


Introduction
The complex ecosystem of the rumen, a major digestive compartment in ruminants, houses high extensive diversity microbes consisting of bacteria, archaea, protozoa, fungi, and phages (Morais and Mizrahi Reference Morais and Mizrahi2019; Newbold and Ramos-Morales Reference Newbold and Ramos-Morales2020). These microbes synergistically contribute to the host’s energy (Huws et al. Reference Huws, Creevey and Oyama2018) and protein supply (Lima et al. Reference Lima, Ingabire and Roehe2023). Bacteria, the most abundant microbes in the rumen (Deusch et al. Reference Deusch, Camarinha-Silva and Conrad2017) and the population is often in excess of 1 × 1010 cells/mL of rumen fluid (Cammack et al. Reference Cammack, Austin and Lamberson2018), are responsible for the cellulose and hemicellulose degradation through carbohydrate hydrolyzing enzymes. Fungi, as a major cellulolytic agent, vary in their cellulose degrading capabilities (Lee et al. Reference Lee, Ha and Cheng2000). Rumen protozoa, comprising up to 50% of the rumen microbial biomass, act as predators in rumen and play a significant role in feed digestion and rumen fermentation, but they can negatively impact intraruminal nitrogen cycling and thereby lowering microbial protein supply to host and reducing dietary nitrogen utilization efficiency (Yu et al. Reference Yu, Yan and Somasundaram2024). Rumen viruses, diverse and abundant microbes in rumen, can regulate rumen microbes at both strain and community levels as well as associated with animal production traits, including feed efficiency, lactation performance, weight gain, and methane emissions (Yan and Yu Reference Yan and Yu2024). While extensive research has focused on the bacterial community using 16S rRNA sequencing, the roles of other rumen microbes’ functions are not negligible. Therefore, whole metagenome shotgun sequencing is essential to understand the comprehensive composition and function of the rumen microbiota.
Numerous studies have highlighted the association between rumen microbiota and ruminant feed intake. For example, Shi et al. (Reference Shi, Guo and Degen2020) observed that the abundance of total bacteria and the proportions of Fibrobactor succinogenes was more abundant in the low feed intake (LFI) yaks than those in high feed intake (HFI) yaks, whereas the relative abundance of Selenomonas ruminantium, Butyrivibrio fbrisolvens, Prevotella brevis, Ruminococcus flavefaciens, and Ruminococcus albus was opposite, revealing that the influence of feed intake on rumen microbial composition. Li et al. (Reference Li, Xi and Wang2020) further observed distinct rumen bacterial profiles in early lactating cows with varying feed intakes. Moreover, Jami et al. (Reference Jami, White and Mizrahi2014) found a strong association between feed intake and the abundance of ruminal Prevotellaceae and Ruminnococcaceae. In transition dairy cows, feed intake, a key trait can significantly boost milk yield. Overall, the rumen microbial composition is responsible for the feed intake of ruminants, while the profile of the rumen metagenome of postpartum dairy cows with low and HFIs is poorly understood.
Our previous study found that feed intake is associated with rumen bacteria in postpartum dairy cows using 16S rRNA sequencing (Huang et al. Reference Huang, Ji and Garret2021). In the present study, we aim to understand the role of rumen microbes, functions, and metabolism in postpartum dairy cows’ feed intake. The results will expand our understanding of the role of rumen microbiota and metabolism in feed intake and may provide new insights into strategies for manipulating rumen microbes to improve ruminant performance.
Materials and methods
Animals, diet, and sample collection
All animals, experiment protocols, and procedures employed in this study were approved by the Animal Care and Use Committee of China Agricultural University (Beijing, China, No. 31772628). As the previous study, a total of 65 healthy multiparous Holstein dairy cows were selected from a commercial dairy farm (Huang et al. Reference Huang, Ji and Garret2021), housed in the same barn, and used for measurement of dry matter intake (DMI). The feed intake data were recorded using an individual feed intake recording system (Insentec B.V., Marknesse, Netherlands). The DMI of each cow was monitored from day 1 to 14 post-calving. Following this period, five cows with the lowest DMI (LFI,15.66 ± 0.60 kg [mean ± standard error of the mean (SEM)]) and five cows with the highest DMI (HFI, 17.45 ± 0.60 kg, Table S1) were allocated to two separate groups. The sample size achieved 98.3% power and a type 1 error of 5% (effect size = 2.78) after a t-test of DMI using power calculations. The dietary composition of the dairy cows was consistent with that of a previous study (Huang et al. Reference Huang, Ji and Garret2021), which included 21.05% corn silage, 20% alfalfa hay, 19.88% steam-flaked corn, 5.32% cottonseed, 4.21% oat hay, 20.23% soybean meal, 2.28 molasses, 1.17% yeast culture XP, 1.46% BergaFat, 0.58% sodium bicarbonate, and 5% premix (comprising 18.08% crude protein, 30.40% neutral detergent fiber, 20.65% acid detergent fiber, 5.04% ether extract). All the cows were free to access diet and fed at 07:30, 14:30, and 19:00, with drinking water ad libitum. No antibiotics or probiotics were administrated to the animals for a period of 3 months prior to the start of the research.
Samples of the rumen fluid were collected pre-feeding using an oral stomach tube (Ancitech, Winnipeg, MB, Canada), filtered through four layers of cheesecloth, and then stored at −80℃ for subsequent DNA extraction.
DNA extraction, metagenome sequencing, and metagenomics data processing
The FastDNA™ Spin kit (MP Bio, Santa Ana, CA) was used to extract the metagenomic DNA from approximately 0.5 mL of rumen fluid according to the manufacturer’s protocol. Briefly, 0.5 mL rumen fluid was added to a Lysing Matrix E tube and mixed with 978 μL sodium phosphate buffer and 122 μL MT buffer. The mixture was subjected to shock for 40 s and then centrifuged at 14,000 rpm for 10 min. The supernate was transferred to a new tube and thoroughly mixed with 900 µL binding matrix. It was briefly centrifuged for 5 s, and the supernatant was discarded. The pellet was washed twice with 500 µL 5.5 M guanidinethiocyanate and SEWS-M, discarding the supernatant after centrifugation at 14,000 rpm for 3 min, and then dried. The dried material was resolved in 100 µL DES at 55℃, allowed to stand for 10 min, and then centrifuged at 14,000 rpm for 2 min. The SPINTM filter was removed and then obtained the total DNA. The extracted DNA’s quality and concentration were assessed by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and 1% agarose gel electrophoresis. DNA fragmentation DNA and adapter ligation were performed with the Covaris M220 kit (Gene Company Limited, China). Library preparation for metagenomic sequencing was carried out with the TrueSeq DNA PCR-Free Library Prep kits (Illumina, San Diego, CA, USA), following the manufacturer’s directions. Sequencing was conducted on the Illumina Hiseq 3000 platform (Illumina, San Diego, CA, USA), generating 2 × 150 bp paired reads. The paired raw metagenomics sequences from all rumen fluid samples have been submitted to the National Center of Biotechnology Information (NCBI) Sequence Read Archive database under the accession number SUB13483300.
