Refine listing
Actions for selected content:
Volume 83 - Issue 3 - June 2019

Contents
The 51st Hallimond Lecture
Time's arrow, time's cycle: Granulite metamorphism and geodynamics
-
- Published online by Cambridge University Press:
- 12 April 2019, pp. 323-338
-
- Article
-
- You have access
- HTML
- Export citation
Article
Constraints on the Equations of State of stiff anisotropic minerals: rutile, and the implications for rutile elastic barometry
-
- Published online by Cambridge University Press:
- 22 April 2019, pp. 339-347
-
- Article
- Export citation
-
We present an assessment of the thermo-elastic behaviour of rutile based on X-ray diffraction data and direct elastic measurements available in the literature. The data confirms that the quasi-harmonic approximation is not valid for rutile because rutile exhibits substantial anisotropic thermal pressure, meaning that the unit-cell parameters change significantly along isochors. Simultaneous fitting of both the diffraction and elasticity data yields parameters of KTR0= 205.14(15) GPa, KSR0= 207.30(14) GPa,
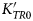 $K_{TR0}^{\prime} $= 6.9(4) in a 3rd-order Birch-Murnaghan Equation of State for compression, αV0= 2.526(16) × 10–5 K–1, Einstein temperature θE = 328(12) K, Anderson-Grüneisen parameter δT = 7.6(6), with a fixed thermal Grüneisen parameter γ = 1.4 to describe the thermal expansion and variation of bulk modulus with temperature at room pressure. This Equation of State fits all of the available data up to 7.3 GPa at room temperature, and up to 1100 K at room pressure within its uncertainties. We also present a series of formulations and a simple protocol to obtain thermodynamically consistent Equations of State for the volume and the unit-cell parameters for stiff materials, such as rutile. In combination with published data for garnets, the Equation of State for rutile indicates that rutile inclusions trapped inside garnets in metamorphic rocks should exhibit negative residual pressures when measured at room conditions.
$K_{TR0}^{\prime} $= 6.9(4) in a 3rd-order Birch-Murnaghan Equation of State for compression, αV0= 2.526(16) × 10–5 K–1, Einstein temperature θE = 328(12) K, Anderson-Grüneisen parameter δT = 7.6(6), with a fixed thermal Grüneisen parameter γ = 1.4 to describe the thermal expansion and variation of bulk modulus with temperature at room pressure. This Equation of State fits all of the available data up to 7.3 GPa at room temperature, and up to 1100 K at room pressure within its uncertainties. We also present a series of formulations and a simple protocol to obtain thermodynamically consistent Equations of State for the volume and the unit-cell parameters for stiff materials, such as rutile. In combination with published data for garnets, the Equation of State for rutile indicates that rutile inclusions trapped inside garnets in metamorphic rocks should exhibit negative residual pressures when measured at room conditions.
Magnesioleydetite and straβmannite, two new uranyl sulfate minerals with sheet structures from Red Canyon, Utah
-
