


Free radicals are produced in mammalian cells during a complex array of physiological processes, which, if left uncontrolled, have the potential to cause metabolic damage(Reference Urso and Clarkson1). Free radical activity is moderated by a complex antioxidant network(Reference Sacheck, Milbury and Cannon2), which has the capacity to temper potentially negative cellular effects(Reference Clarkson and Thompson3). The human body depends on the intake and recycling of dietary antioxidant vitamins such as vitamins C, E and β-carotene in order to support the enzymatic systems(Reference Finaud, Lac and Filaire4); however, a systemic increase in free radical production may overwhelm antioxidant defences leading to a state of oxidative stress(Reference Bloomer, Fisher-Wellman and Hammond5) in the form of molecular damage to DNA, cellular lipid membranes and protein molecules(Reference Halliwell and Whiteman6).
It has been established that various modes and intensities of exercise can lead to an exacerbated state of oxidative stress, although the exact mechanism of free radical production remains elusive(Reference Vollaard, Shearman and Cooper7). Despite this, pharmacological interventions have frequently been investigated for a prophylactic effect against oxidative stress induced by exercise(Reference Clarkson and Thompson3, Reference Mastaloudis, Morrow and Hopkins8–Reference Kerksick, Kreider and Willoughby11), and although research has shown a prophylactic effect with exogenous antioxidant consumption, in some cases, large doses of antioxidants are required. As it has been shown that an overconsumption of oral antioxidants may lead to a pro-oxidant state, causing a disturbance in redox biochemistry(Reference Vina, Gomez-Cabrera and Borras12), it is therefore imperative that food sources naturally high in antioxidant vitamins are considered, due to their capacity to provide increased systemic and cellular protection without excessively elevating in vivo antioxidant vitamin concentrations(Reference Lyall, Hurst and Cooney13).
Cruciferous vegetables, in particular cauliflower, cabbage, broccoli and watercress, are known to reduce oxidative DNA damage during in vitro experimentation in human cells(Reference Steinkellner, Rabot and Freywald14). More specifically, Gill et al. (Reference Gill, Haldar and Boyd15) demonstrated that watercress, which contains per gram weight one of the highest concentrations of glucosinolates and carotenoids of any vegetable, protects lymphocyte DNA against damage in smokers, while Boyd et al. (Reference Boyd, McCann and Hashim16) suggested that crude watercress extracts are also known to provide increased protection against DNA damage associated with human colon cancer cells and H2O2 exposure.
Given that exhaustive exercise promotes oxidative stress, causing severe perturbations to cell structure and function, there is a need to ascertain an antioxidant vitamin-rich vegetable that has the potential to counteract exercise-induced oxidative stress. Although recent research has indicated that watercress may play a role in lymphocyte cell protection(Reference Gill, Haldar and Boyd15), the efficacy of short- and long-term watercress ingestion on exercise-induced oxidative stress, to the best of our knowledge, is not yet known.
We therefore hypothesise that acute and chronic watercress consumption will provide effective prophylaxis against exercise-induced oxidative stress. Thus, the primary aim of the present investigation is to examine the efficacy of acute and chronic watercress ingestion against exercise-induced oxidative stress. A randomised controlled experimental design incorporating a comprehensive assessment of oxidative stress indices was used to test this hypothesis.
Methods
Human subjects and experimental design
A total of ten apparently healthy male subjects were recruited to participate in the chronic phase of the experiment first. The study was conducted according to the guidelines laid down in the Declaration of Helsinki and all procedures involving human subjects were approved by the University of Ulster Research Ethics Committee. It is noted that randomised assignment of participants to a supplemented or control group and incorporating a cross-over element is the most robust method of testing food supplements. However, due to the availability of this naturally occurring product and the quantities required to complete the experiment, delivery of product could only be guaranteed during the optimum growing months. For this reason, the participants completed 8 weeks of chronic watercress ingestion before washing out for an additional 8 weeks and then subsequently completing 8 weeks of no ingestion (control). Upon the completion of the chronic phase (lasting in total 24 weeks), eight out of the original ten participants volunteered to take part in the acute phase with an additional two participants recruited. A randomised cross-over design was utilised in the acute phase with participants washing out for a 2-week period between the intervention and the control. Please see Table 1 for participant characteristics. Participants with a family history of any known haematological or cardiovascular-related condition were excluded from the study. Subjects were also excluded if they smoked or were taking any form of antioxidant supplement. All subjects were assigned to complete two phases of experimental testing, of which one was a chronic interventional phase lasting 24 weeks and the other an acute experimental phase, consisting of a randomised cross-over assignment to either watercress supplementation 2 h before exhaustive exercise or the control group. All commercially available watercress was supplied by Vitacress (Vitacress Limited). All participants were instructed to refrain from altering their usual dietary habits throughout the experimental phases of testing. Written informed consent was obtained from all subjects before participation. A schematic overview of each experimental phase is provided in Figs. 1 and 2.