The raw paired-end reads generated from samples were trimmed and filtered to eliminate low quality (quality value ≤ 20), short length (< 50 bp), or containing ambiguous N bases using fastp 0.20.0 (Chen et al. Reference Chen, Zhou and Chen2018). The high quality sequences were aligned to the bovine genome (bosTau8 3.7, https://doi.org/10.18129/B9.bioc.BSgenome.Btaurus.UCSC.bosTau8) using Burrows Wheeler Alignment 0.7.9a (http://bio-bwa.sourceforge.net, Li and Durbin Reference Li and Durbin2009) to filter out host DNA sequences. Subsequently, Megahit 1.1.2 was used to assemble the high quality metagenomic clean reads (Li et al. Reference Li, Liu and Luo2015). Open reading frames (ORFs) were predicted using Prodigal 2.6.3 (https://github.com/hyattpd/Prodigal, Hyatt et al. Reference Hyatt, Chen and LoCascio2010) with lengths >300 bp of assembled contigs. Cluster Database at High Identity with Tolerance (CD-HIT) (http://www.bioinformatics.org/cd-hit/, Fu et al. Reference Fu, Niu and Zhu2012) with parameters set as 95% identity and 90% coverage was used to cluster the predicted genetic sequence of all the samples and the longest gene was used as the representative sequences to create the nonredundant gene catalog. Reads were then mapped to this catalog with identity >95% using SOAPaligner (http://soap.genomics.org.cn/), and the relative abundance of each gene was calculated (Li et al. Reference Li, Yu and Li2009).
Taxonomic and functional annotation from rumen metagenomes
The contigs were aligned against the NCBI Non-Redundant Protein Sequence Database (NR) database using Diamond 0.8.35 for taxonomic annotation (Buchfink et al. Reference Buchfink, Xie and Huson2015). The profiling and calculating of the relative abundance were examined at various taxonomic levels, including domain, phylum, class, order, family, genus, and species using HUMAnN 2.0 (https://huttenhower.sph.harvard.edu/humann2/). Principal coordinates analysis (PCoA), an unsupervised chemometric method, was employed in R 4.1.0 for visual representation.
The sequences from the NR gene catalog were subjected to Basic Local Alignment Search Tool (BLAST) analysis and annotated against functional databases using Diamond 0.8.35 with default parameters (e-value < 1 × 10−5, Buchfink et al. Reference Buchfink, Reuter and Drost2021). A cluster of orthologous groups of proteins (COG) annotation was performed using Diamond 0.8.35 against the eggNOG database with an e-value of 1 × 10−5 (Jensen et al. Reference Jensen, Julien and Kuhn2008). Carbohydrate-active enZYmes (CAZymes) annotation was conducted using HMMscan 3.1b2 against the CAZymes database (http://www.cazy.org/) with an e-value of 1 × 10−5 (Lombard et al. Reference Lombard, Golaconda Ramulu and Drula2014). Kyoto encyclopedia of genes and genomes (KEGG) were annotated BLASTP 2.2.28 (http://blast.ncbi.nlm.nih.gov/Blast.cgi, Buchfink et al. Reference Buchfink, Xie and Huson2015) against KEGG database with an e-value cutoff of 1 × 10−5. Pathway annotation was conducted using KOBAS 2.0 (KEGG Orthology Based Annotation System, Xie et al. Reference Xie, Mao and Huang2011) according to the BLAST results.
The annotation process involved the integration of the KEGG (http://www.genome.jp/kegg/) database and the CAZymes database (http://www.cazy.org/), which specializes in CAZymes for broader functional classification.
Rumen metabolomics
Metabolite levels in rumen were determined using an untargeted liquid chromatography tandem mass spectrometry platform (LC-MS/MS, Agilent Technologies, Santa Clara, CA, USA) as described in a previous study by Huang et al. (Reference Huang, Zheng and Men2022). We converted the MS raw data files to mzXML format using ProteoWizard software 3.0.81 (Kessner et al. Reference Kessner, Chambers and Burke2008) and processed them with the XCMS program to convolute peak, align, correct retention time, and identify and calculate area. We set the rumen metabolite peaks and the relative standard deviation to 0.5 and 0.3, respectively, and removed unidentified peaks for further analysis. We calculated the relative intensity of the 340 identified rumen metabolites using the proportion of each area of identified metabolites and quality control samples for downstream analysis.
The online platform MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml) was used for metabolic pathway (Xia and Wishart Reference Xia and Wishart2010a) and pathway enrichment analysis on target metabolites (Xia and Wishart Reference Xia and Wishart2010b).
