


Introduction
In recent decades, genome sequencing technologies have matured and spread rapidly across nearly all areas of the biological sciences [Reference Heather and Chain1]. Pathogen genomic information can enhance traditional epidemiology methods and disease surveillance programmes. At the international, national, or regional level, genomic surveillance can be used to study broad evolutionary trends in infectious diseases over time and space and to monitor changes in the genome that potentially impact medical countermeasures. However, local outbreak investigations can also benefit from additional genomic information to elucidate potential sources of infection, identify disease clusters, study individual transmission events, and guide the deployment of public health interventions [Reference Gardy and Loman2, Reference Behrmann and Spiegel3].
Previous investments by governments, academia, and industry in genome sequencing for pathogen surveillance – such as foodborne pathogen surveillance in Europe and the Americas, pandemic influenza monitoring globally, and large-scale outbreak responses like those to the Zika virus in the Americas and Ebola virus in West Africa – were made in both high-income countries (HICs) and low- to middle-income countries (LMICs) [Reference Pollett4–Reference Davedow7]. However, by 2019, the analysis and interpretation of genomic sequence data within the context of local public health were not widely practiced globally. Additionally, the field of genomic (or molecular) epidemiology had not yet been fully implemented by traditional epidemiologists working in infectious disease outbreaks, particularly in applied or lower-resource settings. As a result, when the coronavirus disease 2019 (COVID-19) pandemic emerged, there was significant untapped potential for the widespread application of genomics in local outbreak investigations.
Studying genomic epidemiology practices during the early phase of the COVID-19 pandemic is necessary for understanding how novel technologies were integrated into the public health response. This review focuses on the application of genomic epidemiology during a period when non-pharmaceutical interventions and contact tracing were the primary tools for controlling SARS-CoV-2 transmission. The findings will inform future pandemic preparedness for novel pathogens and illustrate opportunities to strengthen local public health capacity and support implementation in lower resourced settings.
Purpose of the review
This review describes the use of SARS-CoV-2 whole-genome sequencing and genomic epidemiology as part of local outbreak investigations and response during the first year of the COVID-19 pandemic. We examined the study setting, methods used, study results, quality and potential facilitators of the use of genomics in these settings. Finally, we used thematic analysis to illustrate the added value of genomic epidemiology for understanding SARS-CoV-2 transmission in the early pandemic response.
Methods
The study followed common systematic review standards, including the Cochrane Handbook for Systematic Reviews of Interventions and updated Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) guidelines (Supplementary Material 2) [Reference Higgins8, Reference Page9]. The protocol was registered in the international prospective register of systematic reviews before abstract screening (https://www.crd.york.ac.uk/prospero/, CRD42022255562).
Systematic search strategy
A search of PubMed, Scopus, and Web of Science was conducted on 23–24 April 2021, using terms for COVID-19, genomics, and epidemiology (Supplementary Material 2). The search was limited to papers published within one year of the World Health Organization (WHO) pandemic declaration (11 March 2020) [10]. Searches of PubMed used keywords and MeSH terms, while searches of Scopus and Web of Science used only keywords. The search results were uploaded to Covidence for screening (Covidence, Veritas Health Innovation, Melbourne, Australia; www.covidence.org).
Abstract and full-text screening
An interdisciplinary team of reviewers with expertise in genomic epidemiology, global public health, virology, laboratory science, and global health security conducted abstract screening in Covidence. Two independent reviewers evaluated each title and abstract using a priori inclusion criteria (Supplementary Table S2). After achieving an inter-rater reliability of 0.80 on a pilot set of ten papers, the team screened the entire set of abstracts. Full-text screening followed the same protocol, and the primary justification for excluding any full-text article was documented. Any disagreements were resolved by consensus or adjudicated by a third reviewer.
Data abstraction and synthesis
Articles remaining after full-text screening underwent data extraction and thematic synthesis. Numeric and qualitative data elements were manually extracted using a standardized template uploaded into Covidence (Supplementary Material 4). Extracted data were cleaned, coded, and analysed using Microsoft Excel, RStudio 2024.09.0, and Datawrapper.de. World regions and country income level designations were determined using World Bank 2020 definitions. Further thematic analysis of extracted genomic epidemiology conclusions was conducted in NotebookLM (Gemini 2.0).
Qualitative framework for study settings
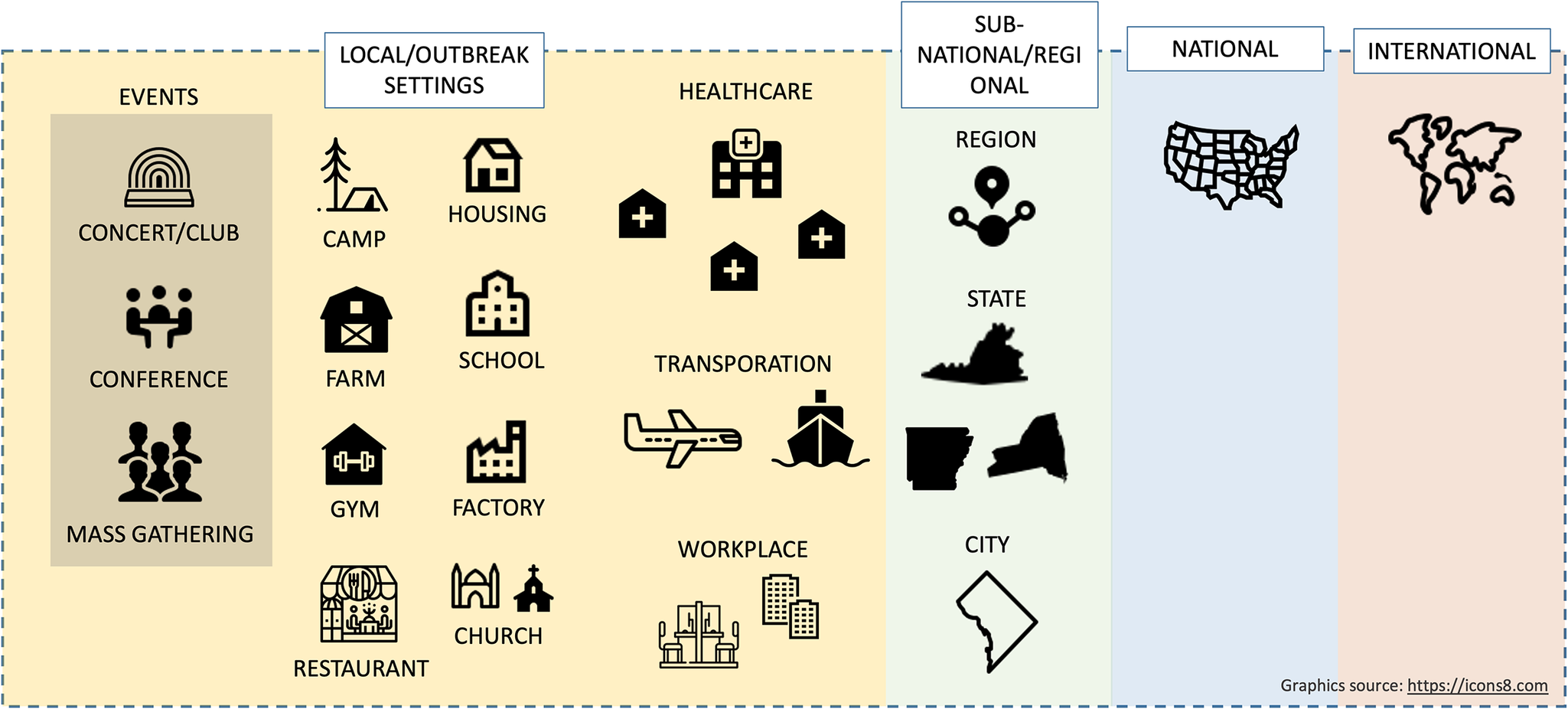
A general qualitative framework was developed to define local-level genomic epidemiology studies, focusing on ‘local outbreak investigations’ or discrete, setting-specific clusters (e.g. healthcare facilities, workplaces, congregate environments, ships, events) where genomic and epidemiologic data were analysed together to understand transmission (Figure 1). Studies involving multiple institutions across broader geographic areas (e.g. multiple hospitals with separate patient populations) were excluded, as they often informed state- or regional-level analyses rather than local contexts.