Table 1 Participant characteristics (Mean values and standard deviations, n 10)

bpm, Beats per min.
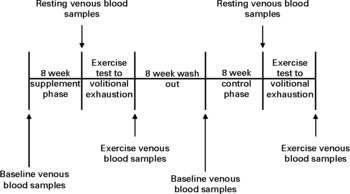
Fig. 1 Experimental time line for the chronic phase.

Fig. 2 Experimental time line for the acute phase.
Chronic interventional phase
Before chronic supplementation and control, a baseline venous blood sample was collected for quantification of lipid-soluble antioxidants. Participants were then required to consume one 85 g portion(Reference Gill, Haldar and Boyd15) of raw watercress on a daily basis for a total of 8 weeks. Watercress was ingested in a single sitting between the hours of 12.00 and 14.00 daily in combination with other foods. Following 8 weeks of supplementation, all participants completed an exercise test to volitional exhaustion while venous blood was drawn at rest and after exercise to determine lipid-soluble antioxidant status and other indices of oxidative stress. Following an 8-week washout period(Reference Gill, Haldar and Boyd15), participants entered an 8-week control phase with no watercress ingestion and then repeated the exercise test to exhaustion.
Acute experimental phase
During the acute experimental phase, participants were randomly assigned using a simple lottery system to either a supplementation or control group. For the supplementation group, participants were asked to consume a single portion of 85 g of raw watercress and 500 ml of water within 30 min between 10.00 and 10.30 hours. Following ingestion, participants rested for 2 h before completing an exercise test to volitional exhaustion. The control group also completed the exercise test to volitional exhaustion, and did not ingest watercress but did consume 500 ml of water. Following a 2-week washout period(Reference Samman, Sivarajah and Man17), all participants crossed over to consume either watercress or nothing and were retested as above.
Exhaustive acute exercise protocol
To avoid the potential effects of diurnal variation, each participant was required to consistently attend the laboratory testing sessions at the same time of day (09.00 hours) for all phases. On each arrival of participants at the laboratory, body mass and stature were measured using standard methods (see Table 1). All participants were required to complete a treadmill test to volitional exhaustion in which the protocol was specifically designed to be progressive and incremental in order to elicit VO2max. Treadmill speed was set at 11 km/h with a 1 % gradient rise at each 1 min interval until volitional fatigue. Validation of VO2max was confirmed when (1) the RER was above 1·15 arbitrary units, (2) a plateau in the oxygen uptake/exercise intensity relationship (>2 ml/kg per min) and (3) a heart rate of within 10 beats per min of age-predicted maximum (220 − age). Oxygen uptake was measured using standard laboratory gas analysis (Cosmed Quark b2), while heart rate was measured via a portable short-angle telemetry device (Polar Sports Tester). All subjects achieved maximum oxygen uptake.
Supplementation compliance
Subjects were asked to complete a weekly journal detailing total amounts of watercress ingested over the chronic interventional phase.
Haematology
Following a standardised 12 h overnight fast, whole blood was drawn from a prominent forearm ante-cubital vein using the vacutainer method (Becton-Dickinson). Blood was collected at baseline (pre-supplementation), at rest (post-supplementation/pre-exercise) and exercise (immediately post-exhaustive exercise) for the quantification of a range of lipid-soluble antioxidants, and at rest and exercise for various indices of oxidative stress. Blood was drawn into either di-potassium EDTA or serum separation tubes. EDTA and serum separation tubes (once clotted) were centrifuged (MSE Centuar 2; Labcare) at 3000 rpm for 10 min at − 4°C. Aliquots of plasma and serum were stored at − 80°C for a maximum of 3 months. All samples from the same participant were analysed within the same batch. Hb (g/dl) was measured using a β-Hb photometer (Hemocure Limited), and packed cell volume (%) was measured using the standard microcapillary reader technique to aid in the correction of all biochemical markers for acute exercise-induced plasma volume shifts using the equations of Dill & Costill(Reference Dill and Costill18).