Omics-explainability calculation
The relative abundances of bacterial genera, KEGG Orthology (KO)s, and the relative concentration of rumen metabolites were normalized to have a zero mean and a unit variance and then were used to build the matrix G, F, and M, respectively (Difford et al. Reference Difford, Plichta and Lovendahl2018). The linear mixed effects model (LMM) utilized to calculate the explainability of three omics on DMI is as follows:
 \begin{equation}{y_{ijk}} = {\text{ }}\mu + {p_i} + {b_k} + {g_i} + {e_{ijk}}\end{equation}
\begin{equation}{y_{ijk}} = {\text{ }}\mu + {p_i} + {b_k} + {g_i} + {e_{ijk}}\end{equation} where y ijk is the recorded DMI in kg/day; μ is the model intercept;  ${p_j}$ is the parity fixed effect (j = 2 levels);
${p_j}$ is the parity fixed effect (j = 2 levels);  ${b_k}$is the body condition score fixed effect (k = 3 levels);
${b_k}$is the body condition score fixed effect (k = 3 levels);  ${g_i}$ is the rumen microbial random effect for the ith animal ∼ NID (0, G
${g_i}$ is the rumen microbial random effect for the ith animal ∼ NID (0, G ${{\sigma }}_g^2$), where
${{\sigma }}_g^2$), where  ${{\sigma }}_g^2$ is the rumen microbial variance and R is the microbial relationship matrix; and
${{\sigma }}_g^2$ is the rumen microbial variance and R is the microbial relationship matrix; and  ${e_{ijk}}$ is the residual effect. The KO variance was estimated by LMM, which was similar to Eq. (1), except for the random effect of
${e_{ijk}}$ is the residual effect. The KO variance was estimated by LMM, which was similar to Eq. (1), except for the random effect of  ${f_i}$, which is the random effect of the KOs for the ith animal ∼ NID (0, F
${f_i}$, which is the random effect of the KOs for the ith animal ∼ NID (0, F ${{\sigma }}_f^2$), where
${{\sigma }}_f^2$), where  ${{\sigma }}_f^2$ is the rumen microbial variance and F is the rumen functional relationship matrix. The rumen metabolic variance was estimated by LMM, which was similar to Eq. (1), except for the random effect of
${{\sigma }}_f^2$ is the rumen microbial variance and F is the rumen functional relationship matrix. The rumen metabolic variance was estimated by LMM, which was similar to Eq. (1), except for the random effect of  ${M_i}$, which is the random effect of the rumen metabolites for the ith animal ∼ NID (0, M
${M_i}$, which is the random effect of the rumen metabolites for the ith animal ∼ NID (0, M ${{\sigma }}_m^2$), where
${{\sigma }}_m^2$), where  ${{\sigma }}_m^2$ is the rumen microbial variance and M is the rumen functional relationship matrix. The DMI variance explained by the rumen microbial variance, functional variance, and rumen metabolic variance was estimated as
${{\sigma }}_m^2$ is the rumen microbial variance and M is the rumen functional relationship matrix. The DMI variance explained by the rumen microbial variance, functional variance, and rumen metabolic variance was estimated as  $\frac{{{{\sigma }}_g^2}}{{{{\sigma }}_d^2}}$,
$\frac{{{{\sigma }}_g^2}}{{{{\sigma }}_d^2}}$,  $\frac{{{{\sigma }}_f^2}}{{{{\sigma }}_d^2}}$,
$\frac{{{{\sigma }}_f^2}}{{{{\sigma }}_d^2}}$,  $\frac{{{{\sigma }}_m^2}}{{{{\sigma }}_d^2}}$, respectively, where
$\frac{{{{\sigma }}_m^2}}{{{{\sigma }}_d^2}}$, respectively, where  ${{\sigma }}_d^2$ is the DMI variance.
${{\sigma }}_d^2$ is the DMI variance.
LMM analysis was conducted using the “lme4” package in R software (https://www.r-project.org, Bates et al. Reference Bates, Mächler and Bolker2015). The P value was calculated using the likelihood ratio tests on the random effect. The random effect will be rejected when the null hypothesis (the variance of the random effect is 0) is a significantly better fit than the random effect of omics data, and vice versa.
Spearman correlation analysis
To identify the associations between rumen metabolites and DMI, we conducted a Spearman correlation analysis to determine the feed intake-associated metabotypes. A p value < 0.05 was considered a significant metabotypes. We then performed the permutation multivariate analysis of variance (PERMANOVA) based on the Bray Curtis distance to assess the relationship between microbial composition and each feed intake-associated metabotypes covariate (McArdle and Anderson Reference McArdle and Anderson2001) using “vegan” package in R software (Oksanen et al. Reference Oksanen, Blanchet and Kindt2015). Next, we correlated KEGG modules with rumen metabolites significantly related to rumen microbiota. The correlation relationship was visualized using a heatmap with the “pheatmap” package in R software.
Statistical analyses
The Wilcoxon rank test was performed to compare the difference in the relative abundance of rumen microbiota, including bacteria, eukaryotes, archaea, and viruses, the abundance of CAZymes gene at class level, and COG gene at category level through “vegan” package in R 4.1.0 with False discovery rate (FDR)-corrected p value (q value) < 0.05 being considered as significantly different. The PCoA based on Bray Curtis distance was made to describe the rumen microbial composition in bacteria, eukaryotes, archaea, and viruses described through the “ggplot2” package, and further performed PERMANOVA to assess the significant difference of rumen microbial composition using “vegan” package in R 4.1.0 (Oksanen et al. Reference Oksanen, Blanchet and Kindt2015).
The linear discriminant analysis effect size (LefSe) was used for the identification of the significantly different microbiota among groups at the phyla and genera level (Segata et al. Reference Segata, Izard and Waldron2011) through the online platform of Majorbio Cloud Platform (http://www.majorbio.com), with the Linear Discriminant Analysis (LDA) score >2.5 for bacteria, LDA score >3 for eukaryota and q < 0.05. Similarly, the difference in the relative abundances of KEGG pathways at level 3, COG at the function level, and CAZy at the family level was determined by LefSe with the criteria of LDA score >2 and q < 0.05.
Rumen metabolome data was imported into MetaboAnalyst 5.0 for the multivariate analysis using the principal component analysis, partial least squares discriminant analysis (PLS-DA), and t-test. The variable importance in projection (VIP) > 1 (generated by the PLS-DA model) and q ≤ 0.05 were considered statistically significant.
Results
Profiling of the rumen metagenome
In this study, we sequenced the whole metagenome of 10 rumen samples of 10 Holstein cows. A total of 142.38 Gb of metagenome data were obtained, with 14.24 ± 0.35 Gb (mean ± SEM) per sample. These data yielded a total of 942,909,840 raw reads, with an average of 94,290,984 ± 2,300,672 reads (Table S2) per sample. After quality control and filtering the host reads, 937,177,972 clean reads were obtained (Table S2). These reads were de novo assembled into 11,843,246 contigs, ranging in length from 300 to 229,430 bp (N50 length of 743 ± 57 bp, Table S2), and then predicted to 16,265,699 ORFs and 5,113,381 nonredundant genes.
Compositional profiles of the rumen microbiome
After taxonomic annotation and mapping to the NCBI NR database, 5 domains, 8 kingdoms, 134 phyla, 281 classes, 638 orders, 1,203 families, and 3,052 genera were obtained. At the domain level, 95.44% of microbes belonged to bacteria (26,769,063 sequences), followed by viruses (2.18%, 591,370 sequences), eukaryotes (1.56%, 382,510 sequences), and archaea (0.58%, 144,380 sequences; Fig. 1A). For bacteria, the rumen samples were characterized by a high relative abundance of Bacteroidetes (53.44 ± 4.03%), Firmicutes (32.5 ± 3.2%), and unclassified d_Bacteria (3.87 ± 0.37%; Fig. S1A). Within these phyla, the most abundant bacterial genera included Prevotella (39.39 ± 3.94%), followed by Bacteroides (7.94 ± 0.37%), Clostridium (5.64 ± 0.88%), and unclassified f_Lachnospiraceae (3.77 ± 0.35%; Fig. S1C). For eukaryota, the main abundant phyla included unclassified d_Eukaryota (46.40 ± 6.65%), Nematoda (17.09 ± 4.71%), Chordata (6.49 ± 1.26%), and Arthropoda (6.25 ± 1.42%; Fig. S1B). Among these phyla, the high relative abundance genera mainly concentrate in Trichuris (15.99 ± 4.71%), Oxytricha (7.67 ± 1.83%), Stylonychia (6.68 ± 1.58%), Trichomonas (5.46 ± 1.11%), and Tetrahymena (5.24 ± 1.13%; Fig. S1D).