- Published online by Cambridge University Press:
- 28 May 2018, pp. 349-360
-
- Article
- Export citation
Gem amphiboles from Mogok, Myanmar: crystal-structure refinement, infrared spectroscopy and short-range order–disorder in gem pargasite and fluoro-pargasite
-
- Published online by Cambridge University Press:
- 14 September 2018, pp. 361-371
-
- Article
- Export citation
Rinkite-(Y), Na2Ca4YTi(Si2O7)2OF3, a seidozerite-supergroup TS-block mineral from the Darai-Pioz alkaline massif, Tien-Shan mountains, Tajikistan: Description and crystal structure
-
- Published online by Cambridge University Press:
- 29 June 2018, pp. 373-380
-
- Article
- Export citation
-
Rinkite-(Y), ideally Na2Ca4YTi(Si2O7)2OF3, is a new rinkite-group (seidozerite-supergroup) TS-block mineral from the Darai-Pioz alkaline massif, Tian-Shan mountains, Tajikistan. The mineral is of hydrothermal origin. It occurs as aggregates (up to 1.5 cm long) of acicular crystals 0.1–1.0 mm thick, and as separate elongated columnar, flattened-prismatic crystals up to 1 cm long with rectangular or rhombic sections up to 0.5 mm across. Associated minerals are quartz, aegirine, microcline, neptunite, pectolite, calcite, eudialyte-group minerals, fluorite, titanite, turkestanite, kupletskite, galena, albite and pyrochlore-group minerals. Crystals are transparent and colourless to occasionally white, with a vitreous lustre. Rinkite-(Y) has a white streak, uneven, conchoidal fracture and does not fluoresce under a cathode or ultraviolet light. Cleavage is very good on {100}, no parting was observed, Mohs hardness is ~5, and it is brittle, Dmeas. = 3.44(2) g/cm3, Dcalc. = 3.475 g/cm3. It is biaxial (+) with refractive indices (λ = 590 nm) α = 1.662(2), β = 1.666(2), γ = 1.685(5); 2Vmeas. = 50(3) and 2Vcalc. = 49.7°. It is nonpleochroic. Rinkite-(Y) is monoclinic, space group P21/c, a = 7.3934(5), b = 5.6347(4), c = 18.713(1) Å, β = 101.415(2)° and V = 764.2(2) Å3. The six strongest reflections in the X-ray powder diffraction data [d(Å), I, (hkl)] are: 3.057, 100, (006,
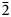 $\bar{2}$12, 210); 2.688, 28, (016); 9.18, 24, (002); 2.929, 17, (
$\bar{2}$12, 210); 2.688, 28, (016); 9.18, 24, (002); 2.929, 17, (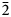 $\bar{2}$13, 211); 3.559, 15, (104, 014) and 2.783, 14, (021). The empirical formula calculated on 18 (O + F) is Na2.11(Ca3.74Sr0.03Mn0.03)Σ3.80(Y0.50Nd0.16Ce0.16Gd0.07Dy0.06Sm0.05Pr0.03La0.03
$\bar{2}$13, 211); 3.559, 15, (104, 014) and 2.783, 14, (021). The empirical formula calculated on 18 (O + F) is Na2.11(Ca3.74Sr0.03Mn0.03)Σ3.80(Y0.50Nd0.16Ce0.16Gd0.07Dy0.06Sm0.05Pr0.03La0.03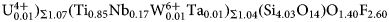 ${\rm U}_{0.01}^{{\rm 4 + }} {\rm )}_{\Sigma 1.07}{\rm (T}{\rm i}_{0.85}{\rm N}{\rm b}_{0.17}{\rm W}^{6+}_{0.01}{\rm T}{\rm a}_{0.01}{\rm )}_{\Sigma 1.04}\left( {{\rm S}{\rm i}_{4.03}{\rm O}_{14}} \right){\rm O}_{1.40}{\rm F}_{2.60}$ with Z = 2. The ideal formula is Na2Ca4YTi(Si2O7)2OF3. The crystal structure was refined on a twinned crystal to R1 = 4.59% on the basis of 1489 unique reflections (F > 4σF) and is a framework of TS (Titanium-Silicate) blocks. The TS block consists of HOH sheets (H – heteropolyhedral, O – octahedral) parallel to (100). In the O sheet, the Ti-dominant [6]MO1 site ideally gives 1 Ti apfu. The [8]MO2 and [6]MO3 sites are ideally occupied by Na and (NaCa) apfu. In the H sheet, the [7]MH site is occupied by Ca1.13Y0.50REE0.37, (REE = rare-earth element), ideally (CaY), <MH–φ> = 2.415 Å and the [7]AP site is occupied by Ca1.81REE0.19, ideally Ca2, <AP–φ> = 2.458 Å. The MH + AP sites ideally give (Ca3Y) apfu. The MH and AP polyhedra and Si2O7 groups constitute the H sheet. Linkage of H and O sheets via common vertices of MH and AP polyhedra and Si2O7 groups with MO1–3 polyhedra results in a TS block. The TS block in rinkite-(Y) exhibits linkage 1 and stereochemistry typical for the rinkite group (Ti = 1 apfu) of the seidozerite supergroup. For rinkite-(Y), the ideal structural formula of the form AP2MH2MO4(Si2O7)2
${\rm U}_{0.01}^{{\rm 4 + }} {\rm )}_{\Sigma 1.07}{\rm (T}{\rm i}_{0.85}{\rm N}{\rm b}_{0.17}{\rm W}^{6+}_{0.01}{\rm T}{\rm a}_{0.01}{\rm )}_{\Sigma 1.04}\left( {{\rm S}{\rm i}_{4.03}{\rm O}_{14}} \right){\rm O}_{1.40}{\rm F}_{2.60}$ with Z = 2. The ideal formula is Na2Ca4YTi(Si2O7)2OF3. The crystal structure was refined on a twinned crystal to R1 = 4.59% on the basis of 1489 unique reflections (F > 4σF) and is a framework of TS (Titanium-Silicate) blocks. The TS block consists of HOH sheets (H – heteropolyhedral, O – octahedral) parallel to (100). In the O sheet, the Ti-dominant [6]MO1 site ideally gives 1 Ti apfu. The [8]MO2 and [6]MO3 sites are ideally occupied by Na and (NaCa) apfu. In the H sheet, the [7]MH site is occupied by Ca1.13Y0.50REE0.37, (REE = rare-earth element), ideally (CaY), <MH–φ> = 2.415 Å and the [7]AP site is occupied by Ca1.81REE0.19, ideally Ca2, <AP–φ> = 2.458 Å. The MH + AP sites ideally give (Ca3Y) apfu. The MH and AP polyhedra and Si2O7 groups constitute the H sheet. Linkage of H and O sheets via common vertices of MH and AP polyhedra and Si2O7 groups with MO1–3 polyhedra results in a TS block. The TS block in rinkite-(Y) exhibits linkage 1 and stereochemistry typical for the rinkite group (Ti = 1 apfu) of the seidozerite supergroup. For rinkite-(Y), the ideal structural formula of the form AP2MH2MO4(Si2O7)2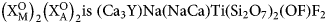 $ \left( {{\rm X}_{\rm M}^{\rm O} } \right)_2\left( {{\rm X}_{\rm A}^{\rm O} } \right)_2{\rm is }\;\left( {{\rm C}{\rm a}_3{\rm Y}} \right){\rm Na}\left( {{\rm NaCa}} \right){\rm Ti}\left( {{\rm S}{\rm i}_2{\rm O}_7} \right)_2\left( {{\rm OF}} \right){\rm F}_2 $ with Z = 2. The mineral is named rinkite-(Y) as it is structurally identical to rinkite-(Ce) and Y is the dominant rare-earth element.
$ \left( {{\rm X}_{\rm M}^{\rm O} } \right)_2\left( {{\rm X}_{\rm A}^{\rm O} } \right)_2{\rm is }\;\left( {{\rm C}{\rm a}_3{\rm Y}} \right){\rm Na}\left( {{\rm NaCa}} \right){\rm Ti}\left( {{\rm S}{\rm i}_2{\rm O}_7} \right)_2\left( {{\rm OF}} \right){\rm F}_2 $ with Z = 2. The mineral is named rinkite-(Y) as it is structurally identical to rinkite-(Ce) and Y is the dominant rare-earth element.