Biochemical analysis
DNA
Lymphocytes were isolated from the whole blood (EDTA) by layering 3 ml onto Histopaque-1077 (3 ml) and centrifuged (MSE Centuar 2; Labcare) at 3500 rpm (relative centrifugal force 1356 g) for 30 min at room temperature. The opaque mononuclear layer was aspirated and washed three times using 15 ml PBS (one tablet added to 200 ml of double-distilled water, 0·2 m, pH 7·2). The comet assay was subsequently carried out on prepared cells using the protocol of Singh et al. (Reference Singh, McCoy and Tice19). Dakin fully frosted slides (3 inch × 1 inch; 1·2 mm thick; Richards Supply Company Limited) were covered with 100 μl of 0·5 % normal-melting point agarose and allowed to solidify under a coverslip (22 × 40 mm, no. 1 thickness; GBH, Laboratory Supplies). Then, 50 μl of cells (1 × 104) were mixed with 50 μl of low-melting point agarose layered on top of the normal-melting point agarose and allowed to solidify under a coverslip. The coverslip was then removed and the slides were placed in lysis buffer (2·5 m-NaCl, 100 mm-Na-EDTA, 10 mm-Tris, 1 % Triton X and 10 % dimethyl sulfoxide, pH 10) for 1 h at 4°C to break down the cell membranes. The slides were removed and placed in a horizontal electrophoresis unit containing electrophoresis buffer (300 mm-NaOH, 1 mm-EDTA, pH 12·5) for 20 min to allow the DNA double helix to relax and unwind. Electrophoresis (25 V, 300 mA, 0·15 V/cm) was performed for 20 min at room temperature. Following electrophoresis, the slides were rinsed with neutralising solution (0·4 m-Tris, pH 7), stained with 50 μl ethidium bromide and a coverslip was applied. A random sample of fifty cells from each slide was analysed using a Hewlett-Packard VGA monitor and Fenestra Comet software program (version 2.22) at magnification of 400 × using an epiflourescent microscope (Olympus BH2). Tail length was selected to report DNA damage, as it acts as a sensitive measure of low levels of DNA damage(Reference Collins, Oscoz and Brunborg20). This assay was performed on each experimental day using fresh lymphocytes. The intra/inter-assay CV were < 9 and < 11 %, respectively.
Lipid hydroperoxides
Serum lipid hydroperoxides (LOOH) were measured spectrophotometrically using the method of Wolff(Reference Wolff21). This ferrous iron/xylenol orange assay quantifies the susceptibility to Fe-induced LOOH formation in the blood. The presence of Fe ions in the assay protocol might therefore yield slightly higher LOOH values compared with other methods. Briefly, 90 μl serum were incubated with 10 μl catalase for 30 min at room temperature. To this solution, 900 μl ferrous iron/xylenol orange reagent 1 (250 μm-ammonium ferrous sulphate, 100 μm-xylenol orange, 100 μm-sorbitol and 25 μm-H2SO4) were added and incubated for a further 30 min at room temperature in the dark. Standard solutions were prepared from H2O2 in the range of 0–5·0 μmol/l and also incubated for 30 min with the ferrous iron/xylenol orange 1 reagent, after which the samples were centrifuged in a Beckman microfuge for 5 min to remove any flocculated material. The absorbance of the supernatant was read spectrophotometrically (U-2001; Hitachi) at 560 nm against the standard curve that was linear in the range of 0–5 μmol/l. The intra/inter-assay CV were < 2 and < 4 %, respectively.