Microbial profiles of rumen microbial composition of HFI and LFI dairy cows. (A) Rumen microbial composition based on the domain level taxonomy. (B) Comparison of microbial domains between HFI and LFI cows. Significantly different domains were tested by t-test with adjusted p value < 0.05. (C) Bacterial compositional profiles of HFI and LFI rumen samples based on species visualized using PCoA. (D) Eukaryota compositional profiles of HFI and LFI rumen samples based on species visualized using PCoA. The PCoA were plotted, and calculated based on the Bray Curtis dissimilarity matrices.

The overall microbial PCoA showed two clusters between the two groups. To further assess the significant difference of the clusters, PERMANOVA tests revealed that the bacterial and eukaryotal communities were both tended to significantly different (p = 0.069 in bacteria, Fig. 1C; p = 0.088 in eukaryota, Fig. 1D), while archaea and virus communities were showed no significant difference (p = 0.224 in archaea, Fig. S2A; p = 0.102 in virus, Fig. S2B).
In addition, the relative abundance of eukaryota, bacteria, and unclassified taxa significantly differed between the two groups (p < 0.05; Fig. 1B), while no significant difference was observed in archaea and viruses among the two test groups (p > 0.05; Fig. 1B) using Wilcoxon rank test. Therefore, the downstream analysis of rumen microbial taxa between the HFI and LFI dairy cows was focused on bacteria and eukaryota.
Differences in bacteria and eukaryota between LFI and HFI cows
The sequenced reads from all samples assigned to 34 microbiota differed markedly in their patterns of bacterial composition (Fig. 2A). Of them, 11 were more enrichment in the LFI group, and 23 were more enrichment in the HFI group (LDA > 2.5, q < 0.05; Fig. 2A). For example, Fibrobacteres and unclassified d_Bacteria were the most enriched phylum in HFI cows (LDA > 2.5, q < 0.05; Fig. 2A). In general, Succinatimonas, Ruminobacter, and Roseburia were significantly enriched in the LFI cows (LDA > 2.5, q < 0.05), while Bacteroides, Fibrobacter, and Paraprevotella were significantly enriched in the HFI cows (LDA > 2.5, q < 0.05; Fig. 2A).
Lefse analysis found the significantly differed bacteria (A) and eukaryota (B) across groups with the cutoff of LDA > 2.5 for bacteria or LDA > 3 for eukaryota, and q < 0.05.
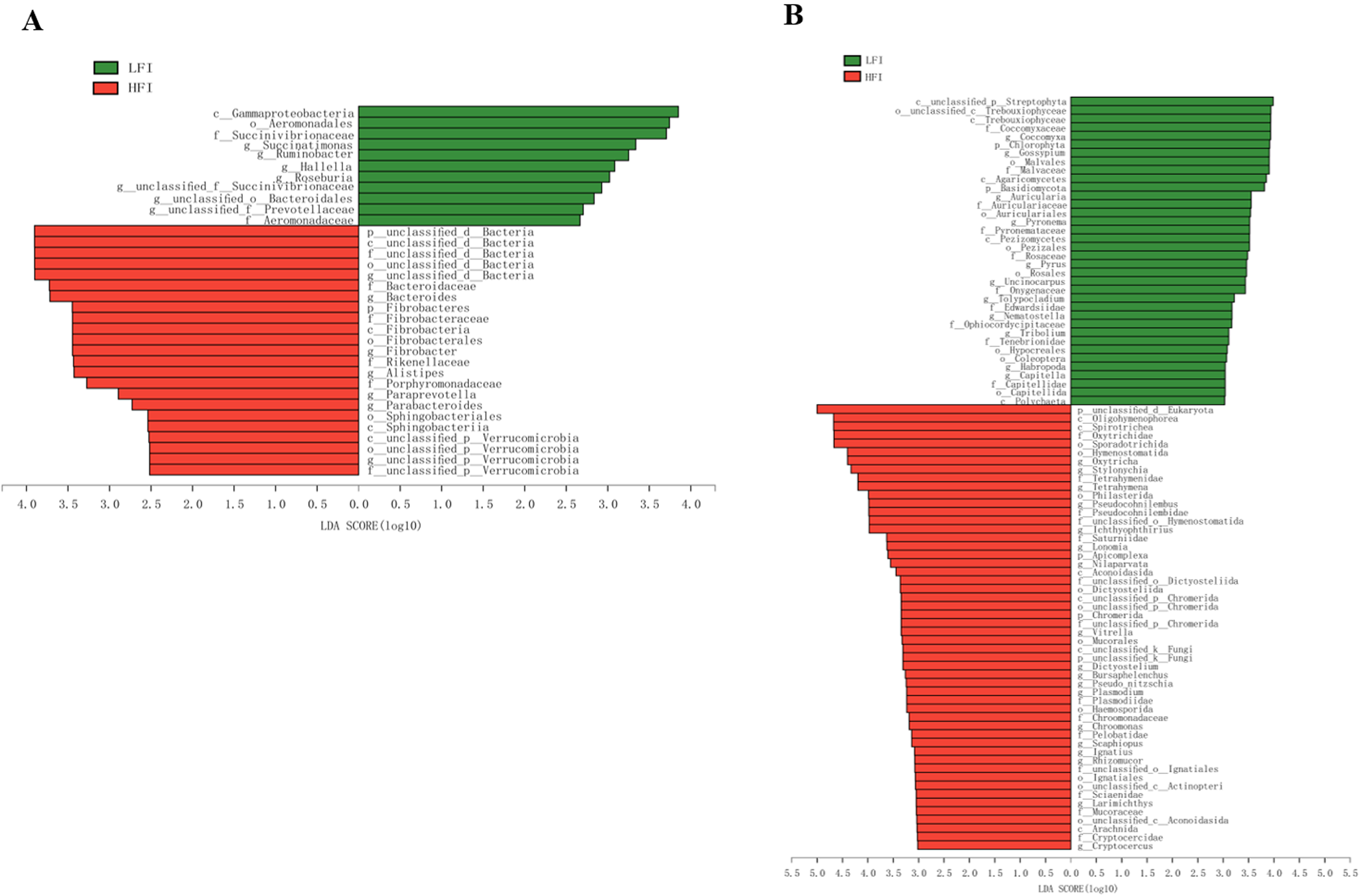
For the comparison analysis of eukaryota, unclassified d_Eukaryota (LFI: 34.55 ± 9.54%; HFI: 58.24 ± 11.23%) was significantly abundant phyla in HFI dairy cows than LFI dairy cows (LDA > 3.0, q < 0.05; Fig. 2B), while Chlorophyta (LFI: 2.51 ± 0.89%, HFI: 0.66 ± 0.95%) and Basidiomycota (LFI: 2.62 ± 0.66%, HFI: 1.27 ± 0.68%) were significantly abundant in LFI cows than HFI cows (LDA > 3, q < 0.05; Fig. 2B). In general, Oxytricha (LFI: 4.69 ± 2.42%, HFI: 10.65 ± 2.73%; Table S3), Stylonychia (LFI: 4.14 ± 2.11%, HFI: 9.22 ± 2.37%; Table S3), and Tetrahymena (LFI: 3.38 ± 1.48%, HFI: 7.10 ± 1.68%; Table S3) was significantly enriched in the rumen of HFI cows than LFI cows (LDA > 3, q < 0.05; Fig. 2B).