Framework for grouping settings and level of analysis for genomic epidemiology studies conducted early in the COVID-19 pandemic. Note: Local outbreak response settings can include event-based, institutional, congregate, workplace, and transportation-related exposures. Several institutions grouped together, or larger geographic units, can inform city, state, and regional-level analyses (source: https://icons8.com).

Quality assessment
Risk of bias was evaluated using a custom tool incorporating complementary criteria from the National Institutes of Health (NIH) Study Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies and the Joanna Briggs Institute (JBI) Critical Appraisal Tools Checklist for Analytical Cross-Sectional Studies [11, 12] (Supplementary Material 5). Studies were also assessed for adherence to the Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID) reporting guidelines [Reference Field13] (Supplementary Material 5). Sub-analyses compared the mean quality and reporting scores between studies from HICs and LMICs, as well as by the extent of genomic epidemiology methods used.
Results
Systematic search and screening results
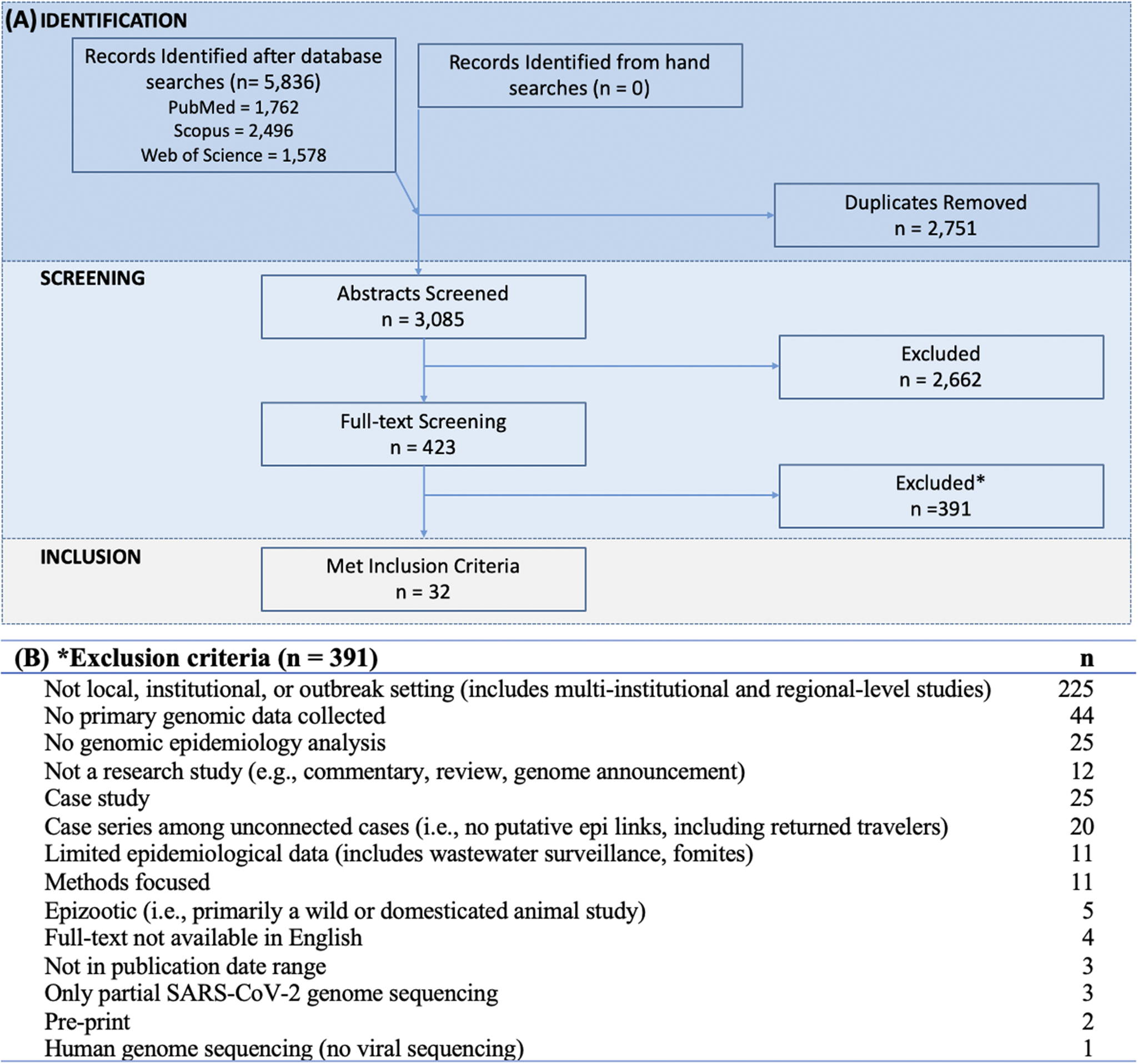
The systematic search identified 5836 abstracts across three databases: 1762 from PubMed, 2496 from Scopus, and 1578 from Web of Science. After removing 2751 duplicate articles, 3085 abstracts were screened with an inter-rater reliability of 88% during the pilot phase and a Cohen’s Kappa of 0.5–0.8 during full screening [Reference McHugh14]. Following exclusion of 2662 abstracts and 391 full-text articles, 32 studies met the full inclusion criteria for final review (Figure 2 and Supplementary Material 6).
PRISMA flow diagram. Note: (a) Depicts studies at each step from identification, screening, to final inclusion. (b) Primary reasons for excluding full-text articles.

Reasons for exclusion
The most common reason for full-text exclusion was a lack of focus on local, institutional, or outbreak settings (225, 58%) (Figure 2). Excluded studies ranged from intra-host viral dynamics to global surveillance trends. Multi-institutional and regional-level analyses were also excluded under this criterion. Additional exclusion reasons included use of only secondary genomic data (44, 11%) and absence of genomic epidemiology methods (25, 6%), which meant the study did not integrate genomic and epidemiological data in the analysis or interpretation. Other excluded articles were case studies (25, 6%), case series among sporadic cases (20, 5%), and articles that contained little to no epidemiological data (11, 3%).