Protein carbonyls
Protein carbonyls (PC) are a reliable measure of protein oxidation and were quantified with an assay kit purchased from Caymen Chemical. After thawing, 20 μl of plasma from each sample were pipetted into 1 ml plastic eppendorfs. The volume was brought up to 200 μl with distilled water and the protein precipitated with 200 μl of 20 % TCA and centrifuged for 1 min at 1300 g at room temperature to remove the supernatant. Then, 500 μl 2,4-dinitrophenyl hydrazine (in 2 m-HCl) were added to each sample, which was subsequently vortexed to resuspend the sample in the solution. The reaction was allowed to proceed for 15 min with occasional vortexing. At the end of the reaction, each sample was re-precipitated with 500 μl TCA vortexed and centrifuged for 1 min at 1300 g. The supernatant was then discarded and the sample was drained by touching the lip of the inverted tube to a clean Kim-wipe. Each sample was washed three times with 1 ml of 1:1 ethanol–ethyl acetate, vortexing each time and centrifuging for 5 min at 16 000 g between washes. After the final wash, the samples were left to briefly air dry. Then, 1 ml of 6 m-guanidine, 500 mm-KPO4 (pH 2·5), was used to redissolve/resuspend each sample with vortexing. The samples were then added to a ninety-six-well plate and read at 370 nm for carbonyl and 276 nm for total protein estimation. The intra/inter-assay CV were < 5 and < 9 %, respectively.
Hydrogen peroxide
H2O2 was measured spectrophotometrically using a commercially available kit (Sigma Chemical Company). After thawing, the serum sample was diluted 1:64 by adding 5 μl of serum to 315 μl of the sample diluent. Then, 50 μl of this diluted sample were added to a ninety-six-well plate in triplicate along with an appropriate standard curve (part no. H2163) and blanks in addition to 100 μl of H2O2 colour reagent (part no. H2288). After 30 min of incubation at room temperature, the plate was read on a Dynatech MRX 650 plate reader with Revelation software (American Instrument Exchange) using 550 nm as the primary wavelength. The intra/inter-assay CV were < 3 and < 6 %, respectively.
Lipid-soluble antioxidants
Plasma retinol, α-tocopherol, γ-tocopherol, β-carotene, lycopene and xanthophyll were analysed by simultaneous determination using the HPLC method of Thurnham et al. (Reference Thurnham, Smith and Flora22). Tocopherol acetate was used as the internal standard during this procedure. Briefly, 200 μl of the internal standard were added to each extraction while recovery was measured by injecting 50 μl tocopherol acetate directly on HPLC with each batch of samples. The internal standard was used to monitor sample recovery and the results were adjusted accordingly. Then, 1000 mg of tocopherol acetate were added to 100 ml of heptane and magnetically stirred. The stock solution (1 ml) was then diluted in 249 ml ethanol (95 %) and stored at 4°C until use. Thereafter, 100 μl of 10 mm-SDS were added to 1 ml of the plasma sample and mixed. Then, 200 μl of the internal standard were added to each sample along with two blanks (for recovery). These were then vortexed for 1 min and 1 ml heptane (containing 50 mg butylated hydroxy-toluene) was added to each sample and vortexed for 4 min. The samples were centrifuged for 10 min at 1000 g at 10°C. Thereafter, 700 μl of the supernatant were discarded and the samples were left to dry under N2 in a fume cupboard. Each sample was reconstituted with 100 μl of the mobile phase (750 ml acetonitrile, 200 ml methanol, 50 ml dichloromethane, 500 mg buthylated hydroxytolulene and sonicated for 30 min to de-gas) and vortexed. The samples were dispensed into HPLC vials and placed on sampling carousel for analysis. For the analysis, the samples were combined with the mobile phase at a flow rate of 1·5 ml/min at 21°C. The HPLC system comprised a Waters 510, Waters 2996 photodiode array detector, connected to a Waters 717 autosampler and a Waters Sunfire™ C18, 3·5 μl, 4·6 × 100 mm column. The temperature of the column was maintained using a Jones chromatography 7900 series HPLC column oven. Samples were analysed using a range of wavelengths (325–450 nm) to detect retinol, tocopherol and carotenoid peaks. The concentration of retinol, α-tocopherol, γ-tocopherol, β-carotene, lycopene and xanthophyll in each sample was corrected in relation to the percentage of the internal standard recovered for each sample. The intra/inter-assay CV were < 6 and < 10 %, respectively.
