


A plethora of data from epidemiological and experimental studies indicates that consumption of long-chain (LC) n-3 PUFA favourably modulates multiple biological processes involved in CVD, cancers, immune diseases, diabetes, brain health and hepatic steatosis(Reference Kim, McMurray and Chapkin1). Among the mechanisms proposed to explain the effects of LC n-3 PUFA, the most often reported include the metabolic interconversion into bioactive eicosanoids and/or docosanoids or the alteration of membrane self-organising lipid raft domains. More recently, LC n-3 PUFA have been identified as powerful gene expression modulators, notably in the liver where they are associated with the modulation of several genes related to lipid and lipoprotein metabolism, explaining their hypolipidaemic effects(Reference Harris, Miller and Tighe2, Reference Davidson3).
The effects of LC n-3 PUFA on gene transcription were thought primarily to be mediated by a single subfamily of orphan nuclear receptors: the PPAR. However, it is now becoming evident that LC n-3 PUFA can affect the expression of several different genes either via direct interactions or indirectly through additional transcription factors including hepatic nuclear factor 4α, retinoid X receptor α, sterol regulatory element-binding protein-1c, liver X receptors α and β, farnesol X receptor (FXR) and NF-κB(Reference Jump4). This made the uncovering of biological pathways underlying the effects of LC n-3 PUFA much more complex than initially thought.
Metabolism of LC n-3 PUFA can influence the nature of the bioactive compounds responsible for the modulation of gene expression. Indeed, LC n-3 PUFA are not only enzymatically metabolised into eicosanoids but they are also very prone to non-enzymatic oxidation, leading to the formation of a variety of peroxidised metabolites(Reference Cosgrove, Church and Pryor5). This reaction, known as lipid peroxidation, is particularly intense in oxidative stress conditions, making peroxidated metabolites potential key modulators of gene expression. Hydroxyalkenals are a family of aldehydes produced in abundance during peroxidation of PUFA. They comprise 4-hydroxynonenal (4-HNE) issued from n-6 PUFA and 4-hydroxyhexenal (4-HHE) generated from n-3 PUFA and, more specifically, DHA (20 : 6n-3)(Reference Vankuijk, Holte and Dratz6) which is the abundant n-3 PUFA in most tissues. Hydroxyalkenals, notably 4-HNE which has been identified and studied much more extensively and earlier than 4-HHE, are not only highly reactive molecules that covalently bind proteins, lipids and DNA but they are also known as signal molecules in cells(Reference Leonarduzzi, Arkan and Basaga7). This makes them interesting targets to focus on when investigating the effects of LC n-3 PUFA and their metabolites on the modulation of gene expression.
In this context, we hypothesised that increasing intake of LC n-3 PUFA could, in a dose-dependent manner, enhance the production of 4-HHE and differentially influence the expression of genes in the liver as a central organ of lipid metabolism and oxidative stress. In order to investigate these relationships, we conducted a dose–response supplementation trial in hypercholesterolaemic rabbits. The choice of this model was dictated by the need to induce a pathological condition (i.e. hypercholesterolaemia and steatosis) known to induce metabolic disturbance and inflammation(Reference Yanni8), which can be modulated by LC n-3 PUFA. Moreover, this animal model presents several advantages, among which the rapidity of the induction of hypercholesterolaemia, a lipid metabolism close to what is observed in humans, and the absence of genetic modifications making easier the understanding of gene expression modulations. The choice of an overall gene expression analysis was dictated by the need to study a complex biological system influenced by many factors including the metabolism of LC n-3 PUFA. This also gives the opportunity to generate new insights about the nutrigenomic effects of LC n-3 PUFA in the context of hypercholesterolaemia.
Materials and methods
Animals and diets
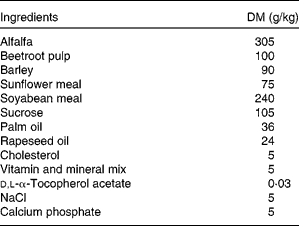
New Zealand White male rabbits (Charles River Laboratories, L'Arbresle, France) of 12 weeks of age (mean weight = 1874 (sem 138) g) were used in the present study. They were individually housed and maintained under conventional conditions with a temperature of 22°C and a 12 h light–12 h dark cycle (07.00–19.00 hours). After 3 weeks of adaptation, rabbits were allocated to four experimental groups (i.e. control; LC n-3 PUFA-dose 1 or group 1; LC n-3 PUFA-dose 2 or group 2; LC n-3 PUFA-dose 3 or group 3) according to their weights and plasma cholesterol concentrations in order to obtain homogeneous groups on these two criteria. Then, animals were fed daily for 7 weeks with 100 g of a cholesterol-enriched diet (Table 1) providing 0·5 g cholesterol/d per rabbit. At the same time 5 d/week, rabbits received 2 ml of either oleic acid-rich sunflower oil (Lesieur, Asnières-sur-Seine, France; control, n 6) or a mix of oleic acid-rich sunflower oil and purified tuna oil (Tuna oil 25DHA, Qualitysilver; Polaris, Pleuven, France) providing 0·1 % (LC n-3 PUFA-dose 1 or group 1, n 8), 0·5 % (LC n-3 PUFA-dose 2 or group 2, n 8) or 1 % (LC n-3 PUFA-dose 3 or group 3, n 7) of energy as DHA (22 : 6n-3). The first dose (dose 1) has been chosen to mimic the nutritional recommendation for DHA in the human diet(Reference Kris-Etherton, Grieger and Etherton9), whereas doses 2 and 3 are supra-nutritional, comparable with what is often used in intervention studies. Oil mixtures were prepared in advance in glass bottles sealed under N2 and stored at 4°C in the dark until use to prevent fatty acid peroxidation. Animals were weighed once per week and food intake was recorded five times per week. There were no statistical differences between the groups for these two parameters (data not shown). All animals were maintained and handled according to the recommendations of the Institutional Ethics Committee of the INRA, in accordance with decree no. 87-848.
Table 1 Composition of the cholesterol-enriched diet given to New Zealand White rabbits during the 7-week experiment
Sample collection
After 7 weeks of the diet, overnight fasted rabbits were anaesthetised with pentobarbital (30 mg/kg, intravenously). Blood was collected from the marginal ear vein in tubes containing EDTA (3 mm, pH 8·2) and butylated hydroxytoluene (0·44 mg/ml fresh blood). Plasma was recovered by centrifugation (2700 g, 10 min, 4°C) and rapidly distributed into 1·5 ml microtubes, flushed with N2 and frozen in liquid N2. Liver was rapidly removed, rinsed with 0·9 % NaCl and a sample (approximately 5 g) was collected from the left lobe, frozen in liquid N2 and stored at − 80°C until further analysis.
