

Congenital myasthenic syndromes (CMSs) are a heterogeneous group of genetic disorders affecting components of the neuromuscular junction (NMJ).Reference Rodriguez Cruz, Palace and Beeson 1 , Reference Engel, Shen, Selcen and Sine 2 Postsynaptic NMJ defects encompass up to 75% of CMS cases and comprise mutations in genes that encode different subunits of the acetylcholine receptor (CHRNA1, CHRNB1, CHRND and CHRNE) or proteins important to maintain the structure or function of the NMJ, such as MUSK, RAPSN or DOK7.Reference Harper 3 Defects in the acetylcholine receptor (AChR) can be primary, resulting from a decreased expression of one of the subunits, or functionally subdivided into fast- (FCCMS) and slow-channel (SCCMS) congenital myasthenic syndromes.
Most cases of primary AChR deficiency involve mutations in the CHRNE gene, because the epsilon subunit can be substituted by the foetal gamma subunit, thus rescuing the phenotype. Compound heterozygous or homozygous null mutations in the CHRNA1, CHRNB or CHRND gene do not benefit from the gamma subunit rescue and are believed to be lethal. We herein present a nonlethal case of a severely affected CMS patient with a CHRNA1 homozygous null mutation.
We describe a boy who was referred to our neuromuscular disorders outpatient clinic at the age of 6 years for evaluation with a diagnosis of severe congenital myopathy. He was born from unaffected first-degree cousins of mixed ancestry (Portuguese, African and native) out of an uneventful pregnancy with reduced foetal movements. The couple had a previous pregnancy of twins with foetal demise at 7 months. Immediately after caesarean section, the patient required orotracheal intubation due to respiratory insufficiency and was admitted to the neonatal ICU, where he stayed for a year and a half. He was discharged to continue home care with gastrostomy and tracheostomy, requiring continuous BiPAP support. Despite having gradually improved, he remained severely impaired, never having acquired head control or the ability to sit. At our first evaluation, he was able to move his arms and legs on a plane, with proximal greater than distal weakness in all four limbs. Physical exam demonstrated bilateral ptosis with complete ophthalmoplegia, absence of facial expression, high-arched palate and marked hypotonia without contractures (Figure 1A). His serum CK level has always remained within the normal range. Muscle biopsy performed at 10 months by another service in the deltoid muscle showed marked predominance and atrophy of type 2 fibres without disruptions of the intermyofibrillar network (Figure 1B).

Figure 1 Main features of the case. (A) Facial aspects included marked ptosis and facial weakness, in addition to a prominent forehead and temporalis atrophy. Respiratory support via tracheostomy can also be seen. (B) Muscle biopsy shows marked predominance and atrophy of type 2 fibres (ATPase 9.4, white bar=50 μm). (C,D,E) NGS data alignment (Integrated Genome Viewer software) showing the mutation p.Gly466Arg in a homozygous state in the proband (C), and in a heterozygous state in the father (D) and mother (E). Coverages were 75, 92 and 129 reads, respectively.
We opted for direct exome sequencing to reach a final molecular diagnosis. Analysis of sequencing data of the trio consisted of filtering variants for known genes implicated in neuromuscular diseases, discarding variants with a minimum allele frequency (MAF) >0.5% in different population variant frequency databases (EVS, ExAC) or with a low pathogenicity prediction score combining evaluations of the PolyPhen-2 and SIFT tools. No variants survived the filtering pipeline for the X-linked and de novo analysis scenarios. Compound heterozygous variants were identified fitting the segregation, but they occurred in genes unrelated to the phenotype of the patient (SYNE1 and SYNE2). BAM files were thoroughly checked for coverage of known genes. In the homozygous recessive analysis scenario, we found the variant c.1396G>A (p.Gly466Arg), in exon 10 of the CHRNA1 gene (NM_001039523) (Figure 1D–F). It has been previously implicated in primary AChR-deficiency CMS.Reference Rahman, Masuda, Ohe, Ito, Hutchinson, Mayeda and Engel 4
A nerve conduction study was performed to demonstrate the NMJ impairment. Repetitive nerve stimulation of the ulnar and peroneal nerves, with recording in the abductor digiti minimi and tibialis anterior muscles, respectively, showed marked decrement of >70% in both the upper and lower limbs (Figure 2A,B). Concentric needle electromyography revealed instability of motor unit action potentials with increased jitter on voluntary contraction. The patient was then started on pyridostigmine and had an almost immediate improvement in limb movement, being able to raise his arms and legs against gravity, and gradual improvement in ventilatory function, reducing BiPAP support to the point where it was only necessary at night.