Subaerial sulfate mineral formation related to acid aerosols at the Zhenzhu Spring, Tengchong, China
-
- Published online by Cambridge University Press:
- 14 January 2019, pp. 381-392
-
- Article
- Export citation
Pampaloite, AuSbTe, a new mineral from Pampalo gold mine, Finland
-
- Published online by Cambridge University Press:
- 04 July 2018, pp. 393-400
-
- Article
- Export citation
-
Pampaloite, AuSbTe, is a new mineral discovered in the Pampalo gold mine, 65 km east of Joensuu, Finland. It forms anhedral grains (up to ~20 μm) intergrown with gold, frohbergite and altaite. Pampaloite is brittle and has a metallic lustre. Values of VHN25 lie between 245 and 295 kg/mm2, with a mean value of 276 kg/mm2, corresponding to a Mohs hardness of ~4–5 (measured on synthetic material). In plane-polarised light, pampaloite is white with medium to strong bireflectance, weak reflectance pleochroism from slightly pinkish brown to slightly bluish white (only visible in grains of synthetic material containing multiple orientations), and strong anisotropy, with blue to light brown rotation tints; it exhibits no internal reflections. Reflectance values of pampaloite in air (R1,R2 in %) are: 60.0, 62.5 at 470 nm, 62.5, 64.8 at 546 nm, 63.2, 65.6 at 589 nm and 63.7, 66.0 at 650 nm. Ten electron-microprobe analyses of natural pampaloite give an average composition: Au 44.13, Sb 27.44 and Te 28.74, total 100.31 wt.%, corresponding to the empirical formula Au1.00Sb1.00Te1.00 based on 3 atoms; the average of eleven analyses on synthetic pampaloite is: Au 44.03, Sb 27.26, and Te 29.08, total 100.38 wt.%, corresponding to Au0.99Sb1.00Te1.01. The density, calculated on the basis of the empirical formula, is 9.33 g/cm3.The mineral is monoclinic, space group C2/c, with a = 11.947(3), b = 4.481(1) Å, c = 12.335(3) Å, β = 105.83(2)°, V = 635.3(3) Å3 and Z = 8. The crystal structure was solved and refined from the single-crystal X-ray-diffraction data of synthetic AuSbTe. The pampaloite crystal structure can be considered as a monoclinic derivative of the CdI2 structure composed of [AuTe3Sb3] octahedra. The strongest lines in the powder X-ray diffraction pattern of synthetic pampaloite [d in Å (I) (hkl)] are: 4.846(24)(
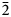 $\bar{2}$02), 3.825(18)(111), 2.978(100)(
$\bar{2}$02), 3.825(18)(111), 2.978(100)(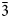 $\bar{3}$11), 2.968(50)(004), 2.242(25)(020), 2.144(55)(313), 2.063(33)(
$\bar{3}$11), 2.968(50)(004), 2.242(25)(020), 2.144(55)(313), 2.063(33)(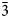 $\bar{3}$15) and 1.789(18)(024).
$\bar{3}$15) and 1.789(18)(024).
Mineralogy of the baotite-bearing Gundrapalli lamproite, Nalgonda district, Telangana, India
-
- Published online by Cambridge University Press:
- 31 January 2019, pp. 401-411
-
- Article
- Export citation
Ferri-fluoro-katophorite from Bear Lake diggings, Bancroft area, Ontario, Canada: a new species of amphibole, ideally Na(NaCa)(Mg4Fe3+)(Si7Al)O22F2
-
- Published online by Cambridge University Press:
- 02 July 2018, pp. 413-417
-
- Article
- Export citation
-
Ferri-fluoro-katophorite is the second species characterised involving the rootname katophorite in the sodium–calcium subgroup of the amphibole supergroup. The mineral and its name were approved by the International Mineralogical Association Commission on New Minerals, Nomenclature and Classification, IMA2015-096. It was found in the Bear Lake diggings, Bancroft area, Ontario, Canada, where coarse euhedral crystals of amphibole, phlogopite, sanidine solid-solution (now coarsely exsolved to microcline perthite), titanite, augite, zircon and fluorapatite crystallised from a low-viscosity silicocarbonatitic magma of crustal origin. Greenish grey prismatic crystals of ferri-fluoro-katophorite generally protrude from the walls into a body of coarsely crystalline calcite, but they also occur away from the walls, completely enclosed by calcite. The empirical formula derived from electron microprobe analysis and single-crystal structure refinement is: A(Na0.55K0.32)Σ0.87B(Na0.79Ca1.18Mn2+0.03)Σ2.00C(Mg3.29Mn2+0.02Fe2+1.19Fe3+0.31Al0.09Ti4+0.08Li0.02)Σ5.00T(Si7.39Al0.61)Σ8.00O22W[F1.23 (OH)0.77]Σ2.00. Ferri-fluoro-katophorite is biaxial (–), with α = 1.640(2), β = 1.652(2), γ = 1.658(2), 2Vmeas. = 68.9(2)° and 2Vcalc.. = 70.1°. The unit-cell parameters are a = 9.887(3), b = 18.023(9), c = 5.292(2) Å, β = 104.66(3)°, V = 912.3(6) Å3, Z = 2 and space group C2/m. The strongest ten lines in the powder X-ray pattern [d values (in Å) I (hkl)] are: 2.708, 100, (151); 2.388, 74, (131); 3.139, 72, (310); 8.449, 69, (110); 2.540, 65, (
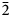 $\bar{2}$02); 2.591, 53, (061); 2.739, 47, (
$\bar{2}$02); 2.591, 53, (061); 2.739, 47, (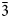 $\bar{3}$31); 2.165, 45, (261); 3.279, 44, (
$\bar{3}$31); 2.165, 45, (261); 3.279, 44, (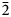 $\bar{2}$40); 2.341, 43, (
$\bar{2}$40); 2.341, 43, (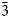 $\bar{3}$51).
