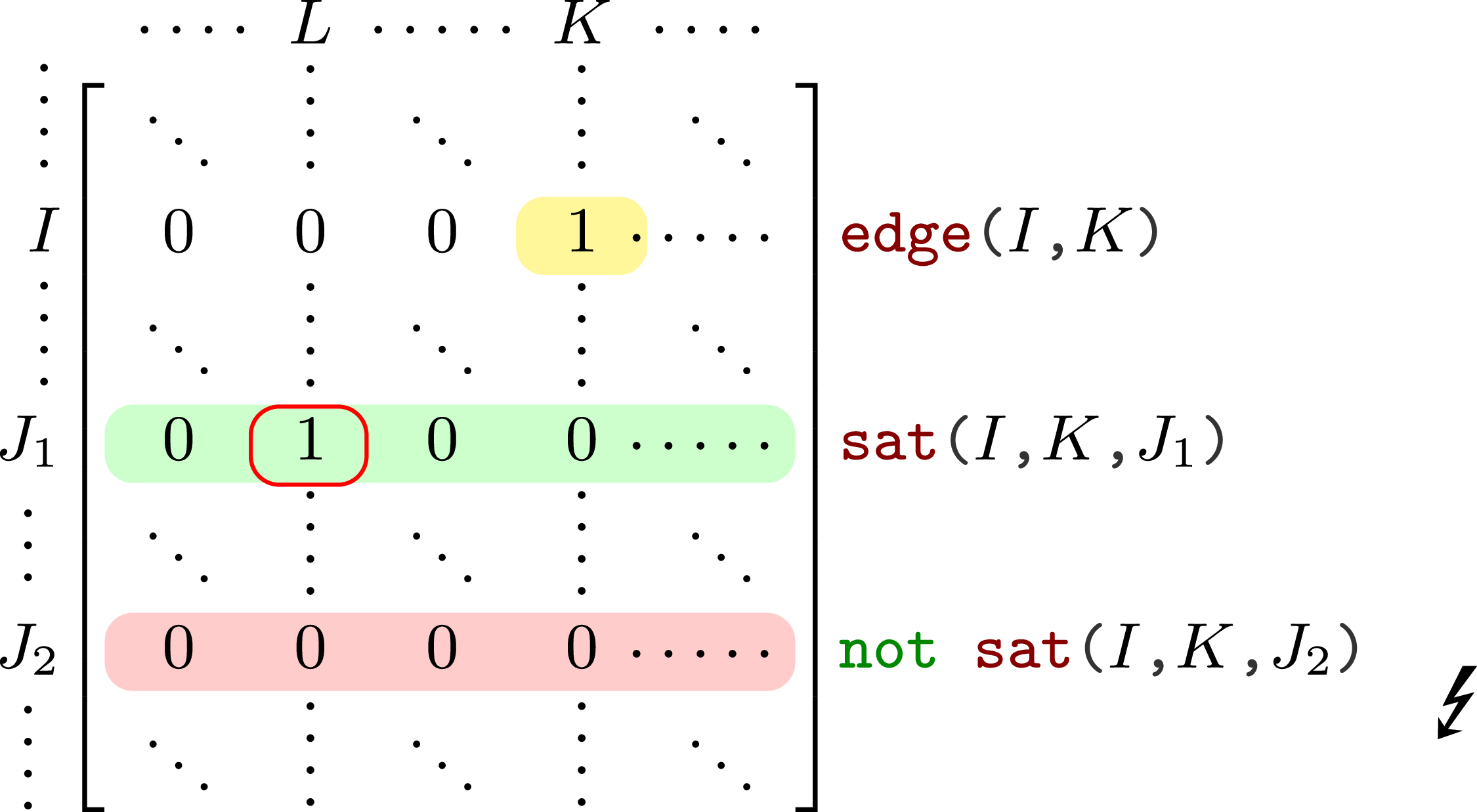
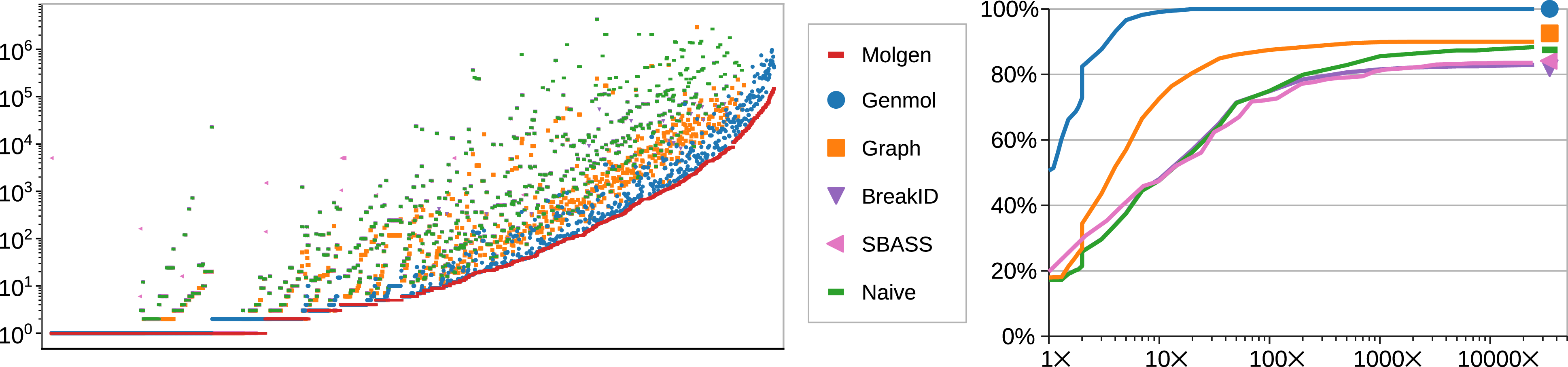