Location and setting
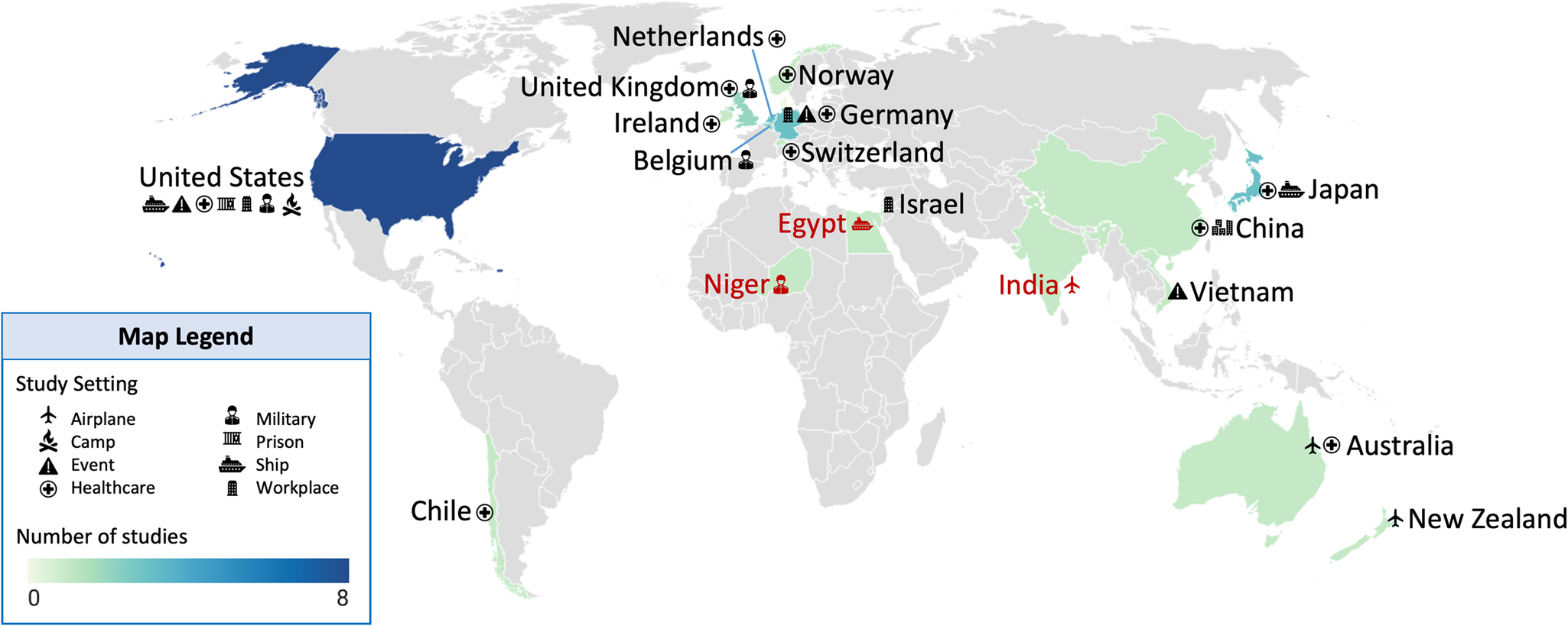
The 32 included studies were conducted in 18 countries (Figure 3). Three of these studies (9%) were conducted in multiple countries (e.g. primary exposure/outbreaks occurred in one country and the investigation occurred in another). Most studies occurred in HICs (29, 91%). Only six studies (19%) were conducted in LMICs (Table 1). In some cases (e.g. Egypt, India, and Niger), SARS-CoV-2 exposure occurred locally, but sample processing and study analysis were conducted elsewhere (Figure 3).
Global map of study locations. Note: Icons indicate the study settings represented within each country, and the colour ramp indicates the number of published studies. A red icon and country name indicate that major exposure or travel history data were from these countries, and sample collection and analysis occurred elsewhere. Figure developed using Datawrapper (source: https://www.datawrapper.de/).

Study characteristics (n = 32)

a Denominator is 32 unless indicated; some categories will sum to over 32 if not mutually exclusive.
b Other, includes housing during an event, outbreak investigation, or quarantine, such as a quarantine hotel, apartment building, or household.
c Cross-sectional includes point and period prevalence studies.
d Cohort includes retrospective and prospective cohort studies.
e Fifteen to sixty days, inclusive.
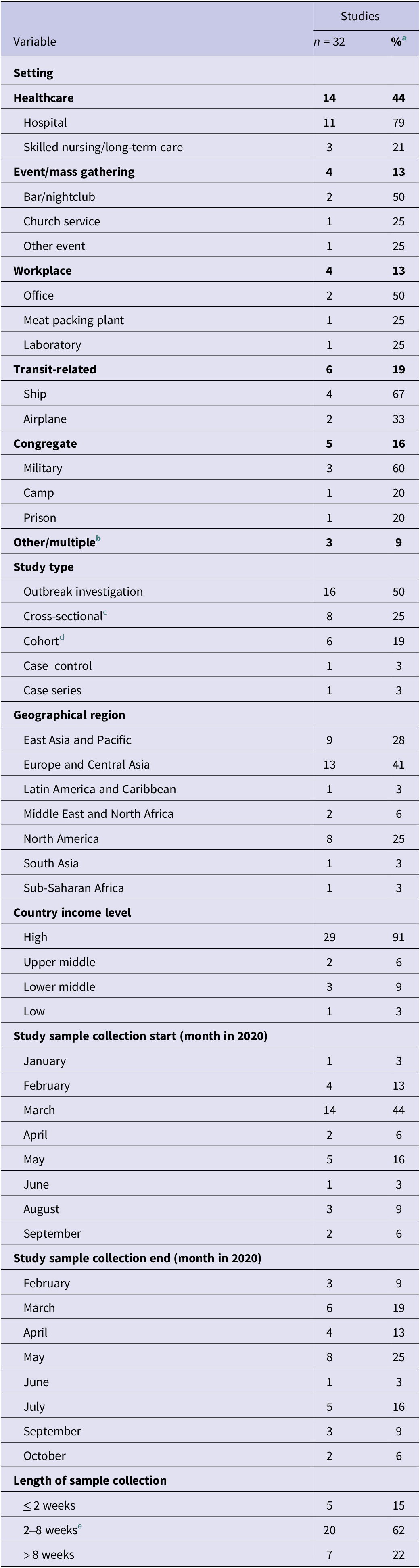
Geographically, most studies were conducted in Europe and Central Asia (13, 41%), East Asia and the Pacific (9, 28%), and North America (8, 28%) (Table 1). The United States had the most studies (n = 8) and the greatest diversity in settings such as healthcare facilities [Reference Arons15–Reference Klompas17], a fishing vessel [Reference Addetia18], a mass gathering [Reference Firestone19], a prison [Reference Hershow20], a summer camp [Reference Szablewski21], and a military training location [Reference Letizia22] (Figure 3). Studies were also conducted in Japan (4), Germany (3), the Netherlands (3), China (2), and the United Kingdom (2).