$\bar{3}$51).
Discovery of Se-rich canfieldite, Ag8Sn(S,Se)6, from the Shuangjianzishan Ag–Pb–Zn deposit, NE China: A multimethodic chemical and structural study
-
- Published online by Cambridge University Press:
- 08 October 2018, pp. 419-426
-
- Article
- Export citation
-
During a study of the ore minerals belonging to the recently discovered Shuangjianzishan Ag–Pb–Zn deposit in NE China, we have discovered exceptional selenium enrichment in canfieldite (up to 11.6 wt.% of Se). Incorporation of Se into canfieldite has been investigated by an integrated approach using field emission scanning electron microscopy, electron microprobe and single-crystal X-ray diffraction. Canfieldite has been identified as one of the dominant Ag-bearing ore minerals in the studied deposit, which occurs mostly in slate-hosted vein type Ag–Pb–Zn ore bodies. Selenium is either homogeneously or, remarkably, heterogeneously distributed in the different canfieldite fragments studied. Chemical variations of Se are mostly attributable to a series of retrograde reactions resulting in diverse decomposition and exsolution of primary phases during cooling, or alternatively, related to influxes of Se-rich fluids during the formation of canfieldite. To evaluate the effects of the Se-for-S substitution in the structure, a crystal of Se-rich canfieldite [Ag7.98Sn1.02(S4.19Se1.81)Σ6.00] was investigated. The unit-cell parameters are: a = 10.8145(8) Å and V = 1264.8(3) Å3. The structure was refined in the space group F
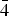 $\bar{4}$3m to R1 = 0.0315 for 194 independent reflections, with 20 parameters. The crystal structure of Se-rich canfieldite was found to be topologically identical to that of pure canfieldite. If the short Ag–Ag contacts are ignored (due to the disorder), the two Ag atoms in the structure can be considered as three-fold (Ag1) and four-fold (Ag2) coordinated. Tin adopts a regular tetrahedral coordination. As in the case of Te-rich canfieldite, the refinement of the site-occupancy factor indicates that Se is disordered over the three anion positions.
$\bar{4}$3m to R1 = 0.0315 for 194 independent reflections, with 20 parameters. The crystal structure of Se-rich canfieldite was found to be topologically identical to that of pure canfieldite. If the short Ag–Ag contacts are ignored (due to the disorder), the two Ag atoms in the structure can be considered as three-fold (Ag1) and four-fold (Ag2) coordinated. Tin adopts a regular tetrahedral coordination. As in the case of Te-rich canfieldite, the refinement of the site-occupancy factor indicates that Se is disordered over the three anion positions.