Differential function of the rumen microbiome between the HFI and LFI cows
To compare the function of the microbiome between LFI and HFI cows, the rumen microbiome nonredundant gene catalog of all sampled cows was annotated to CAZymes, COG, and KEGG databases. The CAZymes annotation results found that genes were mainly encoding glycoside hydrolases (GHs, 57.69 ± 0.68%), followed by glycosyltransferase (GTs, 24.37 ± 0.69%), carbohydrate esterases (CEs, 11.31 ± 0.19%), and carbohydrate-binding modules (CBMs, 2.91 ± 0.18%); a few genes encoding polysaccharide lyases (PLs, 2.71 ± 0.15%), and auxiliary activities (AAs, 1.00 ± 0.06%; Fig. S3A). For the family level, GT2 was the most highly represented, followed by GH2, GT4, GH25, GT41, CE1, CE10, GH97, GH24, GH31, GT73, GH3, and GH28 (Fig. S3B). At the class level, among the LFI and HFI groups, the proportion of genes encoding AAs tended to increase with feed intake, while others showed no significant difference among LFI and HFI groups (q < 0.05; Fig. 3A). At the family level, LefSe analysis showed that among genes encoding CAZymes that deconstruct cellulose, hemicellulose, starch, and protein, of them, 21 were significantly characterized in the HFI group, including 6 GHs, 6 CBMs, 4 GTs, 3 CEs, and 2 AAs; while 19 CAZymes were characterized in the LFI group, including 11 GHs, 5 GTs, 2 PLs, and 1 CEs (LDA > 2, q < 0.05; Fig. 3B).
Functional difference of ruminal microbiome of dairy cows. Comparison of CAZyme function at class level (A) and COG function at category level (C). Difference analysis of CAZyme function at family level (B) and COG function at function level (D) using LefSe analysis with q value < 0.05. (E) Histogram of the LDA scores computed for differently abundant KEGG function at level 3 with LDA > 2 and q < 0.05. GH: glycoside hydrolases; GT: glycosyltransferase; CE: carbohydrate esterases; CBM: carbohydrate binding modules; PL: polysaccharide lyases; AA: auxiliary activities; A: RNA processing and modification; O: Posttranslational modification, protein turnover, chaperones; Y: Nuclear structure.
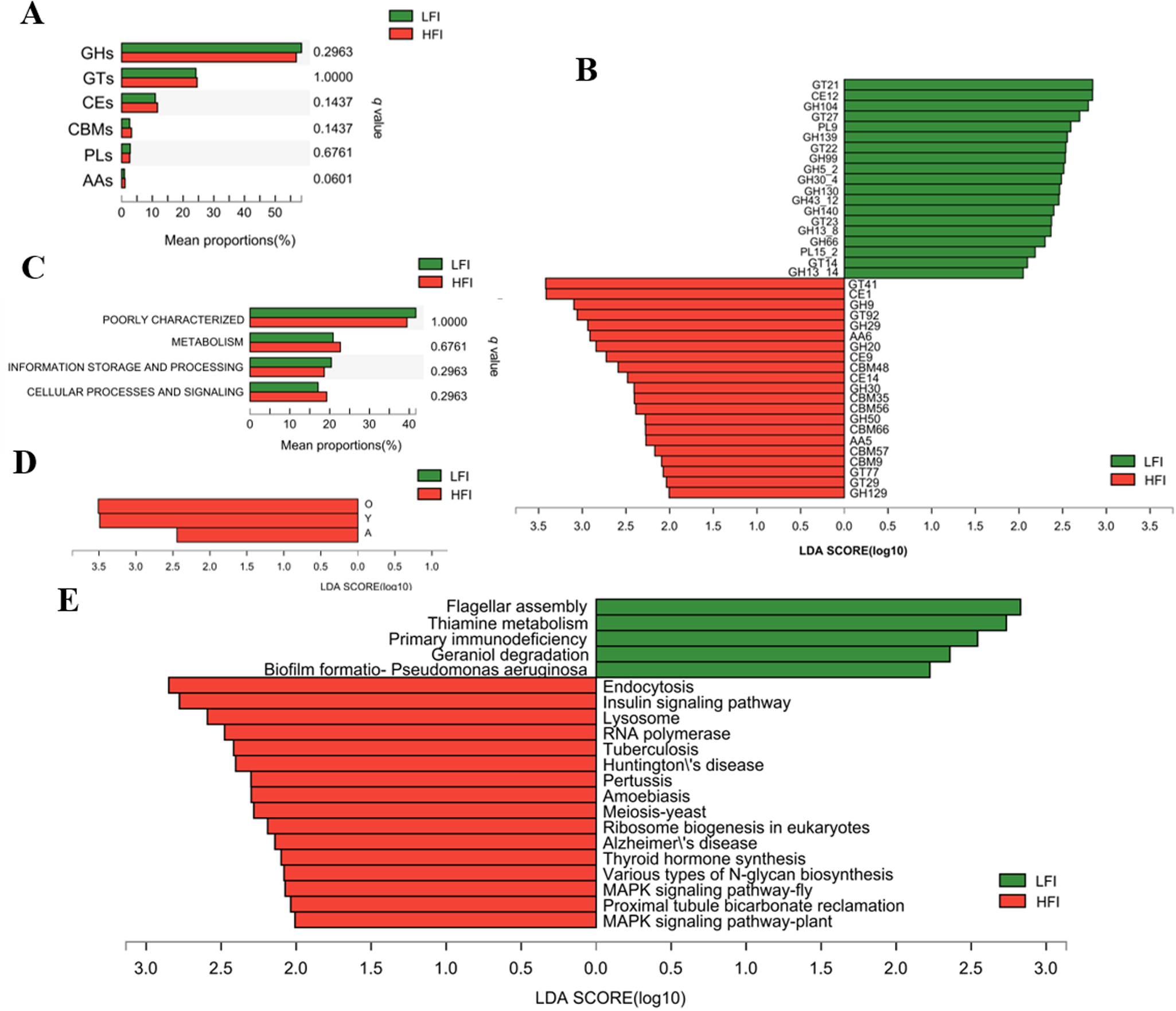
Meanwhile, COG annotation revealed that functions unknown (40.25 ± 1.77%), recombination, and repair (12.63 ± 1.83%), cell wall/membrane/envelope biogenesis (6.65 ± 0.30%), carbohydrate transport and metabolism (5.63 ± 0.48%), and amino acid transport and metabolism (3.98 ± 0.30%) were more abundant among the functions (Fig. S3). At the category level, the proportion of gene functions showed no significant differences among groups (q > 0.05; Fig. 3C). At the functional level, LefSe analysis revealed that gene functions of posttranslational modification, protein turnover, nuclear structure, and RNA processing and modification were significantly abundant in HFI cows when compared to LFI cows (LDA > 2, q < 0.05; Fig. 3D).