Fatty acid composition of liver phospholipids
Total lipids were extracted from liver samples, as described by Folch et al. (Reference Folch, Lees and Sloane Stanley10), and subsequently separated into neutral and polar lipids by solid-phase extraction (C18 SPE cartridge; Macherey-Nagel, Hoerdt, France). The solid phase extraction cartridges were washed with chloroform (4 ml) to elute neutral lipids followed by 8 ml methanol to elute polar lipids(Reference Juaneda and Rocquelin11). Phospholipids were evaporated to dryness under a gentle stream of N2 and dissolved in methanol and toluene (4:1, v/v) for methylation. Phospholipid fatty acid methyl esters (FAME) were obtained after trans-esterification with sodium methoxide in methanol (Sigma-Aldrich, St Louis, MO, USA) followed by acid trans-esterification with boron trifluoride in methanol (14 %; Sigma-Aldrich)(Reference Ledoux, Chardigny and Darbois12).
GC analysis of FAME was performed using a gas chromatograph GC Trace (Thermo Fischer Scientific, Courtaboeuf, France), equipped with a fused silica CP-Sil 88 capillary column (100 % cyanopropyl-polysiloxane, 100 m, 0·25 mm in inner diameter, 0·20 μm in film thickness; Varian S.A, Les Ulis, France), a programmed temperature vaporisation injector (250°C) and a flame-ionisation detector. The sample (1 μl) was injected in the splitless mode. The oven temperature programme ran between 70 and 225°C in four separate steps. He gas was used as a carrier, with a constant pressure (264 kPa). The identities of sample methyl esters were determined by comparing their relative retention times with those of external well-known FAME standards (Supelco™ 37 Component Fatty Acid Methyl Esters Mix and Menhaden Oil; Sigma Aldrich, St Quentin Fallavier, France). Other standard FAME mixtures were obtained from Nu-Chek-Prep (Elysian, MN, USA).
Plasma lipid profiles
Plasma samples were analysed for their contents in total cholesterol, HDL-cholesterol, TAG and apoB using a Konelab 20 analyser (Thermo Electron SA, Cergy-Pontoise, France). LDL-cholesterol concentration was calculated by the Friedewald equation as follows:
TAG and cholesterol concentration in liver
Liver samples (200 mg) were homogenised in a saline solution (500 μl) with a Polytron homogeniser PT-MR2100 (Kinermatica AG, Littau/Luzern, Switzerland) and lipids were extracted by chloroform–methanol (2:1, v/v) with overnight agitation. The chloroform phase was recovered after centrifugation and evaporated under dry air. Lipids (including TAG) were then saponified with 0·5 mm-KOH–ethanol at 70°C for 30 min followed by the addition of 0·15 mm-MgSO4 to neutralise the mixture. After centrifugation (2000 g, 5 min), glycerol from TAG in the supernatant was estimated by an enzymatic assay (TG PAP 150 kits; Bio-Merieux, Marcy-l'Etoile, France). Cholesterol in the lipid residue was dissolved in isopropanol and measured enzymatically (Cholesterol RTUTM; Bio-Merieux).
Glutathione system
Liver contents of reduced glutathione and glutathione disulphide (oxidised glutathione) were measured according to the method of Griffith(Reference Griffith13). The enzymatic activities of glutathione peroxidase, glutathione reductase and glutathione-S- transferase were assessed as already described by Flohe & Gunzler(Reference Flohe and Gunzler14), Carlberg & Mannervik(Reference Carlberg and Mannervik15) and Habig et al. (Reference Habig, Pabst and Jakoby16), respectively.
Lipoperoxidation biomarkers
Concentrations of thiobarbituric acid-reactive substances (TBARS), as indices of global lipid peroxidation, were measured in liver homogenates according to the method of Sunderman et al. (Reference Sunderman, Marzouk and Hopfer17). Moreover, to determine the specific peroxidation of n-6 and n-3 PUFA, we measured the hepatic concentrations of thioether protein adducts with HNE (4-HNE-P) and HHE (4-HHE-P), respectively. Because of the high reactivity of hydroxyalkenals, the measurement of protein adducts is a more reliable lipoperoxidation biomarker. These adducts were assessed by GC/MS. Sample preparation was done according to the method described by Asselin et al. (Reference Asselin, Bouchard and Tardif18). Briefly, 100 mg of liver tissue were mixed with 1 ml of cold buffer (pH 7·0) containing 39 mm-HEPES, 0·4 mm-EDTA and 0·9 mm-butylated hydroxytoluene and immediately treated with 1·14 mm-NaB2H4 to reduce aldehydes into their chemically stable 2H-labelled alcohol derivatives. Then, proteins were precipitated with saturated sulfosalicylic acid (0·2 ml). After centrifugation, the protein pellet was resuspended into 8 mm-guanidine buffer (0·5 ml), 0·25 nmol of the labelled internal standard [2H11](TBDMS) were added, and an incubation with Raney nickel (approximately 2 g) was performed (overnight, 55°C) in order to cleave the thioether linkages and reduce the C–C bonds. Free saturated derivatives were then extracted three times with ethyl acetate. The extract was evaporated under a gentle stream of N2 and treated with 75 μl N-methyl-N-(t-butyldimethylsylyl)-trifluoroacetamide 1,4-dihydroxynonene (DHN) for 2 h at 80°C for derivatisation. Samples were analysed using a triple quadrupole mass spectrometer Quattro Micro (Waters Corporation, Manchester, UK) coupled with a GC Agilent 6890N system (Agilent Technologies, Palo Alto, CA, USA), operating in the positive chemical ionisation mode using isobutane as the reagent gas. Injections (1 μl) were made at 250°C in the split mode (1/20). The carrier gas was high-purity He at a constant flow rate of 0·7 ml/min. Chromatographic separation was performed using an HP-5MS Agilent Technologies capillary column (50 m × 0·2 mm in inner diameter × 0·33 μm in phase thickness; Agilent Technologies) under the following conditions: 170°C for 1 min, increased by 10°C/min until 220°C, 2°C/min until 235°C, 5°C/min until 250°C and then by 30°C/min until 300°C. At the end of each run, temperature was kept at 300°C for 5 min to purify the column. MS parameters were optimised using the standard solutions. Maximum sensitivity was obtained for an ion-source temperature set at 120°C, an electron energy at 90 eV and an emission current at 300 μA.
Quantification was achieved by measuring product ions (multiple reaction monitoring) from the fragmentation of the protonated [M+H]+molecules. Collision energy potentials were then adjusted to optimise the signal for the most abundant product (daughter) ions: m/z 390>258 for [2H]DHN (reduced 4-HNE), m/z 348>216 for [2H] 1,4-dihydroxyhexene (reduced 4-HHE) and m/z 400>268 for the internal standard [2H11]DHN using argon as collision gas. Quantification was performed using calibration curves with external standards. Quantities of 4-HNE- and 4-HHE-P adducts that are reported in the present study represent averages of duplicate sample injections.
RNA isolation
Total RNA from liver tissue was isolated using the Norgen RNA Purification kit (Norgen Bioteck Corporation, Thorold, ON, Canada), according to the manufacturer's instruction. RNA was quantified with a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and RNA integrity assessed with a RNA 6000 Nano Labchip kit using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Only total RNA with an optical density 260/280 ratio >1·8, an optical density 260/230 ratio >1·7 and a RNA integrity number >8 was used for microarray hybridisation.