Middlebackite, a new Cu oxalate mineral from Iron Monarch, South Australia: Description and crystal structure
-
- Published online by Cambridge University Press:
- 02 July 2018, pp. 427-433
-
- Article
- Export citation
-
Middlebackite is a new supergene mineral formed in the upper levels of the Iron Monarch quarry, South Australia. It occurs as aggregates of blue, prismatic crystals up to 0.3 mm across comprising individual crystals up to 0.05 mm in length associated with atacamite and mottramite. Crystals are translucent with a vitreous lustre and have a pale blue streak. Middlebackite is brittle with one perfect cleavage and uneven fracture. Mohs hardness is ~2. The calculated density is 3.64 g cm–3. Crystals are biaxial (+) with α = 1.663(4), β = 1.748(4) and γ = 1.861(4) (measured in white light). The calculated 2V is 86.7°. Pleochroism is X = colourless, Y = very pale blue and Z = dark sky blue; Z > Y > X. The empirical formula unit, based on six oxygen atoms per formula unit is Cu2.00(C2O4)Cl0.02(OH)1.98. Middlebackite is monoclinic, space group P21/c with a = 7.2597(15), b = 5.7145(11), c = 5.6624(11) Å, β = 104.20(3)°, V = 227.73(8) Å3 and Z = 2. The five strongest lines in the powder X-ray diffraction pattern are [d(Å), (I), (hkl)]: 7.070 (16) (100), 3.739 (100) (11
 $\bar{1}$), 2.860 (18) (020), 2.481 (12) (12
$\bar{1}$), 2.860 (18) (020), 2.481 (12) (12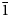 $\bar{1}$) and 2.350 (9) (300). The crystal structure was refined from synchrotron single-crystal X-ray diffraction data to R1 = 0.0341 for 596 observed reflections with F0 > 4σ(F0). The structure is based on sheets of edge- and corner-sharing octahedra parallel to the bc plane. Sheets link in the a direction via oxalate anions.
$\bar{1}$) and 2.350 (9) (300). The crystal structure was refined from synchrotron single-crystal X-ray diffraction data to R1 = 0.0341 for 596 observed reflections with F0 > 4σ(F0). The structure is based on sheets of edge- and corner-sharing octahedra parallel to the bc plane. Sheets link in the a direction via oxalate anions.
Substitution mechanisms in In-, Au-, and Cu-bearing sphalerites studied by X-ray absorption spectroscopy of synthetic compounds and natural minerals
-
- Published online by Cambridge University Press:
- 04 March 2019, pp. 435-451
-
- Article
- Export citation
New arsenate minerals from the Arsenatnaya fumarole, Tolbachik volcano, Kamchatka, Russia. IX. Arsenatrotitanite, NaTiO(AsO4)
-
- Published online by Cambridge University Press:
- 04 July 2018, pp. 453-458
-
- Article
- Export citation
-
The new durangite-group mineral arsenatrotitanite, ideally NaTiO(AsO4), was found in the Arsenatnaya fumarole at the Second scoria cone of the Northern Breakthrough of the Great Tolbachik Fissure Eruption, Tolbachik volcano, Kamchatka, Russia. It is associated with orthoclase, tenorite, hematite, johillerite, bradaczekite, badalovite, calciojohillerite, arsmirandite, tilasite, svabite, cassiterite, pseudobrookite, rutile, sylvite, halite, aphthitalite, langbeinite and anhydrite. Arsenatrotitanite occurs as prismatic, tabular, lamellar or acicular crystals up to 0.3 mm × 0.8 mm × 2 mm. They are separated or combined in open-work aggregates up to 2 mm across or interrupted crusts up to 2 mm × 5 mm in area and up to 0.3 mm thick. Arsenatrotitanite is transparent, brownish red to pale pinkish-reddish or almost colourless, with vitreous lustre. It is brittle and the Mohs’ hardness is ~5½. Cleavage is perfect on {110} and the fracture is stepped. Dcalc is 3.950 g cm–3. Arsenatrotitanite is optically biaxial (+), α = 1.825(5), β = 1.847(6), γ = 1.896(6) (589 nm) and 2Vmeas. = 70(5)°. Chemical composition (wt.%, electron-microprobe) is: Na2O 12.26, CaO 3.10, Al2O3 4.39, Fe2O3 9.57, TiO2 17.11, SnO2 1.03, As2O5 50.17, F 3.29, O = F –2.39, total 99.53. The empirical formula based on 5 (O + F) apfu is (Na0.91Ca0.13)Σ1.04(Ti0.49Fe3+0.27Al0.20Sn0.02)Σ0.98(As1.00O4.00)(O0.60F0.40). Arsenatrotitanite is monoclinic, C2/c, a = 6.6979(3), b = 8.7630(3), c = 7.1976(3) Å, β = 114.805(5)°, V = 383.48(3) Å3 and Z = 4. The strongest reflections of the powder X-ray diffraction (XRD) pattern [d,Å(I)(hkl)] are: 4.845(89)(
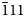 $\bar{1} {11}}$), 3.631(36)(021), 3.431(48)(111), 3.300(100)(
$\bar{1} {11}}$), 3.631(36)(021), 3.431(48)(111), 3.300(100)(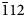 $\bar{1} {12}}$), 3.036(100)(200), 2.627(91)(130) and 2.615(57)(022). The crystal structure was solved from single-crystal XRD data with R = 1.76%. Arsenatrotitanite belongs to the titanite/durangite structure type. It is named as an arsenate of sodium (natrium in Latin) and titanium isostructural with titanite.