In terms of setting, nearly half of the studies (14 of 32, 44%) investigated healthcare-associated outbreaks, including hospitals, long-term care facilities, and a psychiatric ward [Reference Arons15–Reference Klompas17, Reference Løvestad23–Reference Weinberger32]. Congregate living environments were the second most common (5, 16%), including military barracks [Reference Letizia22, Reference Pirnay33, Reference Taylor34], a prison [Reference Hershow20], and a sleep-away camp [Reference Szablewski21]. Six studies (19%) focused on transmission during travel or in transit-related environments, including cruise ships [Reference Plucinski35–Reference Sekizuka37], commercial flights [Reference Eichler38, Reference Speake39], and fishing vessels [Reference Addetia18]. Event-based (4, 13%) and workplace-related outbreaks (4, 13%) were less common but represented diverse contexts like nightclubs [Reference Muller40], mass gatherings [Reference Firestone19, Reference Voeten30, Reference Muller40], a meat-processing plant [Reference Günther41], and a laboratory [Reference Zuckerman42] (Table 1).
Major characteristics of LMIC studies
Many studies from LMICs were excluded during full-text screening. Common reasons included reliance solely on secondary genomic data, a focus on broader national or international trends; the studies were an evolutionary analysis of SARS-CoV-2 and other coronaviruses or were a ‘snapshot’ of the first imported cases in a country. The limited number of included papers from LMICs prevented extensive sub-analyses for country-income-level effects.
Six studies were either conducted entirely in LMICs (3, 9%) or focused on major SARS-CoV-2 exposure events in LMICs (3, 9%). For the three LMIC-led studies, a study in Vietnam investigated a potential superspreading event at a bar and leveraged resources from Oxford University and the Wellcome Trust [Reference Chau43]. Two studies were conducted in China, entirely utilizing local institutions and resources. One analysed nosocomial transmission of SARS-CoV-2 in a Wuhan hospital [Reference Wang31], while the other examined community transmission in Beijing [Reference Zhang44]. For studies of groups of individuals with extended travel in LMICs, this included Belgian military personnel training in Niger, Japanese tourists on Nile River cruises in Egypt, and an international flight from India to New Zealand [Reference Pirnay33, Reference Sekizuka36, Reference Eichler38]. Notably, the authors, institutions, and funding sources for these three LMIC-travel-related studies were primarily based in HICs (Supplementary Material S3-1).
Study type and characteristics
Outbreak investigations were half (16, 50%) of the included studies, often employing contact tracing, snowball sampling, or elements of multiple observational epidemiology study designs (Table 1). A quarter of papers (8, 25%) were cross-sectional studies, including point prevalence and period prevalence studies. Cohort (6, 19%), case–control (1, 3%), and case series (1, 3%) comprised the remaining quarter of publications.
Nearly half of the studies (14, 44%) started sample collection in March 2020, coinciding with the declaration of the COVID-19 pandemic (Table 1). The earliest collection period began in January, and the latest was September 2020. While a quarter of studies ended sample collections in May 2020, end dates were more evenly distributed across the year compared to start dates. Most studies collected samples over 2 weeks to 2 months (22, 68%), with durations ranging from 2 to 113 days.
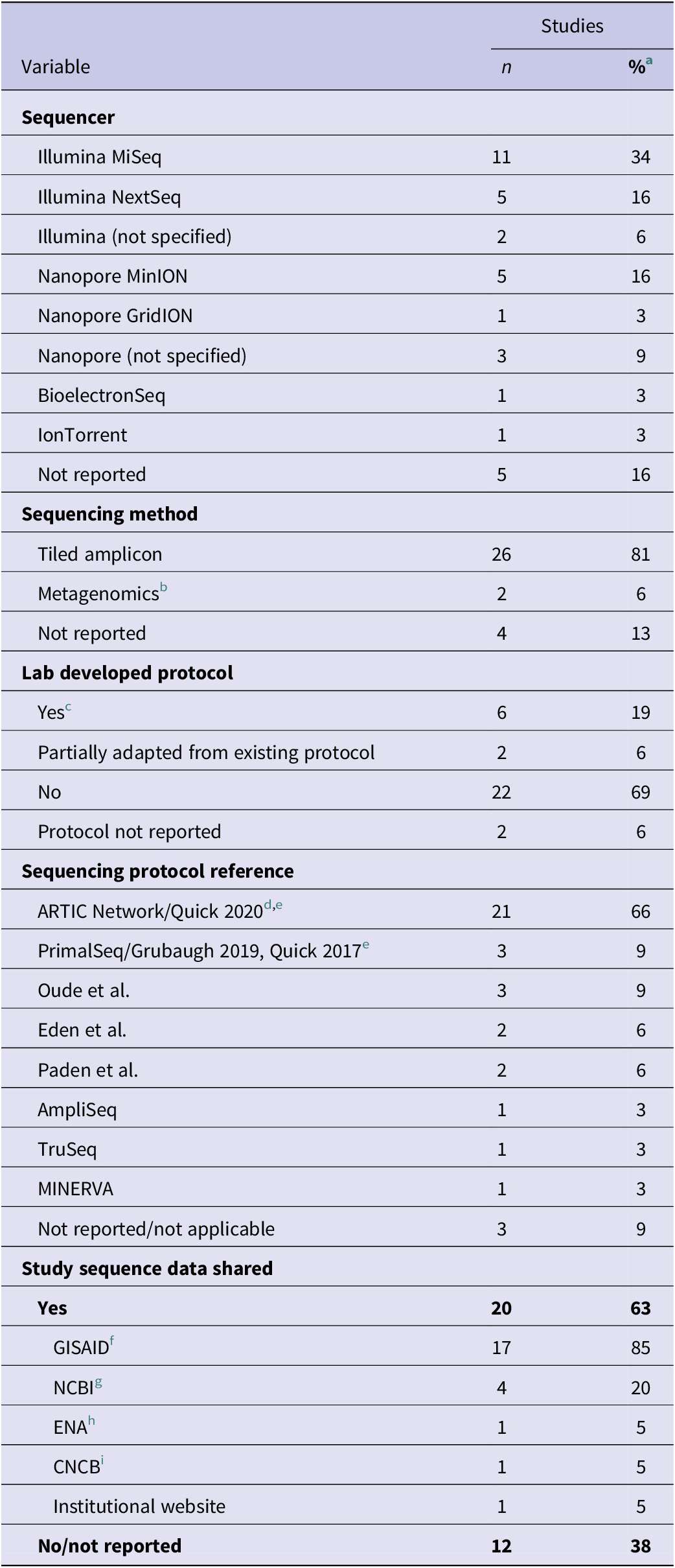
Sequencing methods
Eighteen studies used Illumina short-read sequencing platforms (56%); the most common were MiSeq and NextSeq. Nine studies used long-read sequencing (28%), primarily the Oxford Nanopore Technologies (ONT) MinION. Less commonly used platforms included the Ion Torrent (1, 3%) and CapitalBio BioelectronSeq (1, 3%), while five studies (16%) did not specify a sequencer. Two studies (6%) used more than one sequencing technology (Table 2).