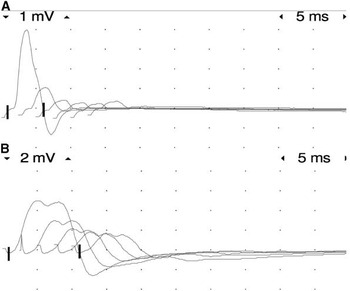
Figure 2 3-Hz repetitive stimulation test on the left ulnar nerve (A) and on the left peroneal nerve (B) showing a decrement greater than 70%.
The variant p.Gly466Arg gives rise to a primary AChR defect by way of reducing the expression of the AChR alpha subunit to a level of around 15% of normal.Reference Rahman, Masuda, Ohe, Ito, Hutchinson, Mayeda and Engel 4 It was previously described in a 53-year-old man with myasthenic symptoms since birth who harboured the mutation (under a different notation, it was referred to as p.Gly421Arg) in a heterozygous state, compounded with an additional heterozygous splicing disrupting mutation. He had partial response to pyridostigmine and amifampridine.
The patient we describe expands our understanding of the complex genetic picture of congenital myasthenic syndromes with the demonstration of a homozygous null mutation expected to give rise to a lethal phenotype but instead originating a severely affected progeny of asymptomatic carriers. The delay in diagnosis of a condition that benefits from immediate symptomatic treatment prompts one to emphasize the need to always consider a CMS diagnosis in severely hypotonic infants, even in patients with mild inconclusive muscle biopsies or suggestive of other diseases.Reference Kinali, Beeson, Pitt, Jungbluth, Simonds and Aloysius 5 In particular, type 2 fibre predominance or atrophy is not a common feature in CMS.Reference Kinali, Beeson, Pitt, Jungbluth, Simonds and Aloysius 5 Repetitive nerve stimulation may point to the diagnosis, and this is an important ancillary exam to order in these cases. On the other hand, the absence of decrement on repetitive nerve stimulation does not rule out NMJ defects. Single-fibre electromyography can be a technical challenge in young children, and normative values are not well defined.Reference Kosac, Gavillet and Whittacker 6 Molecular diagnosis and knowledge of the mutation effect in CMS is important to enable defining an effective and safe symptomatic treatment. Primary AChR deficiency and FCCMS may respond to pyridostigmine and amifampridine, cholinergic agents that must be avoided in SCCMS and in deficiency of Dok-7.Reference Engel, Shen, Selcen and Sine 2 Ideally, the choice of treatment should be withheld until a genetic diagnosis is reached, or done with close observation in a monitored setting.Reference Engel, Shen, Selcen and Sine 2 Of note, there are reports of NMJ involvement and myasthenic symptoms with response to acetylcholinesterase inhibitors in different forms of confirmed or nonspecific congenital myopathies,Reference Rodriguez Cruz, Sewry, Beeson, Jayawant, Squier and McWilliam 7 , Reference Robb, Sewry, Dowling, Feng, Cullup, Lillis and Abbs 8 an important differential diagnosis of CMS. Our present report illustrates how personalized genomics can quickly lead to personalized treatment in nonspecific cases.
ACKNOWLEDGMENTS
OAN was supported by a fellowship from the CAPES Foundation, Ministry of Education of Brazil (process no. 1286/51-2). This work was supported by grants from Fondation Maladies Rares to JL, as well as by the France Génomique National infrastructure, funded as part of the “Investissements d’Avenir” program managed by the Agence Nationale pour la Recherche (contract no. ANR-10-INBS-09).
DISCLOSURES
Osorio Abath Neto has the following disclosure: CAPES: researcher, grant.
Jocelyn Laporte has the following disclosures: FMR (Fondation Recherche Medicale): researcher, grant; ANR–FGN: researcher, grant.
Carlos Otto Heise, Cristiane de Araújo Martins Moreno, Eduardo de Paula Estephan, Lilia Mesrob, Doris Lechner, Anne Boland, Jean-François Deleuze, Acary Souza Bulle Oliveira, Umbertina Conti Reed, Valérie Biancalana and Edmar Zanoteli do not have anything to disclose.
STATEMENT OF AUTHORSHIP
OAN and EZ were responsible for the clinical description and follow-up of the patient, as well as for preparation of the first drafts of the manuscript. CAMM, EPE and UCR helped in the clinical description and follow-up of the patient. COH performed nerve conduction studies and helped discuss the electrophysiological findings. ASBO was responsible for the muscle biopsy of the patient and its report. LM, DL, AB and JFD performed exome sequencing. VB and JL helped in the analysis of the exome data. OAN, EZ and JL reviewed and prepared the final version of the manuscript.