Microarrays
RNA samples were labelled using the Agilent Quick-Amp Labelling kit (5190-0424; Agilent Technologies, Wilmington, DE, USA), according to the manufacturer's instructions. Briefly, 500 ng of purified total RNA from each sample were amplified and reverse-transcribed in vitro to complementary DNA using the T7-polymerase, which was subsequently labelled with either cyanine 3-CTP or cyanine 5-CTP dyes (5188-1170-P; Agilent Technologies). Nanodrop ND-1000 was used to monitor the yield of amplification and dye incorporation; all samples had a yield >825 ng complementary RNA (cRNA) and a specific activity >8·0 pmol Cy3 or Cy5 per μg cRNA. The fluorescently labelled cRNA was hybridised using the Agilent gene expression hybridisation kit (5188-5242-A; Agilent Technologies) following the manufacturer's instructions. Briefly, 825 ng cyanine 3-labelled, linearly amplified cRNA were mixed with 825 ng cyanine 5-labelled, linearly amplified cRNA. The mix was loaded onto the Agilent Rabbit Gene Expression Microarrays (4 × 44k; Agilent Technologies) following a loop design and hybridisation proceeded in a hybridisation oven set to 65°C for 17 h. Then, the slides were washed in solutions I, II and III (Agilent Technologies) and air-dried.
Slides were scanned immediately after washing using the Agilent DNA Microarray Scanner (G2565AA/G2565BA; Agilent Technologies), at photomultiplier tube voltages red and green. Spot identification and quantification were performed using Agilent Feature Extraction Software 7.0 (Agilent Technologies). The complete formatted dataset is deposited as Gene Expression Omnibus accession GSE27621, which can be accessed at http://www.ncbi.nlm.nih.gov/geo/.
Analysis of microarray data
Statistical analyses were performed using linear models for microarray analysis (Limma) from the Bioconductor project(Reference Smyth, Michaud and Scott19). Quality of the microarray data was assessed on diagnostic plots (box plots and density plots) and spatial images generated from the raw (non-processed) data. All twelve arrays passed these strict criteria and were included in the analyses. Intensity ratio values for all microarray spots were normalised using a global loess smoothing procedure to remove the effect of systematic variation in the microarrays and no background correction was necessary due to homogeneous hybridisation(Reference Smyth and Speed20). The following three experimental comparisons were performed: (1) control v. group 1, (2) control v. group 2 and (3) control v. group 3. For each experimental comparison, a candidate list of differentially expressed probe sets was generated by calculating a moderated t statistic for each probe set using the Limma package. The Limma library implements an empirical Bayes approach to assign differential gene expression. Probe sets that satisfied the criterion of ≥ 1·2-fold change (FC) with a moderated P < 0·05 were considered to be significantly different. This FC was chosen because previous studies have shown that the use of a level ≥ 1·6 can underestimate the number of genes that are differentially regulated by a dietary treatment(Reference Sulzle, Hirche and Eder21, Reference Taniguchi, Miura and Iwao22).
Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Inc., Redwood City, CA, USA; www.ingenuity.com) was used to relate changes in gene expression levels to functional changes. The full dataset from the microarray analysis (including Entrez gene identification and gene expression values) was uploaded into IPA. The IPA library of canonical pathways identified those pathways that were most significant to the dataset. Genes differentially expressed and associated with the top 10 canonical pathways in the Ingenuity Pathways Knowledge Base are presented in the present paper. The significance of the association between a dataset and a specific canonical pathway was estimated in two ways: (1) the proportion of genes in the dataset included in the canonical pathway and (2) Fisher's exact test is used to calculate a P value determining the probability of the association between the dataset and the canonical pathway.
A complementary analysis was performed using EASE (software version 2.0; National Institutes of Health, Bethesda, MD, USA) to identify enriched biological themes within gene lists using Gene Ontology (GO) category over-representation analysis(Reference Hosack, Dennis and Sherman23). The same dataset as the one uploaded in IPA was used. An EASE score (adjusted Fisher's exact test for statistical significance) was calculated for the likelihood of over-representation of hierarchical categories based on biological processes, molecular functions and cellular components using the GO public database. Gene categories with an EASE score < 0·05 were considered to be significantly over-represented.
Statistics
Statistical analyses of fatty acid composition, lipoperoxidation parameters and liver glutathione level parameters were performed using GraphPad InStat version 3.06 (GraphPad Software, San Diego, CA, USA; www.graphpad.com). Data are presented as means with their standard errors (n 6–8) and followed a Gaussian distribution (Kolmogorov–Smirnov test), and differences between standard deviations (Bartlett's test) were not significant except for TBARS, TC:HDL-cholesterol, SFA and n-3 PUFA, which were then analysed using a non-parametric test (Kruskal–Wallis test). All other data were analysed using a one-way ANOVA, and all significant differences among means at the level of P < 0·05 were tested with Tukey–Kramer's post hoc analysis. Differences were considered as a trend when 0·05 < P < 0·1.
Results
Effects of long-chain n-3 PUFA on fatty acid composition of liver phospholipids
Fatty acid composition of liver phospholipids was measured to assess the incorporation of dietary LC n-3 PUFA. Table 2 shows that the proportion of LC n-3 PUFA (i.e. EPA (20 : 5n-3), docosapentaenoic acid (DPA) (22 : 5n-3) and DHA (22 : 6n-3)) in the liver of rabbits, given the tuna oil supplements, was substantially increased in a dose-dependent manner in comparison with the control group (4 × , 13 × and 22 × , P < 0·01 in groups 1, 2 and 3, respectively). All the dietary treatments raised the DHA contents, reaching 15 % of total fatty acids in group 3 and 100 times higher than the levels in the control group (P < 0·01). The incorporation of LC n-3 PUFA was positively correlated with the peroxidisability index (r 2 0·81, P < 0·001), which was twice higher in group 3 than in the control group (P < 0·0001). Whatever the dose of LC n-3 PUFA, arachidonic acid (20 : 4n-6) concentration was unchanged but linoleic acid (18 : 2n-6) proportion decreased by 3, 20 and 44 % in groups 1, 2 and 3, respectively (P < 0·01), when compared with the control condition. More generally, the progressive incorporation of LC n-3 PUFA in liver phospholipids (17 % of total FAME in group 3 v. control) induced a similar reduction of the incorporation of n-6 PUFA ( − 14 % of total FAME in group 3 v. control). The concentrations of most other fatty acids were significantly changed by the LC n-3 PUFA supplementations but to a lesser extent.
Table 2 Fatty acid composition (relative percentage of fatty acid methyl ester) of liver phospholipids from rabbits fed daily for 7 weeks either oleic sunflower oil (control) or a mixture of oils providing 0·1 % (group 1), 0·5 % (group 2) or 1 % (group 3) of daily energy intake as DHA
(Mean values with their standard errors, six to eight animals per group)

LC, long-chain; FAME, fatty acid methyl esters; PI, peroxidisability index.
a,b,c,d Mean values with unlike superscript letters were significantly different (P < 0·05).
* Data were analysed using a one-way ANOVA, and all significant differences among means at the level of P < 0·05 were tested with the Tukey–Kramer post hoc analysis.
† PI = (% dienoic × 1)+(% trienoic × 2)+(% tetraenoic × 3)+(% pentaenoic × 4)+(% hexaenoic × 5).