Study laboratory methods (n = 32)

a Denominator is 32 unless indicated otherwise; some categories will sum to over 32 if not mutually exclusive.
b Includes shotgun metagenomics and RNA-enrichment metagenomics.
c Yes, indicates the study was the first publication of the sequencing protocol, or the lead author of the sequencing protocol was a co-author on the study.
d ARTIC Network’s SARS-CoV-2 tiled-amplicon sequencing primer scheme, Versions 1, 2, and 3; Quick is the primary author on the first ARTIC SARS-CoV-2 protocols published in early 2020.
e Although closely related, PrimalSeq (Grubaugh 2019, Quick 2017) refers to the broader precursor tiled-amplicon-sequencing framework for viral genomes, while ARTIC (Quick 2020) refers specifically to the ARTIC Network’s SARS-CoV-2 sequencing protocols built on the same tiled-amplicon idea.
f GISAID; Global Initiative on Sharing All Influenza Data.
g NCBI; National Center for Biotechnology Information, U.S. National Institutes of Health, includes both GenBank and SRA (sequence read archive) databases.
h ENA; European Nucleotide Archive.
i CNCB; China National Center for Bioinformation, National Genomics Data Center.
Most studies used a whole-genome-tiled-amplicon method to sequence SARS-CoV-2 (26, 81%), while two studies (6%) used either shotgun metagenomics or RNA-enrichment metagenomics [Reference Addetia18, Reference Zhang44]. Notably, four studies (13%) either did not report a sequencing protocol or the specific method could not be determined from the reference (Table 2). Six studies (19%) used in-house protocols or cited those developed by a coauthor (Table 2) [Reference Pirnay33, Reference Oude Munnink45–Reference Addetia48]. Two studies (6%) did not specify a sequencing protocol. Most studies (22, 69%) referred to common or previously published protocols, while two studies (6%) reported adapting a published protocol. Twenty-one studies (66%) used the ARTIC Network tiled-amplicon-whole-genome-sequencing protocol for SARS-CoV-2 published in January 2020 [Reference Quick49], while three studies (9%) used methods similar or derived from ARTIC (PrimalSeq) [Reference Grubaugh50, Reference Quick51] (Table 2).
Twenty studies (63%) reported openly sharing genomic data, while 12 (37%) did not. Of the 20 studies that shared data, most deposited sequences in the Global Initiative on Sharing All Influenza Data (GISAID) (17, 85%) or the National Center for Biotechnology Information (NCBI) (4, 20%). Other databases included European Nucleotide Archive (ENA) (1, 5%), Chinese National Center for Bioinformation (CNCB) (1, 5%), and an institutional website (1, 5%) (Table 2).
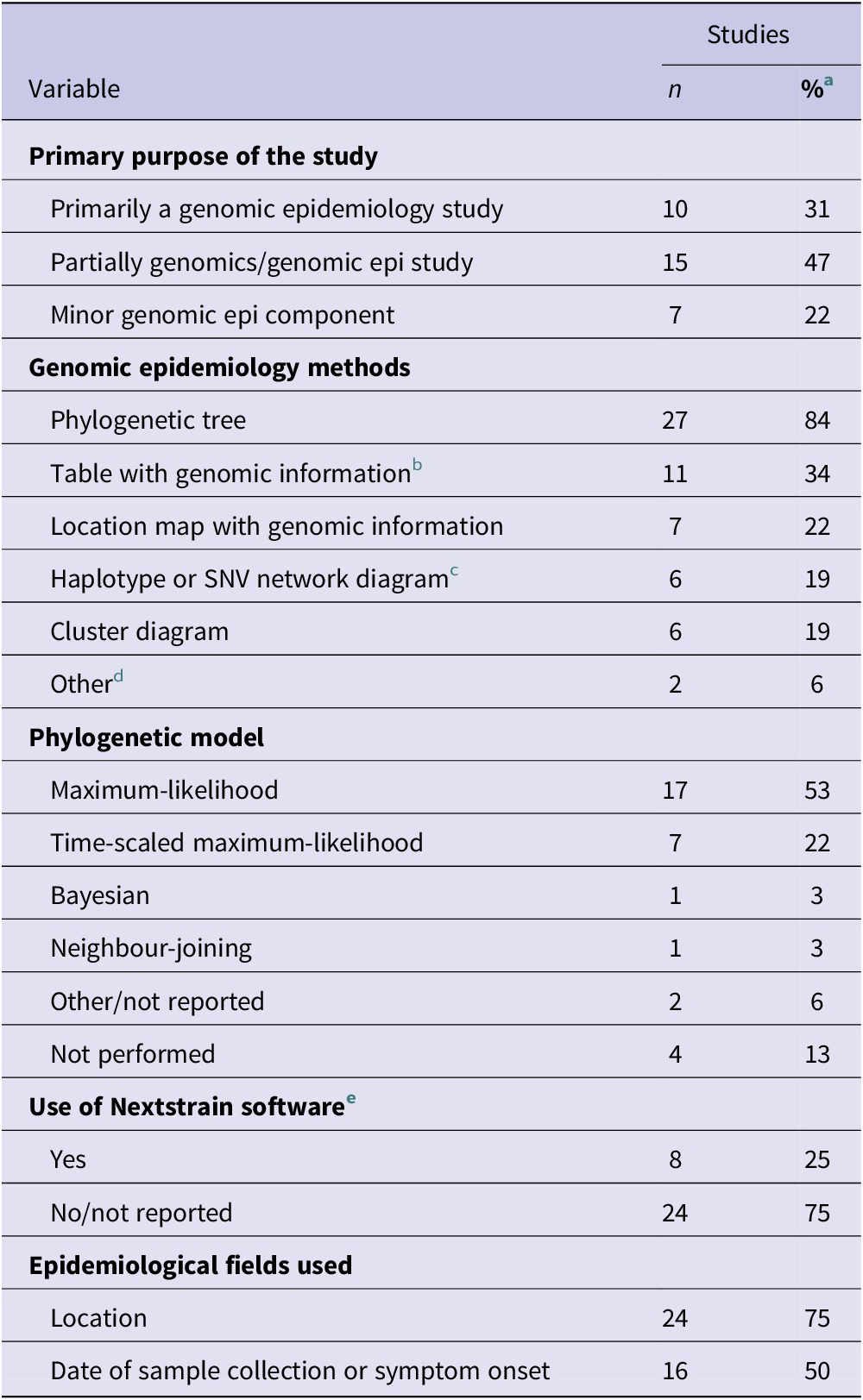
Genomic epidemiology methods
The use of genomic epidemiology methods was grouped into three categories: primary/extensive, moderate, and minor. Only ten studies (31%) included genomic epidemiology as a primary objective of the study. These studies extensively utilized genomics and genomic epidemiology in the methods and analyses and often had genomic epidemiology in the title or abstract. Fifteen studies (47%) used genomic epidemiology to a moderate extent, meaning that while these methods were used, they were not the primary purpose of the study. Finally, seven studies (22%) used genomics as a minor component of a more traditional epidemiological study (e.g. seroconversion study) (Table 3).