Statistical analysis
Statistical analysis was performed using the SPSS statistics package (version 15.0; SPSS). A prospective calculation of power was performed using the equations of Altman(Reference Altman23) and based on experimental DNA data by Gill et al. (Reference Gill, Haldar and Boyd15). Data were analysed using parametric statistics following mathematical confirmation of a normal distribution using Shapiro–Wilk W tests. Baseline, pre- and post-intervention data were analysed using a two-way (A × B) mixed ANOVA which incorporated one between-subject (group: watercress v. control) and one within-subject factor (state: rest v. exercise). When a significant interaction effect was detected, within-participant factors were analysed using Bonferroni-corrected paired-sample t tests. Between-participant differences were analysed using a one-way ANOVA with an a posteriori Tukey's honestly significant difference test. The α level was established at P< 0·05 and all values are reported as means and standard deviations, unless otherwise stated.
Results
Compliance
A total of ten participants (100 %) completed the chronic phase of the study, and compliance to supplementation throughout was 98 % (548 consumed doses from 560 possible doses). The ten participants also completed the acute phase (100 %) and compliance with the supplementation protocol was 100 % (ten consumed from ten possible doses).
Indices of oxidative stress
DNA damage increased following exercise in both control conditions (chronic-phase increase of 61 % from rest, P< 0·05 v. supplementation; acute-phase increase of 63 % from rest, P< 0·05 v. supplementation; Table 2); however, watercress consumption attenuated DNA integrity following exercise in both experimental phases (chronic phase, P< 0·05 v. control; acute phase, P< 0·05 v. control; Table 2). Similarly, there was a rise in LOOH from rest to exercise in both control conditions (chronic-phase increase of 15 % from rest, P< 0·05 v. supplementation; acute-phase increase of 16 % from rest, P< 0·05 v. supplementation; Table 2), which was conversely decreased as a function of watercress supplementation (chronic phase, P< 0·05 v. control; acute phase, P< 0·05 v. control; Table 2). PC concentration remained unchanged following exhaustive exercise in both chronic and acute supplementation phases (P>0·05; Table 2).
Table 2 Indices of oxidative stress at rest and exercise for the watercress-supplemented and control groups for both experimental phases (Mean values and standard deviations)

Δ%, Change from rest.
* Between-group differences at exercise (P< 0·05).
† Within-group differences between rest and exercise (P< 0·05).
Reactive oxygen species
There was a comparatively higher production of H2O2 as a function of exhaustive exercise in both control conditions (chronic phase, P< 0·05 v. supplementation; acute phase, P< 0·05 v. supplementation; Fig. 3), while watercress ingestion decreased H2O2 concentration in both experimental groups (chronic phase, P< 0·05 v. control; acute phase, P< 0·05 v. control; Fig. 3).

Fig. 3 Hydrogen peroxide concentrations at rest and following exhaustive exercise within both supplemented (n 10) and non-supplemented experimental phases (n 10). * Within-group difference (P< 0·05 v. rest); † between-group difference (P< 0·05 v. rest).
Lipid-soluble antioxidants
Table 3 shows the effect of watercress and exercise on plasma antioxidant concentration. There was an increase in α-tocopherol concentration following chronic watercress ingestion (P< 0·05 v. control), while α-tocopherol, γ-tocopherol and xanthophyll all increased in the acute phase (P< 0·05 v. control). Following exhaustive exercise, α-tocopherol increased in both acute watercress and control states (P< 0·05 v. rest); however, exercise decreased α-tocopherol in the chronic watercress group (P< 0·05 v. control). Exercise also lowered γ-tocopherol concentration across the watercress and control groups for both phases (P< 0·05 v. rest) and β-carotene decreased in the acute and chronic watercress groups (P< 0·05 v. control), with no change observed in the control state (P>0·05).
Table 3 Plasma lipid-soluble antioxidants at baseline, rest and exercise for the watercress-supplemented and control groups for both experimental phases (Mean values and standard deviations)

* Between-group differences (P< 0·05).
† Within-group difference (P< 0·05).
‡ Change from baseline to rest.
§ Change from rest to exercise.
Discussion
Strong experimental evidence indicates that exhaustive exercise may activate the production of free radical species and cause damage to important biological molecules such as DNA, lipid and protein(Reference Fogarty, Hughes and Burke24). Watercress is a green leafy vegetable that possesses one of the highest concentrations of antioxidant vitamins, and is known to protect cells against oxidative stress, in particular DNA damage(Reference Gill, Haldar and Boyd15). The primary purpose of the present study was to ascertain the effects of acute and chronic watercress ingestion against exercise-induced oxidative stress. The study demonstrates that exhaustive aerobic exercise may cause DNA damage and lipid peroxidation; however, these perturbations are attenuated by either short- or long-term watercress supplementation, possibility due to the higher concentration of lipid-soluble antioxidants following watercress ingestion.