Effects of long-chain n-3 PUFA on plasma and liver lipids and apoB100
Plasma and liver lipid profiles were determined to investigate the effects of LC n-3 PUFA on cholesterol and TAG metabolism. Table 3 shows that the concentrations of plasma biomarkers were unchanged by none of the LC n-3 PUFA doses. In liver, total cholesterol was not modified by LC n-3 PUFA supplementation but there was a strong increase in TAG with dose 3 (3 × in comparison with the control, P < 0·01). This effect on TAG accumulation in liver was not dose-dependent but occurred only with the highest dose of LC n-3 PUFA.
Table 3 Concentration of total cholesterol, HDL-cholesterol, LDL-cholesterol, apoB100 and TAG from rabbits fed either oleic sunflower oil (control) or a mixture of oils providing 0·1 % (group 1), 0·5 % (group 2) or 1 % (group 3) of daily energy intake as DHA
(Mean values with their standard errors, six to eight animals per group)

* Data were analysed using a one-way ANOVA, and all significant differences among means at the level of P < 0·05 were tested with the Tukey–Kramer post hoc analysis.
Effect of long-chain n-3 PUFA on liver glutathione metabolism
Glutathione, one the main endogenous and intracellular antioxidant, and biomarkers of its metabolism were quantified in the liver in order to estimate the antioxidant status of rabbits and the effects of LC n-3 PUFA. None of the different parameters analysed was significantly affected by LC n-3 PUFA, as shown in Table 4. However, the data show that group 3 tended to have a distinct profile in comparison with the other groups. In contrast to groups 1 and 2, when comparing group 3 with the control, reduced glutathione concentration was 12 % lower while oxidised glutathione 27 % higher, leading to a 36 % higher reduced glutathione:oxidised glutathione ratio (NS).
Table 4 Liver concentration of reduced (GSH) and oxidised (GSSG) glutathione (nmol/g fresh tissue) and activities (U/g protein) of glutathione peroxidase (GPX), glutathione reductase (GR) and glutathione-S-transferase (GST) from rabbits fed daily either oleic sunflower oil (control) or a mixture of oleic sunflower oil and a DHA-enriched tuna oil providing 0·1 % (group 1), 0·5 % (group 2) or 1 % (group 3) of daily energy intake as DHA
(Mean values with their standard errors, six to eight animals per group)

* Data were analysed using a one-way ANOVA, and all significant differences among means at the level of P < 0·05 were tested with the Tukey–Kramer post hoc analysis.
† Redox potential (E hc), referring to the half-cell reduction potential of the GSSG/2GSH couple, was calculated using the Nernst equation (25°C, pH 7·0)(60).
Effect of long-chain n-3 PUFA on liver lipid peroxidation
Several biomarkers of lipid peroxidation were measured to assess the dose–response effects of LC n-3 PUFA supplementations on the endogenous production of peroxidised metabolites (Fig. 1). As expected with regard to the peroxidisability index determined in liver phospholipids (Table 2), lipid peroxidation was substantially increased in a dose-dependent manner in the liver of rabbits given the tuna oil supplements (Fig. 2). TBARS concentration rose by 38, 119 and 632 % in groups 1, 2 and 3, respectively. This dose–response relationship was confirmed by the positive correlation between LC n-3 PUFA incorporation into liver phospholipids and the levels of TBARS (r 2 0·65, P < 0·0001; Fig. 2). The analysis of specific metabolites produced from n-6 and n-3 PUFA peroxidation, namely 4-HNE-P and 4-HHE-P, was also performed. Because of the high reactivity of these molecules as free, we chose to quantify the aldehyde–protein adducts. Fig. 1(B) and (C) shows that lipid peroxidation was mainly associated with n-3 PUFA. Indeed, 4-HHE-P hepatic levels increased by 16, 84 and 227 % in groups 1, 2 and 3, respectively (P = 0·003), whereas concentrations of 4-HNE-P were unchanged. In the control group, 4-HNE-P levels were almost ten times higher than 4-HHE-P levels and despite the substantial increase in 4-HHE-P levels of group 3, they still remain twice lower than 4-HNE-P levels. Moreover, linear regression presented in Fig. 2 shows that the hepatic contents of LC n-3 PUFA phospholipids were positively correlated with those of 4-HHE-P (r 2 0·48, P = 0·001), suggesting again a dose-dependent response; however, only group 3 was significantly different from the control group. Overall, Figs. 1 and 2 show that dietary LC n-3 PUFA led to a raised level of lipoperoxidation, essentially due to n-3 PUFA peroxidation without significant modification of n-6 PUFA peroxidation. Finally, the positive correlations existing between dietary LC n-3 PUFA and their incorporation into liver phospholipids (r 2 0·97, data not shown), and LC n-3 PUFA incorporation into liver phospholipids and the production of TBARS and 4-HHE-P, both confirm the dose-dependent relationships between the intake of LC n-3 PUFA and liver lipoperoxidation.

Fig. 1 Dose–response effects of dietary long-chain n-3 PUFA on lipoperoxidation measured as (A) thiobarbituric acid-reactive substances (TBARS), a general biomarker of lipoperoxidation, and thioether aldehyde–protein adducts, namely (B) 4-hydroxynonenal–protein (4-HNE-P) and (C) 4-hydroxyhexenal–protein (4-HHE-P), by-products issued specifically from peroxidation of n-6 PUFA and n-3 PUFA, respectively. Values are means (six to eight animals per group), with standard errors represented by vertical bars. Data were analysed using a one-way ANOVA, and all significant differences among means at the level of P < 0·05 were tested with the Tukey–Kramer post hoc analysis. a,b,c Mean values with unlike letters were significantly different (P < 0·05). MDA, malondialdehyde.

Fig. 2 Correlations between long-chain (LC) n-3 PUFA incorporation into liver phospholipids and lipoperoxidation biomarkers. Correlation coefficients between a set of data correspond to the Pearson coefficient (r) and were calculated using GraphPad InStat version 3.06 (GraphPad Software, San Diego, CA, USA; www.graphpad.com). TBARS, thiobarbituric acid-reactive substances; MDA, malondialdehyde; FAME, fatty acid methyl esters; HHE-P, hydroxyhexenal–protein.
Effects of long-chain n-3 PUFA on liver gene expression
Global gene expression changes in the liver of rabbits fed the three different doses of LC n-3 PUFA were compared with control rabbits. The microarray analyses revealed 314, 387 and 370 genes differently expressed (FC ≥ 1·2, P < 0·05) in groups 1, 2 and 3, respectively. As shown in the Venn diagram (Fig. 3), thirty-eight genes were common between the three groups. Most genes had increased levels of expression with LC n-3 PUFA except with dose 3 where mRNA levels of hepatic genes were mostly reduced. The uploading of datasets into IPA reveals that more than 80 % of genes differentially expressed were eligible for network generation and most of them were also associated with pathways and functions.

Fig. 3 Venn diagram of significantly differentially expressed genes in groups 1, 2 and 3, providing 0·1, 0·5 and 1 % of daily energy intake as DHA, respectively.