The KEGG function analysis identified a total of 10,242 pathways based on a total of 5,113,381 nonredundant genes. In the first level category, “organismal systems” was the most abundant pathway (59.41 ± 1.79%), followed by “metabolism” (19.8 ± 1.23%), “human diseases” (5.95 ± 0.21%), and “cellular processes” (5.63 ± 0.41%). At the second level, the “replication and repair” (13.85 ± 1.31%), “nucleotide metabolism” (10.94 ± 0.8%), “global and overview maps” (8.07 ± 0.39%), and “amino acid metabolism” (7.75 ± 0.34%), being most dominant (average relative abundance of >5% for all samples). LefSe analysis revealed that one pathway belonging to glycan biosynthesis and metabolism, two pathways belonging to signal transduction, two pathways belonging to environmental information processing of signal transduction, the pathway belonging to the organismal systems of the endocrine system (insulin signaling pathway and thyroid hormone synthesis), and excretory system (proximal tubule bicarbonate reclamation) were all enriched in HFI group than LFI group (LDA > 2, q < 0.05; Fig. 3E). Furthermore, one pathway of the thiamine metabolism attached to the metabolism of cofactors and vitamins, and two pathways associated with the cellular processes were both more abundant in the rumen of the LFI group’s cows (LDA > 2, q < 0.05; Fig. 3E).
Rumen metabolome and metabolic pathway analysis
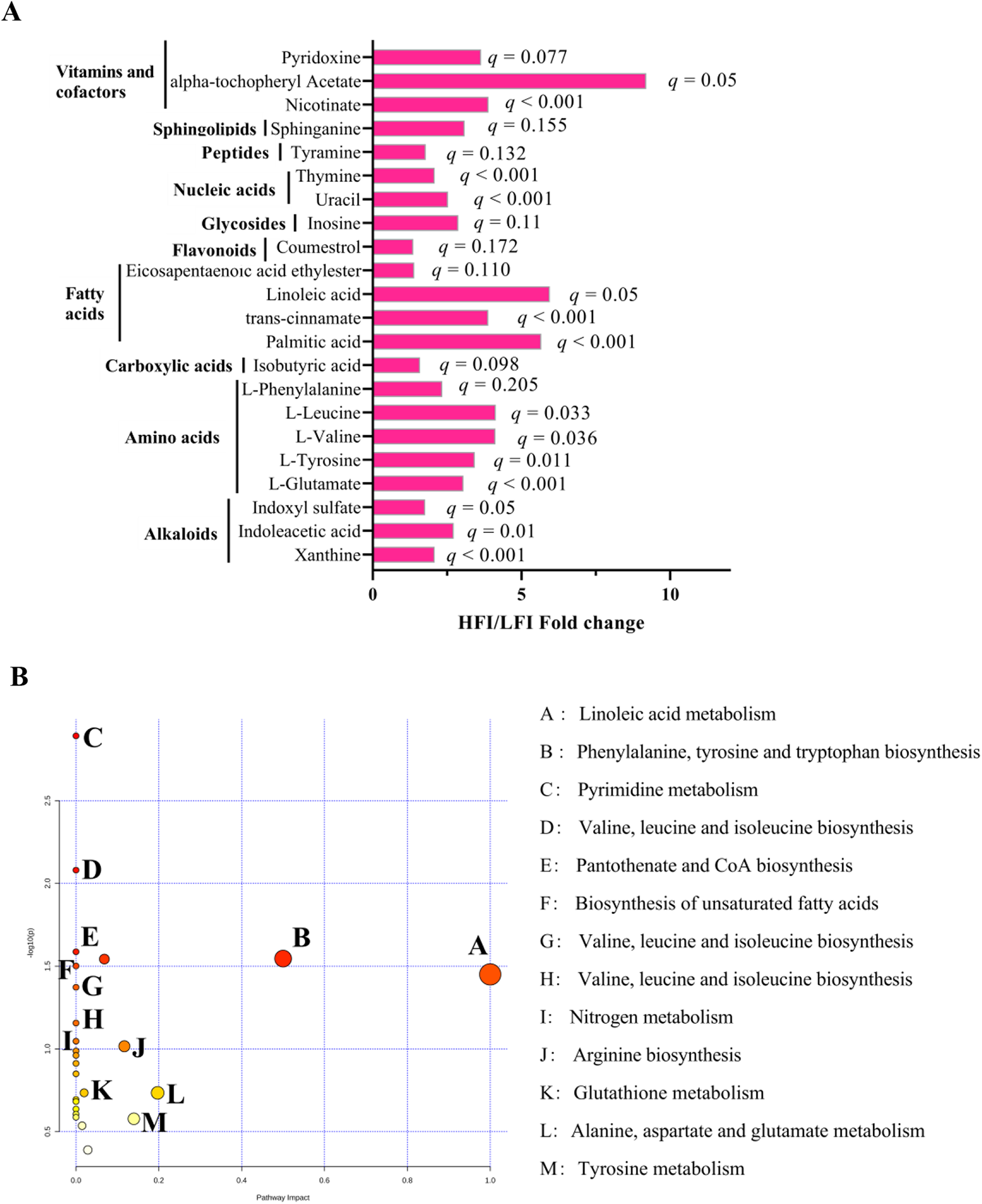
To further understand the differences in rumen microbiota that caused the change in rumen metabolism, the nontarget metabolome of all collected samples was performed to analyze the rumen metabolite profiles. We identified 320 rumen metabolites in the rumen metabolome. T-test suggests that HFI cows contained a higher relative proportion of 22 rumen metabolites than the LFI cows (p < 0.05, VIP > 1; Fig. 4A), of them 14 were significantly (q ≤ 0.05, VIP > 1; Fig. 4A). Metabolic pathway enrichment analysis revealed that 14 significantly differed rumen metabolites were enriched to 10 pathways (Fig. 4B), of them, the linoleic acid metabolism and (p = 0.04, impact = 1) and phenylalanine, tyrosine, and tryptophan biosynthesis was the different pathway (p = 0.03, impact = 0.5). For phenotype (DMI) association analysis, we found that 24 rumen metabolites were significantly associated with DMI, and those metabolites were determined to be the DMI-associated metabotypes (Table S4). PERMANOVA analysis showed that among 24 DMI-associated metabotypes, 14 DMI metabotypes (all were DMI-positive metabotypes) were associated with variations in the rumen microbiome (p < 0.05; Table S4).