Exercise-induced oxidative stress
Previous work from our laboratory and, indeed, others demonstrates that intense exhaustive exercise can increase damage to circulating lymphocyte DNA and lipid membranes(Reference Davison, Hughes and Bell10, Reference Fogarty, Hughes and Burke24). The comet assay is a sensitive, valid and versatile tool for the measurement of single- and double-strand breaks in DNA(Reference Collins, Oscoz and Brunborg20) and has been previously used to detect DNA damage following exhaustive exercise(Reference Mastaloudis, Morrow and Hopkins8, Reference Davison, Hughes and Bell10, Reference Hartmann, Plappert and Raddatz25–Reference Radak, Apor and Pucsok27). Moreover, many of these reports attribute an increase in free radical production as the main cause of exercise-induced DNA damage; however, the exact mechanism(s) for the production of primary free radical species during exercise remains elusive, although free radicals generated within the skeletal muscle are often proposed as the main protagonist(Reference Gomez-Cabrera, Vina and Ji28). Considering the vast majority of human investigations that have quantified oxidative stress in plasma, serum or leucocytes, it is imperative to attempt to identify the primary source of free radical generation during exercise(Reference Nikolaidis and Jamurtas29). Evidence from the present investigation portrays a potential primary mechanism of cellular exercise-induced DNA damage which may be mediated by H2O2. The up-regulation of leucocytes and neutrophil enzyme activation is known to cause an increased release of superoxide(Reference Pedersen, Rohde and Zacho30, Reference Niess, Baumann and Roecker31), and superoxide has the capacity to directly damage DNA(Reference Guetens, De Boeck and Highley32) and/or be dismutated to H2O2 by superoxide dismutase(Reference Powers and Jackson33). H2O2 can readily diffuse across cell membranes(Reference Nikolaidis and Jamurtas29), and depending on the availability of transition metal ions, hydroxyl radicals may be generated which can cause single- and double-strand breaks in DNA and base-pair modification(Reference Radak, Goto and Radak34). The change in H2O2 as a function of exercise in both control groups provides a tentative explanation for DNA damage observed. An elevated H2O2 concentration may also be a direct mediator of DNA damage through the disruption of DNA repair pathways(Reference Boyd, McCann and Hashim16). Furthermore, H2O2 can be generated from a number of extracellular pathways during exercise and may also be independent of superoxide formation. The auto-oxidation of neurological chemicals such as catecholamine has been shown to increase following exercise resulting in H2O2 formation(Reference Bracken, Linnane and Brooks35). In addition, the oxidation of dopamine by monoamine oxidase releases H2O2 as a metabolic by-product(Reference Halliwell and Gutteridge36).
Superoxide and hydroxyl radical production may also initiate vascular cell membrane lipid peroxidation, which may generate LOOH and facilitate further DNA damage. PUFA decomposition can produce an array of metabolic by-products including mutagenic compounds and lipid peroxidation intermediates(Reference Davison, Morgan and Hiscock37), including alkoxyl free radicals(Reference Davison, Ashton and Davies38) and malondialdehyde(Reference Marnett39). Both molecules are potentially capable of directly damaging DNA, while alkoxyl free radicals, in particular, may perpetuate the process of lipid peroxidation(Reference Davison, Morgan and Hiscock37). Unsaturated alkenals are also by-products of lipid peroxidation, which can lead to the production of PC(Reference Marnett39). PC are normally produced following the oxidation of amino acids and have previously been used as a sensitive measure of protein damage during high-intensity exercise(Reference Lushchak40). Exhaustive exercise has been found to have no effect on protein oxidation throughout any condition, which perhaps suggests that protein oxidation has a greater affinity with exercise duration rather than intensity(Reference Alessio, Hagerman and Fulkerson41, Reference Bloomer, Goldfarb and Wideman42).