Differentially expressed genes were classified into GO categories. The top ten GO classifications for biological processes for the three groups are represented in Fig. 4. As shown in the overlapping circles, groups 1 and 2 shared almost half of the identified biological processes and most of them were associated with lipid metabolism: lipid transport, steroid metabolism, lipid metabolism, sterol biosynthesis, steroid biosynthesis, lipid biosynthesis and sterol metabolism. Group 3 is differentiated by the low number of biological processes in common with groups 1 and 2 and the absence of links with lipid metabolism. The most over-represented biological processes in group 3 are related to urea metabolism and blood coagulation.

Fig. 4 Venn diagram depicting the top ten Gene Ontology (GO) classifications for biological processes and indicating the specificities and the commonalities (overlapping circles) between the three groups. JNK, c-Jun NH2-terminal kinase; TGF-β, transforming growth factor-β.
The top ten metabolic and signalling pathways showing significant changes in gene expression levels for the three experimental groups are reported in Fig. 5 and Table 5. Pathways related to inflammation and coagulation were over-represented in all groups, whereas lipid metabolism pathways were only associated with groups 1 and 2. Pathways related to energy metabolism were also well represented in all groups.

Fig. 5 Top ten canonical pathways significantly modulated in (A) group 1, (B) group 2 and (C) group 3. ‘Ratio’ indicates the number of molecules in a given pathway that meet cut-off criteria, divided by the total number of molecules that make up that pathway. P value is calculated using the right-tailed Fisher's exact test. LPS, lipopolysaccharide; RXR, retinoid X receptor; FXR, farnesol X receptor.
Table 5 Top 10 metabolic and signalling pathways significantly modulated in each comparison: control v. group 1, control v. group 2 and control v. group 3; the doses of DHA in the three different groups being 0·1, 0·5 and 1 % of daily energy intake, respectively

ID, identification; FC, fold change; SDR, short-chain dehydrogenase/reductase; TCR, T-cell receptor; FXR, farnesol X receptor; RXR, retinoid X receptor; MDR, multidrug resistance; TAP, transporter associated with antigen presentation; RWD, ring finger and WD repeat domain; LPS, lipopolysaccharide.
Effects of long-chain n-3 PUFA on inflammation
The ‘acute-phase response signalling’ pathway is one of the most significant pathways associated with datasets. It is also one of the top three pathways in the three groups. Interestingly, the association of this pathway to the dataset increases with the dose of LC n-3 PUFA. Among the twenty-three genes differently expressed, three were common between the three groups. These included the serine peptidase inhibitors serpin peptidase inhibitor, clade A (α1 antiproteinase, antitrypsin), member 1 (SERPINA1) and serpin peptidase inhibitor, clade D (heparin cofactor), member 1 (SERPIND1) genes as well as the suppressor of cytokine signalling. Some of the genes, whose abundance was significantly modulated, include gene-encoding proteins involved in signal transduction such as v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue, mitogen-activated protein kinase kinase 4 and Neuroblastoma RAS viral (v-ras) oncogene homologue. Other molecules were specific to either group 1, 2 or 3. In group 1, the expression level of protease inhibitor and cytokine transporter α2-macroglobulin genes was lowered in comparison with the control group. The abundance of several genes was specifically changed in group 2. Among them, the reduced expression levels of the TNF receptor TNF receptor superfamily, member 11b gene and the increased expression levels of ApoA2 and kallikrein B, plasma (Fletcher factor) 1 (KLKB1) genes are directly linked to the inflammatory response. Other genes significantly modulated in group 2 were associated with the regulation of gene transcription such as v-fos FBJ murine osteosarcoma viral oncogene homologue and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α, a member of the IκB family, whose abundance was increased. In group 3, the expression levels of the gene coding for the major inflammatory protein C-reactive protein were substantially lowered and the transcription factor Jun oncogene was up-regulated.
Among the top ten pathways of groups 1, 2 and 3, other pathways were linked to inflammation such as ‘CCR5 signalling in macrophages’, airway pathology in chronic obstructive disease', ‘calveolar-mediated endocytosis’, ‘lymphotoxin b-receptor signalling’, ‘thyroid cancer signalling’ and ‘complement system’. They were mainly associated with group 2 and contain genes directly related to inflammation such as the chemokine ligands chemokine (C–X–C motif) ligand 3 (CXCL3) and chemokine (C–C motif) ligand 4 (CCL4), the interleukin IL-8, the integrins integrin, α D, integrin, α V (vitronectin receptor, α polypeptide, antigen CD51) and integrin, β 5, and the TNF-associated factor TNF receptor-associated factor 3. Moreover, the top first networks generated from the overall datasets of groups 1, 2 and 3 were all characterised by a common main central node: the NFκB complex, a major transcription factor involved in inflammation.
Effect of long long-chain n-3 PUFA on coagulation
The ‘coagulation system’ was the second common pathway between the three groups. The serine protease inhibitors SERPINA1 and SERPIND1, also found in the ‘acute-phase response signalling’ pathway, were the only genes in common between groups 1, 2 and 3. These serine protease inhibitors, together with serpin peptidase inhibitor, clade C (antithrombin), member 1 (FC = − 1·30 in group 3), are all involved in the inhibition of the F2A factor (thrombin). In group 2, the mRNA levels of two coagulation factors were significantly increased: the coagulation factor III, also known as the tissue factor, and KLKB1, also known as the Fletcher factor, both of which are involved in the initiation of coagulation. In group 3, these factors were unchanged but the subunit B of the coagulation factor XIII was significantly down-regulated (FC = − 1·55).
Effects of long long-chain n-3 PUFA on lipid metabolism
Among all the pathways significantly modulated by LC n-3 PUFA, three were directly in relation to the metabolism of lipids. These include ‘fatty acid metabolism’ (top 8 pathway for group 1), ‘arachidonic acid metabolism’ (top 3 pathway for group 2) and ‘FXR/retinoid X receptor activation’ (top 5 pathway for group 2). In the ‘fatty acid metabolism’ pathway, there were significant changes in several genes coding for the enzymes such as acetyl-CoA acetyltransferase 2, acyl-CoA oxidase 2, acyl-CoA synthetases such as acyl-CoA synthetase long-chain family member 3, and hydroxyacyl-CoA dehydrogenase. The ‘arachidonic acid metabolism’ pathway was mainly modulated in group 2. Basically, in group 2, there was an inhibition of the arachidonate metabolism via the reduced mRNA levels of unspecified mono-oxygenases (cytochrome P450, family 2, subfamily C, polypeptide 18 and cytochrome P450, family 4, subfamily B, polypeptide 1) and PG synthase PG E synthase 3 genes. LC n-3 PUFA-dose 2 also induced the expression of the detoxification enzyme glutathione peroxidase 4 gene, which is involved in the metabolism of lipid peroxides. This increase in expression level was not associated with an increase in glutathione peroxidase activity (Table 4). The ‘FXR/retinoid X receptor activation’ pathway was the main signalling pathway linked with lipid metabolism altered by LC n-3 PUFA. Among the genes whose abundance was significantly changed by LC n-3 PUFA, the microsomal TAG transfer protein (MTTP) and the lipoprotein receptor VLDLR are of interest because of their crucial role in lipoprotein metabolism. Their expression level was differently modulated with the three doses of LC n-3 PUFA. Indeed, MTTP mRNA level was decreased with doses 1 and 2 but not with dose 3. On the contrary, the abundance of the VLDLR gene was raised in a dose-dependent manner, leading to a FC of almost 3 in group 3.