Rumen metabolome of HFI and LFI cows. (A) HFI/LFI fold change of significantly different rumen metabolites between HFI and LFI cows. (B) Pathway analysis was performed using the significantly different rumen metabolites between HFI and LFI cows.
Association of the rumen microbiome, rumen metabolome, and their contribution to DMI variation
To understand the contribution of rumen microbiome and metabolome on DMI variations, Spearman’s rank correlations analysis was performed. The analysis found that there are 248 significant correlations existed in rumen microbiota and metabolites (R > 0.631, p < 0.05; Fig. 5A). Of them, 135 negative correlations were found between 3 Succinivibrionaceae (unclassified_f_Succinivibrionaceae, Succinatimonas, and Ruminobacter) and amino acids and peptides (−0.828 < R < −0.693, p < 0.05). Furthermore, to uncover the co-occurrence patterns of rumen microbiota in DMI-associated metabotypes, a network was established based on significant correlations (−0.828 < R < −0.693, p < 0.05; Fig. 5B). The network showed 35 significant correlations between the rumen microbiota and DMI-associated metabotypes. Among the 35 correlations, 3 Succinivibrionaceae (unclassified_f_Succinivibrionaceae, Succinatimonas, and Ruminobacter) also significantly correlated to the DMI-associated metabotypes, including rumen metabolites involved in fatty acids and conjugates, favonoids, and gycerophosphocholines.
Interactions between rumen metagenome, metabolome, and serum metabolome. (A) Spearman’s rank correlations between rumen microbiota and rumen microbial metabolites. (B) Spearman’s correlation network showing relationships between rumen microbiota and microbial MPY-associated metabotypes. Only strong correlations (R > 0.631 or R < −0.693; p < 0.05) were showed in the correlation networks.

The contribution degree of rumen microbial composition, microbial functions, and rumen metabolites on DMI were estimated individually using the linear mixed-effect model. The rumen metagenome, function, and rumen metabolome were responsible for 29.63, 27.30, and 33.50% of the DMI variation (Fig. 6).
Consolidated results and model. Rumen microbial genus and functions (CAZymes and KEGG functions) were compared between two DMI groups. Rumen metabolites were separated into two groups that were either positively or negatively correlated with DMI; and then PERMANOVA was performed based on the microbial abundance profiles to assess the effect of each metabolites (metabolites with p < 0.05 were considered to associate with rumen microbiota). The rumen metabolome was also separated into two groups that were significantly different between two DMI groups; and the key rumen metabolic pathways were enriched based on the significantly different metabolites. The proportion of variance in DMI explained by the rumen microbial genera and functions, and rumen metabolome (defined as biome-explainability) were estimated using linear mixed effects model.

Discussion
The main purpose of this work was to assess the contribution of rumen microorganisms, functions, and rumen metabolome of dairy cows on DMI variations. Dairy cows are a highly important livestock species providing high quality proteins via milk for humans. DMI is an important productive trait for dairy cows. It is a well-documented fact that feed intake affects the rumen microbiome of cattle (Li et al. Reference Li, Xi and Wang2020; Shi et al. Reference Shi, Guo and Degen2020). Furthermore, previous studies demonstrated that the rumen microbiome is associated with the feed intake of dairy cows (Jami et al. Reference Jami, White and Mizrahi2014; Wallace Reference Wallace2019), including our previous study for transition dairy cows (Huang et al. Reference Huang, Ji and Garret2021), indicating that feed intake is an important factor for the rumen microbiome. Therefore, rumen fluid samples of postpartum dairy cows with low and high DMI levels were collected to explore the explanation of rumen microbial composition, functions, and rumen metabolites on DMI variations in postpartum dairy cows.
The present study revealed that the rumen metagenome and its function all influenced by the postpartum cows’ DMI. DMI is an important productive trait for transition dairy cows, especially for postpartum cows (0–21 days after calving), low DMI can increase the risk of metabolic diseases and decrease the milk production and profits of the farm (Allen and Piantoni Reference Allen and Piantoni2013). Previous studies reported the variations of rumen bacteria with different feed intake levels in fresh dairy cows, and some specific bacteria, such as Prevotellaceae_UCG-001, Lachnobacterium, and Olsenella, were directly correlated to DMI (Huang et al. Reference Huang, Ji and Garret2021). In our study, we found the relative abundance of the genus Fibrobacter was significantly higher in HFI cows. Fibrobacter is a gram-negative anaerobic bacteria, and previous studies found that Fibrobacter has an excellent ability to degrade cellulose in the herbivore rumen (Kobayashi et al. Reference Kobayashi, Shinkai and Koike2008). A previous study found that GH9 and GH30 were produced by members of the genera Fibrobacter (Wu et al. Reference Wu, Elekwachi and Bai2021). GH9, a member of the xylanase family and can cleave β-1,4-xylosidic bonds between xylose monomers, is important for cellulose degradation. GH30, a endo-beta-1,4-xylanase, plays a role in hemicellulose digestion. Moreover, Neves et al. (Reference Neves, Yu and Suzuki2021) found that high-efficient cattle exhibited an increased relative abundance of Fibrobacter succinogenes and enzymes for xylanases and endoglucanases production compared to low-efficient cattle. In our study, CAZymes function analysis found that GH9 and GH30 were significantly enriched in HFI cows. Collectively, the significant abundance of Fibrobacter, GH9 and GH30 in the rumen of HFI cows, implies that the HFI cows may have a higher cellulose-degrading ability for forage than LFI cows.