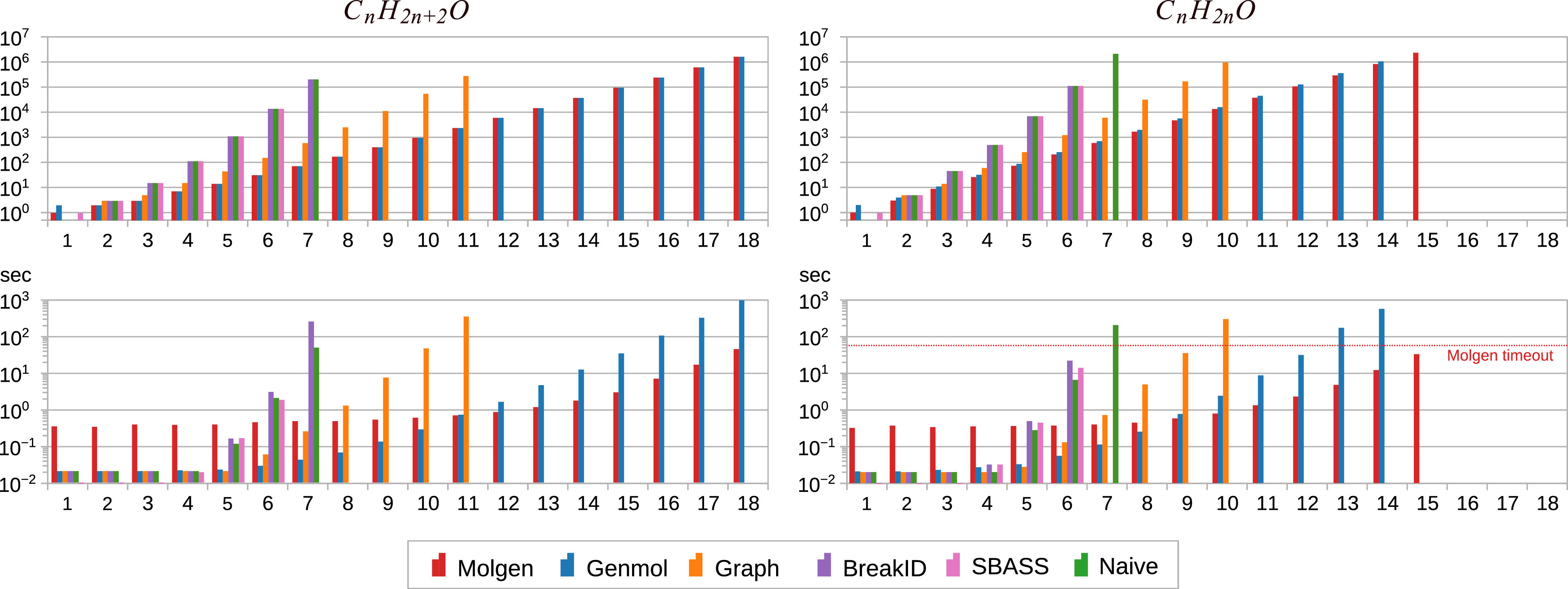

 Open access
Open access