Effects of long long-chain n-3 PUFA on energy metabolism
Several of the top ten pathways in groups 1, 2, and 3 were associated with energy metabolism. These include ‘synthesis and degradation of ketone bodies’ with the down-regulation of 3-hydroxy-3-methylglutaryl-CoA synthase, ‘pyruvate metabolism’ and ‘glycolysis/gluconeogenesis’.
Specific effects of LC n-3 PUFA-dose 3
As shown in Figs. 1 and 2, group 3 was associated with a significantly increased lipid peroxidation level. As peroxidised metabolites produced from LC n-3 PUFA are potential modulators of gene expression, we focused our analysis on genes that were specifically and substantially changed (i.e. FC >1·5) in group 3 (Table 6). Among the thirty-four genes differentially expressed at a FC >1·5 or < − 1·5, ten genes had increased expression levels. Among them, two molecules were once again central in the metabolism of lipoprotein and cholesterol. These include the LDL-receptor (LDLR) that contributes to the uptake of circulating LDL and subsequently to hepatic metabolism of cholesterol. The abundance of the insulin-induced gene 1 was also increased in group 3. This gene encodes an endoplasmic reticulum membrane protein that plays a critical role in regulating cholesterol concentration in cells. Indeed, insulin-induced gene 1 binds to the sterol-sensing domains of sterol regulatory element-binding protein and 3-hydroxy-3-methylglutaryl-CoA reductase and is essential for the sterol-mediating trafficking of the two proteins. It is also interesting to note the increased abundance of the gene coding for ubiquitin-conjugating enzyme (ubiquitin-conjugating enzyme E2T) involved in the proteasome-dependent proteolysis. In group 3, twenty-four genes had decreased levels. These include a serine protease inhibitor (serpin peptidase inhibitor. clade F (α2 antiplasmin, pigment epithelium-derived factor), member 1) and the coagulation factor XIII genes already described. Other genes altered such as haemopexin, glutathione peroxidase 3 or the metallothioneins (metallothionein 3, Metallothionein 2A, Metallothionein 1X) are directly linked to oxidative stress.
Table 6 List of genes significantly up- and down-regulated at a fold change >1·5 or <−1·5, only with dose 3 of LC- n-3 PUFA (1 % of daily energy intake as DHA)
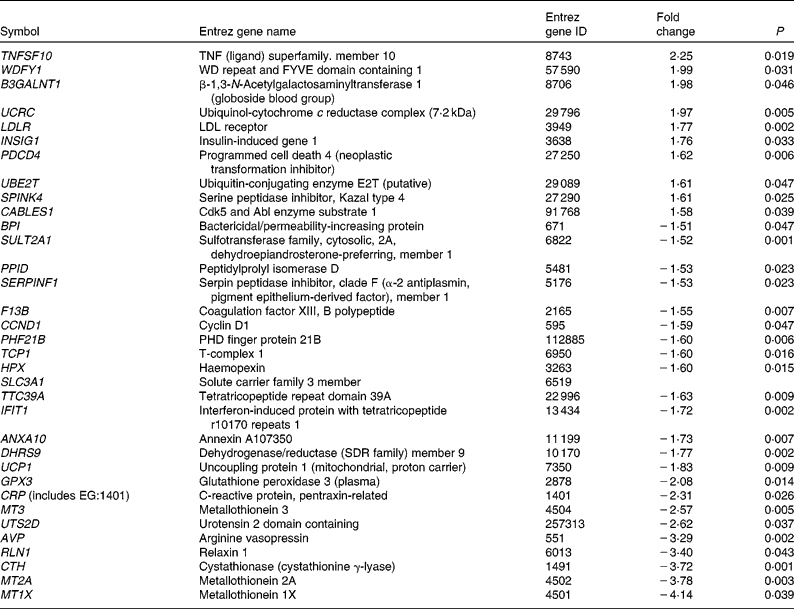
ID, identification; WD, tryptophan-aspartate; FYVE, domain identified in Fab1p, YOTP, VAC1p and EEA1 domain; PHD, plant homeo domain.
Discussion
The biological effects of LC n-3 PUFA can be mediated in part by their actions at the gene expression level but the complexity of altered biological pathways and the nature of the bioactive molecules are still under investigation. The present experiment was conducted to investigate the dose–response effects of LC n-3 PUFA on liver lipid peroxidation and gene expression. For the first time, we demonstrated that increasing intake of LC n-3 PUFA enhances, in a dose-dependent manner, the hepatic production of 4-HHE, a specific peroxidised metabolite having cell signalling properties(Reference Riahi, Cohen and Shamni24). At the gene expression level, the different doses of LC n-3 PUFA tested induced contrasting effects on inflammation pathways and dose-dependently modulated the expression of major molecules involved in hepatic lipoprotein metabolism, possibly through a mechanism involving the FXR signalling pathway.
Dietary LC n-3 PUFA modified lipid metabolic profiles
The present results show for the first time that dietary LC n-3 PUFA increases, in a dose-dependent manner, not only the incorporation of LC n-3 PUFA in liver phospholipids but also the hepatic production of 4-HHE. As expected, the diets used here increased the content of DHA and, to a lesser extent, of EPA. The dose-dependent incorporation of DHA has previously been reported in various human tissues including liver(Reference Arterburn, Hall and Oken25). This is usually associated with an equivalent decrease of arachidonic acid content because of their competition on the sn-2 position of phospholipids. This competition determines part of the biological effects of DHA since the relative proportion of arachidonic acid and DHA in phospholipids influences their availability for phospholipase cleavage and their subsequent metabolism into eicosanoids. These fatty acids and/or their metabolites are also involved in gene expression regulation. The present data did not show any significant change in arachidonic acid proportion despite the substantial increase of DHA in liver phospholipids, suggesting that the administrated doses were not sufficient to lower arachidonic acid levels. Indeed, in studies reporting a shift between arachidonic acid and DHA contents, the dose of LC n-3 PUFA (EPA or DHA) was higher than what is presently used: about 2 % of daily energy intake(Reference Arterburn, Hall and Oken25, Reference Berger and German26). Besides the effects of LC n-3 PUFA supplementation on the native fatty acids, we focused our attention on peroxidised metabolites, potential bioactive molecules involved in the modulation of gene expression. Interestingly, the dose-dependent production of 4-HHE was not associated with a decrease of 4-HNE. Overall, these modifications of lipid metabolic profiles suggest that the changes in gene expression levels are unlikely due to the reduced effects of arachidonic acid and/or 4-HNE, which were unchanged, but more probably due to DHA and/or 4-HHE, which were dose-dependently produced in response to LC n-3 PUFA supplementation.