Study genomic epidemiology methods (n = 32)

Note: SNV, single-nucleotide variant.
a Denominator is 32 unless indicated otherwise; some categories will sum to over 32 if not mutually exclusive.
b Results included a table(s) with genomic comparison methods such as genetic distances, SNPs, lineage information, SARS-CoV-2 variant information, or genomic cluster assignment.
c Includes haplotype networks, minimum-spanning trees using SNV differences, or median-joining networks using haplotype differences.
d Includes narrative comparisons or other qualitative comparisons.
e Nextstrain indicates study used Nextstrain open-source software resources, including Auspice and Augur, for bioinformatic or phylogenetic analyses.
Nearly all studies used a phylogenetic tree to integrate genomic and epidemiological data (28, 88%) (Table 3). The most common models were maximum-likelihood (17, 53%) and time-scaled maximum-likelihood trees (7, 22%). The most sophisticated method was Bayesian time-scaled tree-building, used in only one paper [Reference Chau43]. Notably, a fifth of studies either did not report the phylogenetic method used (2, 6%) or did not produce a phylogenetic tree (4, 14%). Other genomic epidemiology methods included cluster diagrams (6, 19%), haplotype or single-nucleotide variant (SNV) networks (6, 19%), genotype classification tables (e.g. SNVs, lineages, clusters) (11, 34%), and location maps annotated with genomic information (7, 22%) (Table 3).
Eight studies (25%) reported using the open-source software Nextstrain for genomic epidemiology analyses (https://docs.nextstrain.org/projects/ncov/en/latest/) [Reference Hadfield52]. Twenty-four studies (75%) used location information, while 16 (50%) used sample collection date. Location data were commonly used to colour nodes on phylogenetic trees or cluster diagrams, while collection dates informed time-scaled phylogenetic trees (Table 3).
Synthesis of genomic epidemiology study findings
Thematic analysis showed that, across studies, genomic epidemiology supported multiple, recurring applications in early SARS-CoV-2 outbreak response (Table 4). First, genomic and phylogenetic analyses were used to confirm epidemiologically suspected transmission clusters by showing close genetic relatedness among viral genomes from individuals within a cluster, often differing by only a small number of single-nucleotide polymorphisms (SNPs) [Reference Arons15, Reference Firestone19, Reference Szablewski21, Reference Chau43]. Second, genomic data revealed transmission patterns not identifiable through traditional epidemiologic methods alone, including cryptic linkages and missed transmission events [Reference Lucey24, Reference Voeten30, Reference Weinberger32, Reference Speake39].
Thematic analysis identified benefits of viral genomics in understanding the transmission of SARS-CoV-2

Note; HCW, healthcare worker; SNP, single-nucleotide polymorphism.
*Graphics source: https://www.freepik.com/icons.
Third, several studies used viral genomics to distinguish multiple independent introductions of SARS-CoV-2 from subsequent local transmission, clarifying whether observed case increases reflected ongoing spread within a setting or repeated importations [Reference Lucey24, Reference Olmos25, Reference Taylor34, Reference Sekizuka37]. Fourth, when combined with contact tracing and sampling of asymptomatic individuals, genomics was also used to detect transmission chains involving asymptomatic, presymptomatic, and subclinical carriers, inferred through near-identical viral genomes (≤1–2 SNP differences) among individuals whose epidemiologic timelines suggested exposure prior to symptom onset [Reference Chau43, Reference Zhang44].
Finally, genomic data were used to evaluate the effectiveness of infection prevention and control interventions by assessing whether genetically linked transmission clusters persisted or ceased following implementation of non-pharmaceutical measures such as universal masking, physical distancing, or cohorting [Reference Karmarkar16, Reference Løvestad23, Reference Lucey24, Reference Paltansing26]. In several investigations, the absence of further spread within defined genomic clusters after intervention implementation was interpreted as molecular evidence consistent with intervention effectiveness.
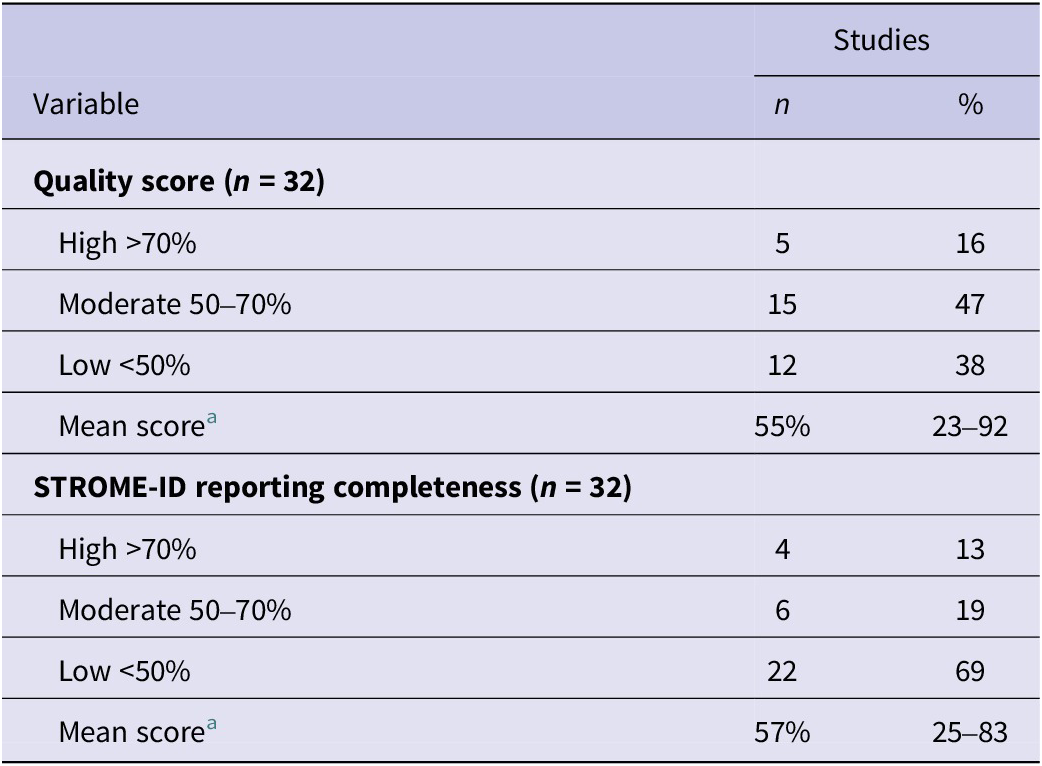
Quality assessment and adherence to reporting guidelines
Across all studies, the average quality score was 55%, and the average completeness of STROME-ID reporting elements was 57% (Table 5). Nearly half of the studies (n = 15) were rated as moderate quality, while only five studies achieved a high-quality score (≥70%). The remaining 12 studies were considered lower quality due to potential bias in study design, analysis, or insufficient methodology details. Only four studies demonstrated high STROME-ID reporting completeness. Six showed moderate (50–70%), and 22 had low completeness scores (<50%).