Rumen protozoa constitute up to 50% of the microbial biomass in the rumen (Andersen et al. Reference Andersen, Altshuler and Vera-ponce de León2023) and exert a profound impact on fiber digestion (Yu et al. Reference Yu, Yan and Somasundaram2024). A recent study of rumen ciliates found that rumen ciliates have an excellent ability to encode CAZymes and degrade all major kinds of plant carbohydrates (Li et al. Reference Li, Wang and Zhang2022). In the current study, we found that genera of Oxytricha and Stylonychia were significantly higher in the rumen of HFI cows than in the rumen of the LFI cows. Thus, the significantly higher relative abundance of Oxytricha and Stylonychia observed in the rumen of HFI cows showed that HFI cows have a higher ability to degrade the plant-source carbohydrates. Furthermore, CAZymes functional analysis showed a greater function of GTs, CEs, and CMBs in the HFI group, which implies that the rumen of HFI cows has more feed degradation ability than LFI cows. Thus, the rumen environment of HFI dairy cows may be preferable for plant digestion and energy production.
Our previous study showed that the rumen bacteria were associated with the DMI of dairy cows during the postpartum period based on 16S rRNA sequencing (Huang et al. Reference Huang, Ji and Garret2021). Meanwhile, a previous study also found that the feed intake of lactating dairy cows was correlated with the rumen bacteria using 16S rRNA sequencing (Li et al. Reference Li, Xi and Wang2020). The ruminal microbiome hosts microorganisms not only bacteria but also archaea, protozoa, and fungi, with all interacting with each other and altering the rumen environment and substrates that will be available for the host digestion and metabolism (Lobo and Faciola Reference Lobo and Faciola2021). Therefore, to profile the whole rumen microbiome of dairy cows among different DMI levels is necessary to know the variations of microbiota and function in dairy cows. In this study, the COG functional annotation showed that the nonredundant genes in groups were mainly related to carbohydrate transport and metabolism, and amino acid transport and metabolism. We found that the number of genes involved in posttranslational modification, and protein turnover was significantly higher in the HFI group as compared to the LFI group (LDA > 2, p < 0.05). It is suggested that the HFI dairy cows have more active material interaction with their microbes. For the KEGG analysis, we found that the various types of N-glycan biosynthesis pathways were significantly higher in the HFI group as compared to the LFI group. Most proteins carry carbohydrate chains-glycans as part of their structure, indicating that the HFI cows have a great ability to produce protein.
Recent studies have reported that the rumen microbiome affects host traits, including feed efficiency (Shabat et al. Reference Shabat, Sasson and Doron-Faigenboim2016) and feed intake (Huang et al. Reference Huang, Ji and Garret2021; Jami et al. Reference Jami, White and Mizrahi2014; Wallace Reference Wallace2019) in dairy cows. Our results found that the rumen metagenome and rumen metabolome all influenced the postpartum cows’ DMI similar to the previous study, which found the rumen metagenome, rumen metabolome, and host serum metabolome of lactation Holstein cows all influenced the milk protein yield (Xue et al. Reference Xue, Sun and Wu2020). In our study, the strong association between the rumen microbiome and metabolome suggests that the rumen microbiome potentially interacts with host metabolism. Notably, Succinivibrionaceae may affect host fatty acids and conjugates, favonoids, and gycerophosphocholines metabolism. The variation in phenotypes could be explained by host gastrointestinal microbial composition, microbial functions, and microbial metabolites, then the estimated proportions of variation in host phenotype due to gastrointestinal microbial composition, microbial functions, and microbial metabolites was defined as omics-explainability (Xue et al. Reference Xue, Sun and Wu2020). Xue et al. (Reference Xue, Sun and Wu2020) reported that omics-explanation of the rumen microbial composition, microbial functions, and rumen metabolome for host milk protein yield were 17.81, 21.56, and 29.76, respectively. In the current study, we found that the omics-explanation of rumen microbial composition (29.63%), microbial functions (27.30%), and rumen metabolome (33.50%) was higher than that reported for milk protein yield. It suggests that the rumen metagenome, microbial function, and metabolome potentially make greater contributions to DMI relative to milk protein yield. Our omics-explainability results further suggest that rumen metagenome and rumen metabolome were good predictions for the DMI of dairy cows, and they may be used for other trait predictions associated with rumen function and metabolism.
The limitation of our study is the small sample size selected for the experimental dairy cows. The rumen is a huge biological resource fermenter that contains numerous enzymes that degrade plant fibers, but the understanding of the rumen enzymes is still relatively lacking. Therefore, according to the previous study (Mogodiniyai Kasmaei and Sundh Reference Mogodiniyai Kasmaei and Sundh2019), it is easier to identify some novel bacterial GHs from the rumen of higher productive performance dairy cows using the whole-genome shotgun metagenomic sequencing. Moreover, ruminal microbial transplantation has been proposed as one of the promising methods of reshaping the rumen microbiota homeostasis to treat rumen disorders, enhance productivity, and improve health (Zhou et al. Reference Zhou, Peng and Chen2018). Thus, HFI dairy cows are a potential candidate for the donor rumen microbial transplantation for lower performance dairy cows or cattle for improvements in both animal welfare and profitability.
Conclusion
This study describes the composition and functions of the rumen microbiome of dairy cows of different feed intakes, and the results demonstrate that Prevotella was the most abundant bacteria genera, and Oxytricha was the predominant protozoa genera in the rumen of HFI cows. The bacterial genera Fibrobacter and Bacteroides were significantly enriched in HFI cows, with the enrichment of plant carbohydrate degradation enzymes, such as GH9 and GH30. Most differed COG and KEGG pathways were enriched in carbohydrate metabolism and protein biosynthesis of HFI dairy cows, indicating that HFI dairy cows might produce more energy. The omics basing explainability of phenotype variations analysis revealed that the rumen microbial metabolites made greater contributions to DMI than rumen microbial composition and functions. These results expanded our understanding of the overall differences in dairy cows’ rumen microbiota at low and HFI levels and provided novel insights into strategies for manipulating rumen microbiota to improve ruminant performance, such as rumen fluid transplantation.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/anr.2024.29.
Acknowledgements
This work was supported by the Hainan Provincial Natural Science Foundation of China (grant number 322QN240), the Foundation of Hainan University (grant number RZ2200001242), and the National Natural Science Foundation of China (grant number 32130100). The authors also would like to thank the staff at the Zhongdi dairy farm (Beijing), F. Wang, J. Huang, and C. Guo for their assistance with sample collection.









 Open access
Open access