Previous work showed increased level of lipoperoxidation in liver in response to fish oil feeding(Reference Sen, Atalay and Agren27) but to the best of our knowledge, none specifically identified 4-HHE and established a dose–response relationship. The signalling properties of hydroxyalkenals have been so far mainly focused on 4-HNE, the major aldehyde produced from n-6 fatty acid peroxidation. Studies have clearly indicated that concentrations of these aldehydes greatly influence their cellular effects, and that their signalling properties are mainly mediated through their capacity to form adducts with macromolecules, modifying their structure and function (recently reviewed in Poli et al. (Reference Poli, Schaur and Siems28)). Several kinases are targeted by 4-HNE, including c-Jun NH2-terminal kinase, p38 mitogen-activated protein kinases, IκB kinase (IKK) complex, extracellular signal-regulated kinase (ERK) and protein kinase C β and γ(Reference Leonarduzzi, Robbesyn and Poli29), showing the involvement of 4-HNE in the regulation of several pathophysiological processes including inflammation, proliferation, apoptosis and matrix remodelling. More recently, 4-HNE has been identified as an activator of PPARγ in 3T3-L1 pre-adipocytes(Reference Coleman, Prabhu and Thompson30), suggesting a different mechanism of gene expression modulation and the capacity of hydroxyalkenals to interact with transcription factors. Due to its low lipophilicity and reduced chemical reactivity, 4-HHE is considered less physiologically active than 4-HNE(Reference Esterbauer, Schaur and Zollner31). However, previous studies(Reference Lee, Je and Jung32, Reference Je, Lee and Jung33) have demonstrated that 4-HHE modulates some kinase activities (p38 mitogen-activated protein kinases, ERK and IKK) leading to the activation of the NF-κB and the subsequent induction of the endothelial NO synthase expression. Altogether, these data suggest that the dose-dependent production of hepatic 4-HHE observed here might contribute to the alteration of gene expression in this tissue.
The doses of LC n-3 PUFA used in the present study did not affect plasma lipid profiles. However, even though hypocholesterolaemic and hypotriacylglycerolaemic effects of LC n-3 PUFA are well described in human subjects(Reference Harris34), those are not always reported in animals(Reference Harris35). It is possible that the high-cholesterol-enriched diet (0·5 %) has induced a hypercholesterolaemia that was unable to be corrected by the doses of LC n-3 PUFA used. This is consistent with the study of Zhu et al. (Reference Zhu, Smith and Sievers36), where, in a similar experimental design (rabbits fed a diet enriched with 0·3 % cholesterol and three different doses of LC n-3 PUFA ranging from 0·9 to 2·7 % of the daily energy intake), no effect was observed on total serum cholesterol and HDL-cholesterol. The hypotriacylglycerolaemic effect of LC n-3 PUFA was only detected with a dose of 1·8 % of the daily energy intake, which is almost twice higher than the highest dose used in the present study. In liver, LC n-3 PUFA and, more precisely, dose 3 substantially increased the TAG content. This unexpected effect was not dose-dependent and is not consistent with previous studies reporting that LC n-3 PUFA have no action on or do not prevent steatosis(Reference Saraswathi, Gao and Morrow37–Reference Tandy, Chung and Wat39). However, discrepancies of experimental designs (i.e. animal models, dose and type of LC n-3 PUFA used) make the comparison difficult. We can hypothesise that our original result could be linked to the specific effect of DHA and the high dose used, two parameters that deeply differ with the other studies. Even though the accumulation of liver TAG is surprising, this is consistent with the present transcriptomic results that are further discussed below.
Dose–response effects of LC n-3 PUFA on pathways related to inflammation
Among the top ten pathways of each group, those related to inflammation were systematically associated with datasets and the impact increased with the dose of LC n-3 PUFA. It is interesting to note that the first networks for each group were systematically centred on NFκB, a major transcription factor involved in inflammation(Reference Hayden and Ghosh40). This suggests that in our conditions, LC n-3 PUFA might have modified the expression of genes related to inflammation by modulating the activity of NFκB as reported previously(Reference Calder41). However, differential effects were observed with the three doses. Group 2 was associated with an increased abundance of many genes encoding for pro-inflammatory cytokines and chemokines including IL-8, CXCL3 and CCL4 possibly through the activation of NFκB which controls the expression of all these genes(Reference Hayden and Ghosh40). In contrast, none of these genes was modified in group 3 but the expression levels of the pro-inflammatory protein C-reactive protein were substantially reduced, suggesting an anti-inflammatory effect with this dose of LC n-3 PUFA. As the expression of C-reactive protein is also under the control of NFκB(Reference Agrawal, Cha-Molstad and Samols42), dose 3 might have then prevented the activation of NFκB. One might hypothesise that the discrepancies between groups 2 and 3 can be related to the production of 4-HHE (twice more with dose 3 than with dose 2), which can be involved in the inactivation of NFκB. This would be consistent with studies showing that hydroxyalkenals can modulate NFκB activity; however, contrasting effects (i.e. activation or inhibition) have been reported(Reference Riahi, Cohen and Shamni24). Indeed, it has been reported that 4-HNE prevents the activation of NFκB by inhibiting the phosphorylation and subsequent degradation of the inhibitory proteins IκBα, β and ɛ(Reference Page, Fischer and Baumgartner43, Reference Donath, Fischer and Page44). The inhibition appears to be caused by the formation of covalent adducts on the IKK complex(Reference Ji, Kozak and Marnett45). In response to upstream stimuli, the IKK complex is activated through phosphorylation operated by IKK, namely protein kinase C, protein kinase B/Akt or mitogen-activated protein kinases/ERK 1, 2 and 3(Reference Zandi and Karin46). On the other hand, others have reported in prostate endothelial cells that 4-HHE can activate NFκB by enhancing the phosphorylation of IκB via NFκB-inducing kinase and the p38 mitogen-activated protein kinases pathways(Reference Je, Lee and Jung33). These contrasting data together with the discrepancies between the groups observed here highlight that the effects of LC n-3 PUFA and/or their metabolites may be different and even opposite depending on the dose/intake and probably the cellular environment in which they operate.
Dose–response effects of LC n-3 PUFA on lipid and lipoprotein metabolism
As expected, several pathways and biological processes related to lipid metabolism were associated with datasets. Modulation of hepatic lipid metabolism by LC n-3 PUFA has been well described and generally involves an up-regulation of fatty acid transport, activation and oxidation (via up-regulation of fatty acid-binding proteins, acyl-CoA synthetases, acyl-CoA oxidase and carnitine palmitoyltransferase I) and a down-regulation of lipogenesis and desaturation of fatty acids (via down-regulation of fatty acid synthase and stearoyl-CoA desaturase 1)(Reference Pegorier, May and Girard47). Several transcription factors have been identified as prospective targets for fatty acid regulation of hepatic gene expression. These include a number of nuclear receptors such as the PPAR family (PPARα, β, γ1 and γ2), retinoid X receptor α, liver X receptor α, FXR and hepatic nuclear factors α and γ, as well as several basic helix-loop-helix leucine-zipper transcription factors such as sterol regulatory element-binding protein-1(Reference Jump, Botolin and Wang48). There are two main mechanisms that characterise fatty acid control of these transcription factors. In the first one, fatty acids bind directly to the transcription factors and control their activity, whereas in the second one, they control the nuclear abundance of key transcription factors such as sterol regulatory element-binding protein-1.