Bias assessment and adherence to reporting guidelines

Note: STROME-ID, Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases.
a Average quality and STROME-ID completeness scores for all 32 studies. Minimum and maximum scores.
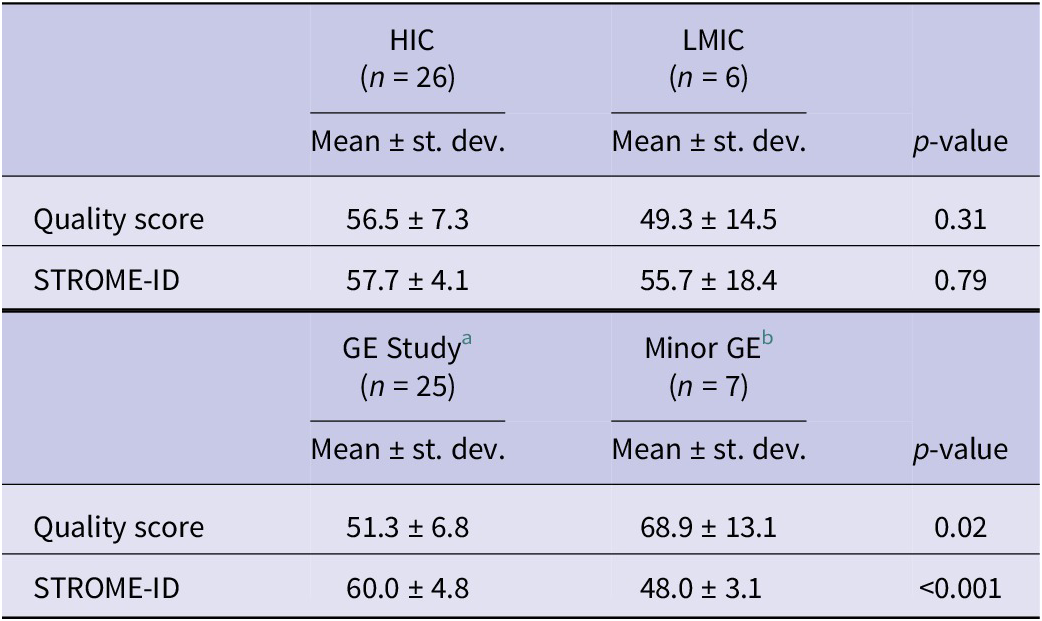
Quality scores were lower for studies from LMICs compared to HICs, though the difference was not statistically significant by two-sample t-test. STROME-ID completeness did not differ between HIC and LMIC studies. Interestingly, studies with limited genomic epidemiology analysis had higher quality scores than those with partial or extensive genomic epidemiology analysis (t-test, p = 0.02). However, these same studies scored significantly lower on STROME-ID completeness compared to studies with more extensive genomic epidemiology analysis (t-test, p < 0.001) (Table 6).
Sub-analysis of mean quality and STROME-ID completeness scores by country income level and extent of genomic epidemiology analysis

Note: HIC, high-income country; LMIC, low- and middle-income country; STROME-ID, Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases.
H0: mean difference = 0; HA: mean difference ≠ 0.
a GE Study means primarily a genomic epidemiology study, or genomics was a major component.
b Minor GE means the study only had a minor genomics or genomic epidemiology component.
Discussion
Features of genomic epidemiology studies published during the early pandemic
The relatively small number of included studies likely reflects the early-pandemic challenges of generating and then integrating genomic and epidemiologic data, as well as the reality that most genomic analyses at the time were designed for broader surveillance purposes rather than outbreak investigation. Reviewing genomic epidemiology studies published early in the COVID-19 pandemic offers insights into the state of infrastructure and local outbreak response capacity at that time. This also limits our focus to early outbreak response practices before widespread availability of vaccines, the emergence of variants of concern, and additional global investments in sequencing [53, 54]. Many studies (44%) involved healthcare-associated outbreaks, reflecting the urgent need early in the pandemic to understand transmission in high-risk settings.
Most local-level genomic epidemiology studies published in the first year of the pandemic were conducted in HICs (91%). In contrast, six studies were considered LMIC-related, and three of those only had exposure events, not the final study analysis, occurring in the LMIC. This imbalance may reflect the limited sequencing infrastructure and bioinformatics expertise in LMICs at the start of the pandemic [Reference Brito55]. Additionally, the diversion of resources to urgent response activities like clinical testing and case ascertainment may have disproportionately impacted LMICs.
Common laboratory methods
The Illumina MiSeq and ONT MinION were the most frequently used sequencing instruments. This observation likely reflects prior global investments in Illumina platforms for pathogen surveillance programmes (e.g. PulseNet [Reference Davedow7]) and the active user community and relatively low price of the MinION [56].
During the early pandemic response, only a small proportion of genomic epidemiology studies (6%) employed shotgun metagenomic sequencing to recover the SARS-CoV-2 genome. In contrast, most (81%) used tiled-amplicon whole-genome sequencing, typically following the open-source ARTIC protocol. This approach is more cost-effective and accessible, particularly for laboratories in resource-limited settings or those newly implementing viral sequencing. However, sequencing protocols were often inadequately described or referenced, limiting the ability to evaluate methodological rigor and the use of quality control measures (see Supplementary Material 5).
Evaluation of genomic epidemiology methods
SARS-CoV-2 genomic data are most useful for public health actions when analysed and visualized in the context of epidemiological data [57]. During the early pandemic, commonly used methods for integrating these data included phylogenetic trees, phylodynamic models, and haplotype and network diagrams. Among the studies reviewed, maximum-likelihood phylogenetic trees were the most frequently employed. This is likely due to the availability of open-source software, like Nextstrain, for exploring SARS-CoV-2 genomic epidemiology data, which utilizes maximum-likelihood methods for rapid tree construction (Nextstrain, https://nextstrain.org/sars-cov-2) [Reference Hadfield52].
However, many studies lacked clarity in their sampling frames, making it difficult to assess the representativeness of sequenced SARS-CoV-2 infections relative to the underlying population (Supplementary Material S3-2). While a few studies followed a well-defined cohort of individuals over time, most relied on contact tracing to identify potential COVID-19 cases. Whole-genome sequencing was typically performed on a fraction of the total PCR-positive samples from a study. Authors rarely reported whether these samples were representative of all SARS-CoV-2 infections in a study or the underlying sample population (See Supplementary Material 5).
Few studies examined potential sources of bias, including confounding or ascertainment bias. Factors influencing which samples were sequenced, the quality of those sequences, and which were ultimately included in analyses were often not reported. For some studies, the number of samples in the phylogenetic tree or other genomic epidemiology analyses did not match the number of successfully sequenced samples. Additionally, the choice of phylogenetic modelling approaches was not consistently described, though a few studies cited tools such as jModelTest.
Several common reasons for excluding samples or sequence data from final genomic epidemiology analyses were noted across studies. These included specimen quality, restrictions on sample sharing, and high CT values indicating low viral load. Often, no specific rationale was provided, or it remained unclear why some samples failed sequencing or were omitted from genomic epidemiology analysis. Each of these issues reduced the representativeness of SARS-CoV-2 cases and the reliability of inferences made from genomic epidemiology analyses.
