Refine search
Actions for selected content:
106129 results in Materials Science
Meeting Report
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, p. 175
-
- Article
- Export citation
Powder Diffraction Notes for Authors
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 63-66
-
- Article
- Export citation
Powder Data for LaCo0.4Fe0.6O3
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 2 / June 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 143-145
-
- Article
- Export citation
X-Ray Powder Data for Tetracycline-urea Tetrahydrate, C23H36N4O13 and Tetracycline Hexahydrate, C22H24N2O8·6H2O
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 3 / September 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 168-171
-
- Article
- Export citation
Lattice Thermal Expansion of Lead Zirconate Titanate Piezoelectrics
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 1 / March 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 23-24
-
- Article
- Export citation
Foreword
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 2 / June 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 148-149
-
- Article
- Export citation
PDJ volume 13 issue 3 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 3 / September 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b3
-
- Article
-
- You have access
- Export citation
X-ray data for triammonium citrate
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 3 / September 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 173-174
-
- Article
- Export citation
-
The lattice parameters of triammonium citrate, (NH4)3C6H5O7, were determined by X-ray single crystal and powder diffraction methods. The crystal structure is orthorhombic with a = 6.187 ± 0.001 Å, b = 14.803 ± 0.004 Å, and c = 10.826 ± 0.003 Å, and from the systematic absences, the space group is Pcc2 (
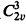 ).
).
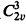 ).
).Author Index to Volume 12
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 4 / December 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 277-280
-
- Article
- Export citation
Structure of the M2CuWO6 System, with M = Ba or Sr
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 228-230
-
- Article
- Export citation
Semiquantitative XRD analysis with the aid of reference intensity ratio estimates
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 3 / September 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 185-187
-
- Article
- Export citation
Variance and centroid optimization in X-ray powder diffraction analysis
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 89-97
-
- Article
- Export citation
Meeting Reports
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 1 / March 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 59-61
-
- Article
- Export citation
Reappraisal of the space group of bafertisite
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 1 / March 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 22-24
-
- Article
- Export citation
Synthesis and cell refinement of Ba0.5+x/2Zr2P3−xSixO12 with x=0 and 0.175
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 2 / June 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 111-114
-
- Article
- Export citation
-
Ba0.5+x/2Zr2P3−xSixO12 or BaZPS compounds were synthesized by the sintering of powders formed by a solid-state reaction. The cell parameters of Ba0.5Zr2P3O12 and Ba0.5875Zr2P2.825Si0.175O12 were determined from X-ray diffraction (XRD) data based on the
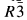 (#148) space group with hexagonal setting. The cell parameters were found to increase with increasing Si content in BaZPS.
(#148) space group with hexagonal setting. The cell parameters were found to increase with increasing Si content in BaZPS.
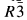 (#148) space group with hexagonal setting. The cell parameters were found to increase with increasing Si content in BaZPS.
(#148) space group with hexagonal setting. The cell parameters were found to increase with increasing Si content in BaZPS.High-resolution X-ray diffraction study of the chromite (Mg0.60Fe0.402+)(Al0.39Cr1.50Fe0.093+)O4
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 2 / June 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 96-99
-
- Article
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 3 / September 1987
- Published online by Cambridge University Press:
- 10 January 2013, p. 207
-
- Article
-
- You have access
- Export citation
Quantitative Analysis of a Textured Coating by a New X-Ray Diffraction Method
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 89-94
-
- Article
- Export citation
PDJ volume 14 issue 4 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 4 / December 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b4
-
- Article
-
- You have access
- Export citation
X-Ray Powder Data for Acetaminophen (C8H9NO2)
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 102-103
-
- Article
- Export citation