In the present experiment, LC n-3 PUFA (mainly DHA) significantly up-regulated acyl-CoA synthetases but most of the other genes involved in fatty acid transport, oxidation, elongation and lipogenesis were not altered. This suggests that PPARα, which controls the expression of almost all the genes involved in these processes(Reference Jump, Botolin and Wang48), was not activated. In a previous study using genome-wide analysis, EPA has been shown as reversing the decrease in colon expression levels of PPARα in IL-10 gene-deficient mice (a model of inflammatory bowel disease), suggesting that EPA activated colon PPARα to produce anti-inflammatory effects(Reference Knoch, Barnett and Zhu49). However, another microarray analysis of hepatic gene expression in response to LC n-3 PUFA, and predominantly DHA, failed to demonstrate a significant activation of the expression of genes controlled by PPARα(Reference Takahashi, Tsuboyama-Kasaoka and Nakatani50, Reference Kramer, LeDeaux and Butteiger51). The absence of PPARα activation in these studies (including the present study) can be explained by the fact that DHA and/or its metabolites (in contrast to EPA and/or its metabolites) are probably weak PPARα ligands. Indeed, LC n-3 PUFA that include EPA, DHA and DPA are often considered as a whole while each fatty acid has specific physical and chemical properties influencing their biological activity. Concerning the ability of LC n-3 PUFA to activate PPARα, receptor–ligand assays have clearly demonstrated that DHA is one of the weakest PPARα, β or γ activators, whereas linoleic acid (a medium-chain n-6 fatty acid) is the strongest inducer of PPARα and β(Reference Krey, Braissant and Lhorset52, Reference Pawar and Jump53). It is then consistent with the present data where the expression levels of PPARα-regulated genes were unchanged since DHA was the most abundant LC n-3 PUFA in liver phospholipids, whereas linoleic acid levels were lowered in a dose-dependent manner in response to DHA incorporation. PUFA's peroxidised metabolites are also recognised PPAR ligands(Reference Krey, Braissant and Lhorset52, Reference Mishra, Chaudhary and Sethi54, Reference Itoh and Yamamoto55) but the only study which specifically investigated hydroxyalkenals showed that contrary to 4-HNE, 4-HHE was not able to induce PPARβ/γ(Reference Coleman, Prabhu and Thompson30). Thus, the use of a DHA-enriched diet here can explain the absence of the effect on PPARα-regulated genes. In contrast, our datasets were clearly associated with the FXR signalling pathway, which suggests that DHA preferentially binds with this nuclear receptor, consistent with recent findings showing that it is a ligand for FXR(Reference Zhao, Yu and Lew56) and that DHA-rich oil consumption was correlated with FXR activation in rat liver(Reference Kramer, LeDeaux and Butteiger51).
Despite the absence of modulation of the usual lipid metabolism pathways, major genes encoding proteins involved in TAG-rich lipoprotein metabolism were altered by LC n-3 PUFA. These include the receptors for LDL and VLDL (LDLR and VLDL-receptor (VLDLR)), whose mRNA abundances were increased, as well as the MTTP, whose expression level decreased with doses 1 and 2. These modulations are consistent with a previous study investigating transcription profiling in rat liver in response to DHA(Reference Kramer, LeDeaux and Butteiger51). The increased expression of LDLR and VLDLR was mostly associated with the highest dose of LC n-3 PUFA and could explain the hepatic accumulation of TAG reported in group 3. Indeed, LDLR and VLDLR are both involved in the clearance of TAG-rich lipoproteins(Reference Daniels, Killinger and Michal57) and therefore enhance the hepatic uptake of circulating lipids. This effect could be interesting to reduced plasma TAG concentration provided that it is associated with a stimulation of fatty acid oxidation and/or an inhibition of lipogenesis. However, this was not the case in the present experiment, which might have contributed to the accumulation of TAG in liver. With doses 1 and 2, the mRNA abundances of VLDLR and LDLR were slightly or not modified but MTTP was down-regulated. This protein is essential for hepatic VLDL secretion because of its involvement in the translocation and maturation of apoB molecules as well as its role in the transfer of TAG upon binding to apoB(Reference Fisher and Ginsberg58). The reduction of the mRNA levels of MTTP might then lead to a reduced VLDL secretion. This is a well-known effect of LC n-3 PUFA but usually attributed to reduced expression levels of genes encoding for fatty acid synthesis together with an increased mRNA abundance of genes involved in fatty acid oxidation and induced by a PPARα-dependent pathway(Reference Harris, Miller and Tighe2). In the present experiment, and consistent with the results reported by Kramer et al. (Reference Kramer, LeDeaux and Butteiger51) in rats, PPARα genes were not regulated, whereas the FXR signalling pathway was induced. Interestingly, LDLR and VLDLR are all induced by FXR(Reference Sirvent, Claudel and Martin59, Reference Claudel, Staels and Kuipers60) and MTTP is indirectly repressed when FXR is activated via the inhibition of hepatic nuclear factor 1 (data from IPA). Therefore, we can hypothesise that in the present experiment, DHA (the most abundant LC n-3 PUFA in diets and in liver phospholipids) and/or its metabolites activated FXR that induced the expression of LDLR and VLDLR and the repression of MTTP expression.
In conclusion, the present study shows that feeding LC n-3 PUFA to hypercholesterolaemic rabbits induced, in a dose-dependent manner, the deposition of these fatty acids (especially DHA) in liver phospholipids but also the production of 4-HHE, a major aldehyde produced from LC n-3 PUFA peroxidation. At the highest dose, LC n-3 PUFA provoked an increased deposition of TAG in liver, which can be directly linked to the increased mRNA levels of lipoprotein hepatic receptors (LDLR and VLDLR). Interestingly, LC n-3 PUFA supplementation induced a dose-dependent activation of pathways related to inflammation, with contrasting effects depending on the dose used. Although the discrimination of the effects attributable to the native DHA or 4-HHE is impossible here, the literature provides evidence suggesting that some of the observed changes in gene expression levels might be induced by 4-HHE. Complementary studies using in vitro models (e.g. hepatic cell lines) and purified molecules are now required for further understanding of the mechanisms of the action of DHA and/or its metabolites on liver gene expression.
Acknowledgements
The authors acknowledge Marie-Anne Verny, Benoit Cohade and Christian Lafarge for their excellent assistance during the animal experiment. We also thank Chrystèle Jouve and Dominique Bayle for the analysis of lipid parameters in plasma and liver. The authors finally wish to thank Anna Russ and Dragan Milenkovic for their advices on the use of bioinformatic software. Finally thanks to Kelly Armstrong and Michelle for their assistance with tissue RNA extraction and array hybridisation. The present study received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. C. G., N. C. R., J.-M. C. and B. C. planned the study, designed the experiment, summarised, discussed and interpreted the results or drafted the manuscript. Other authors were involved in the analysis of biological samples and interpretation of the results as well as participated in the writing of the manuscript. The authors have no potential financial or personal conflict of interest.