The extent and quality of accompanying epidemiological metadata also varied. Some studies, typically traditional observational designs, employed comprehensive exposure and demographic questionnaires. Unfortunately, many studies collected minimal metadata or experienced substantial non-response. Even when rich epidemiological metadata were available, only sample collection dates and geographic locations were consistently used in genomic epidemiology analyses.
Study quality and reporting adherence
Overall, studies using traditional observational epidemiology methods, rather than outbreak investigation or genomic analyses, were of higher quality. Most did not provide information on how missing data were handled, whether and how confounding was addressed, and how sources of bias may have impacted the validity of overall findings (Supplementary Material 5A). These quality issues may reflect how common study appraisal tools assess risk of bias using criteria that are more easily addressed in conventional epidemiology study designs.
We also evaluated adherence to established reporting standards for molecular epidemiology studies. Low adherence to STROME-ID guidelines may be due to accelerated publication timelines during the pandemic, restrictive word limits, or lack of familiarity with these guidelines among authors and journal editors. Despite some criteria being outdated or not well-suited for outbreak-focused genomic studies, greater adherence would have improved overall methods and interpretability across publications (Supplementary Material 5B). More detailed reporting on sample selection, genomic representativeness, laboratory quality control procedures, and justification for analytic approaches would have strengthened the robustness and reproducibility of the genomic epidemiology findings included in this systematic review.
Limitations
This review assumes that the included peer-reviewed studies, authors, and institutions are broadly representative of local or outbreak-related genomic epidemiology investigations conducted during the first year of the COVID-19 pandemic. However, several limitations may have impacted generalizability or comprehensiveness:
Publication bias
Potential publication bias may be high for reasons common to all systematic reviews and reasons unique to the COVID-19 pandemic. Null or seemingly less impactful findings from local outbreak investigations may not have been prioritized for publication. Additionally, the significant demands of the pandemic response may have limited the capacity of public health practitioners to prepare manuscripts. Finally, some countries or institutions may have imposed data and sample sharing restrictions that delayed or prevented peer-reviewed publication of outbreak results altogether.
Language restrictions
Although no language restrictions were applied during the abstract screening stage, only full-text articles available in English were included in the final review. Four full-text studies (two Chinese and two Spanish) were excluded, though they failed other inclusion criteria (e.g. case studies or non-local investigations). The use of English search terms could have missed some locally published studies in LMICs, particularly China. However, ad hoc searches of a common Chinese scientific database (CNKI, https://oversea.cnki.net/index/) did not yield additional eligible publications in English or Chinese. To capture additional local-level investigations, future reviews could include peer-reviewed and grey literature and be conducted in Chinese, Spanish, French, or other common scientific publication languages.
Narrow inclusion criteria
The specificity of our inclusion criteria excluded many valuable broader genomic studies published early in the pandemic. This narrow focus, however, allowed for a meaningful comparison across genomic epidemiology studies conducted in similar operational contexts and enabled the synthesis of the discrete ways genomics informed local outbreak responses.
Future directions
To further inform future outbreak preparedness, we recommend qualitative studies with public health professionals to determine how SARS-CoV-2 genomic data were used in decision-making and what barriers or facilitators influenced the uptake of genomic epidemiology.
Conclusion
Our analysis revealed that genomic epidemiology enhanced outbreak investigations during the early pandemic and likely transformed local public health response [Reference Knyazev58]. Whole-genome sequencing enabled high-resolution analysis to confirm or reject suspected transmission links, uncover hidden transmission chains, and distinguish between single and multiple introductions into populations. Genomic epidemiology studies provided critical early evidence of asymptomatic and presymptomatic transmission of SARS-CoV-2, ultimately influencing the selection and implementation of important mitigation strategies [Reference Qiu59].
As the pandemic progressed, genomic tools were rapidly adapted to track emerging variants and investigate outbreaks across a wider range of local settings. These findings highlight the importance of integrating genomics with traditional epidemiological data to strengthen infectious disease surveillance and inform effective local-level public health interventions. Notably, investments in sequencing infrastructure and analytic capacity accelerated the broader application of these methods globally [53, 54].
Our study also revealed several key factors that enabled the early implementation of genomic epidemiology in local SARS-CoV-2 public health response, specifically:
-
1. Open-access genomic databases and standardized genotyping schemes (e.g. Pangolin and Nextclade)
-
2. Community-vetted sequencing protocols (e.g. ARTIC)
-
3. Open-source analysis tools (e.g. Nextstrain)
-
4. Established outbreak investigation protocols and data collection tools
-
5. Strong research collaborations and emergency response funding
This systematic review provides a global synthesis of how SARS-CoV-2 genomic epidemiology was applied to local outbreak investigations during the first year of the pandemic. This body of work demonstrates how genomic methods contributed to understanding transmission dynamics and evaluating control measures. Moving forward, continued integration of genomic data into surveillance systems and genomic epidemiology practices in local public health institutions will strengthen global preparedness to respond rapidly and effectively to future infectious disease threats.
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1017/S0950268826101381.
Data availability statement
All data to support the results are included in supplemental materials, and the extracted data table will be made publicly available on figshare (https://figshare.com/) upon publication.
Acknowledgements
We thank the librarians at the George Washington University Himmelfarb Health Sciences Library for their assistance with selecting databases, refining search terms, and training on Covidence. We thank the authors of these studies for publishing their work in peer-reviewed journals and for their contributions to the COVID-19 pandemic response. NotebookLM (Gemini 2.0) was used to identify and code common themes from manually extracted results and/or conclusions from included studies. ChatGPT 4o was used for copyediting and reducing word count in the final manuscript to meet the journal requirements. The corresponding author reviewed and modified the output and takes full responsibility for the content presented in the manuscript. Thank you to Dr. Kathleen Creppage and Dr. Shayne Gallaway for their thoughtful comments and feedback on the manuscript drafts. The opinions expressed in this article are the author’s own and do not reflect the view of the U.S. Public Health Service, the Department of Defense, the Department of Health and Human Services, or the United States Government
Author contribution
Conceptualization: L.C.M., B.M.F., and A.A.R.; Data Curation and Screening: K.K., Z.T., A.R., L.H., C.S., B.M.F., L.C.M., and A.A.R.; Formal Analysis: L.C.M.; Project Administration: L.C.M.; Supervision: A.A.R. and B.M.F.; Visualization: L.C.M.; Writing – Original Draft: L.C.M.; Writing – Review and Editing: L.C.M., A.A.R., B.M.F., Z.T., L.H., A.R., C.S.
Funding statement
No specific grant or other funding was used for this project. George Washington University Himmelfarb Health Sciences Library provided access to Covidence software and peer-reviewed article databases. Article processing fees and open access were covered through the George Washington University agreement with Cambridge University Press. L.C.M. was partially supported through the George Washington University Graduate Teaching Assistantship Program. L.C.M. was a mentee of C.S. and worked on early versions of the project as part of the Next Generation Global Health Security Network mentorship programme.
Competing interests
The authors declare none.
Ethical standard
The systematic review did not involve human subjects research and did not require approval from an institutional review board.




 Open access
Open access