Prophylactic effect of watercress
Watercress is a cruciferous vegetable of the Brassica variety and contains, per gram weight, one of the highest concentrations of glucosinolates and carotenoids of any vegetable(Reference O'Neill, Carroll and Corridan43). Glucosinolates can be hydrolysed by the enzyme myrosinase (released from plant cells during the chewing process) to form isothiocyanates which have been shown to possess anticarcinogenic properties and reduce DNA damage properties in both animal models and humans(Reference Conaway, Yang and Chung44). Cellular exposure to isothiocyanates and antioxidants may represent a possible mechanism for the observed protection of cell DNA following both chronic and acute supplementation phases. LOOH also reduced following exercise in both supplemented conditions, providing further evidence of the additional cellular protection provided by the constitutes of watercress. In the present investigation, serum H2O2 decreased following exhaustive exercise under both supplemented conditions but increased in the control groups. These data would lend support to the suggestion that watercress may provide effective in vivo protection against H2O2 production as a function of exercise. It is also plausible that the elevated lipid-soluble antioxidants (under both supplemented protocols) are directly scavenging superoxide and therefore result in a net decrease in H2O2 production(Reference Sies and Stahl45). The observed increase in lipid-soluble antioxidants, as that demonstrated following exercise, may also play a key role in the protection against cell membrane lipid peroxidation.
Watercress has been shown to increase important lipid-soluble antioxidants and, in particular, the tocopherols and carotenoids. Gill et al. (Reference Gill, Haldar and Boyd15) identified a 33 % increase in β-carotene, a 26 % increase in α-tocopherol and a 35 % increase in ascorbic acid, thus highlighting watercress's potential to elevate lipid- and aqueous-soluble systemic antioxidants. In contrast, we observed no change in β-carotene or retinol under either experimental condition. However, the lipid-soluble antioxidants α-tocopherol, γ-tocopherol and xanthophyll were all elevated following supplementation with watercress, and, in doing so, indicates the potential for watercress to act as a source of blood-rich antioxidants. Xanthophyll is an effective scavenger of singlet oxygen(Reference Packer, Witt and Tritschler46) and has shown antioxidant activity that is particularly effective in protecting DNA(Reference Haegele, Gillette and O'Neill47). The increased concentration of xanthophyll following the acute dose of watercress may therefore have played a contributory role in the increased protection of lymphocyte DNA in this supplemented group. Moreover, an increase in lipid-soluble antioxidants occurred as a function of exercise and is perhaps as a result of membrane-bound antioxidants being released into the vascular medium during the lipolysis of adipose tissue(Reference Long, Wells and Englert48). The notion that exhaustive exercise may increase systemic levels of lipid-soluble antioxidants comes from a plethora of evidence suggesting that an increased mobilisation of fatty acids may give rise to α-tocopherol(Reference Fogarty, Hughes and Burke24, Reference Pincemail, Deby and Camus49, Reference Davison, George and Jackson50) in addition to other fat-soluble antioxidants such as retinal and lycopene(Reference McClean, Clegg and Shafat51).
PC are generated following the oxidation of amino acids and have previously been used as a sensitive measure of protein damage during high-intensity exercise(Reference Lushchak40). However, we observed no change in PC following exhaustive exercise with or without watercress ingestion, and this is in contrast to the observed DNA damage and lipid peroxidation. As previously reported, protein oxidation may have a greater affinity with exercise duration rather than intensity(Reference Alessio, Hagerman and Fulkerson41, Reference Bloomer, Goldfarb and Wideman42), and it is also conceivable that muscle damage may be a potential mediating factor in protein oxidation following exercise.
Conclusion
The present study suggests that dietary watercress intervention provides effective protection against exercise-induced oxidative stress, with no additional benefits gained from long-term watercress consumption. The prophylactic effect of watercress may be due in part to the high concentration of antioxidants or other cytoprotective compounds.
Acknowledgements
The authors would like to express sincere thanks to Dr Steve Rothwell, Vitacress Limited, and to Dr Edel Keaveney for her technical help in analysing the lipid-soluble antioxidants. This study received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. There are no conflicts of interest associated with this paper. The watercress used in this study was supplied by Vitacress (Vitacress Limited, Southampton, UK). M. C. F. participated in the experimental design, undertook subject recruitment and data collection, conducted all biochemical analyses and prepared the manuscript. C. M. H. and G. W. D. conceived the study, participated in its design, performed the statistical analysis and prepared the manuscript. G. B. and J. C. B. performed the standard biochemistry analyses. All authors read and approved the final manuscript.