$\bar{1} {12}}$), 3.036(100)(200), 2.627(91)(130) and 2.615(57)(022). The crystal structure was solved from single-crystal XRD data with R = 1.76%. Arsenatrotitanite belongs to the titanite/durangite structure type. It is named as an arsenate of sodium (natrium in Latin) and titanium isostructural with titanite.
The crystal structure of Ni-rich gordaite–thérèsemagnanite from Cap Garonne, France
-
- Published online by Cambridge University Press:
- 14 March 2019, pp. 459-463
-
- Article
- Export citation
Potassic-magnesio-arfvedsonite, KNa2(MgFe2+Fe3+)5Si8O22(OH)2: mineral description and crystal chemistry
-
- Published online by Cambridge University Press:
- 02 July 2018, pp. 465-472
-
- Article
- Export citation
-
The complete mineral description of potassic-magnesio-arfvedsonite, a recently approved (IMA2016-083) new species of the amphibole supergroup is provided using electron microprobe analysis (EMPA), laser ablation inductively coupled plasma mass spectrometry, single-crystal structure refinement, Mössbauer and Raman spectroscopy, as well as measurement of optical and physical properties. The holotype material was found in syenitic and granitic dyke rocks in association with quartz, potassium feldspar and aegirine–augite from the Buhovo–Seslavtsi pluton, Bulgaria. Potassic-magnesio-arfvedsonite is monoclinic C2/m, with unit-cell parameters: a = 9.9804(11), b = 18.0127(19), c = 5.2971(6) Å, β = 104.341(2)° and V = 922.61 Å3. In transmitted plane-polarised light (λ = 590 cm–1), potassic-magnesio-arfvedsonite is pleochroic: X = yellow pale-green, Y = green and Z = dark-violet brown. It is biaxial (–), α = 1.645(2), β = 1.655(2), γ = 1.660(2) and 2Vmeas. = 60° and 2Vcalc. = 70°. The empirical unit formula obtained from EMPA and structure refinement is A(K0.86Na0.0.08)0.94B(Na1.74Ca0.25 Mn2+0.01)2.00C(Mg2.67Fe2+1.42Fe3+0.76Ti0.12Mn2+0.03)5.00TSi8O22W(OH1.58F0.22O0.20)2.00. The Fe3+/Fetot ratio (0.35) is consistent with both the Mössbauer spectra and the single-crystal structure refinement. The 10 strongest X-ray powder reflections [d values (in A°), I, (hkl)] are: 8.519, 80.5, (110); 3.402, 67.3, (131); 3.295, 41.0, (240); 3.173, 65.0, (310); 2.752, 35.6, (
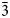 $\bar{3}$31); 2.715, 100.0 (151); 2.591, 44.1, (061); 2.542, 73.2, (
$\bar{3}$31); 2.715, 100.0 (151); 2.591, 44.1, (061); 2.542, 73.2, (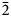 $\bar{2}$02); 2.348, 38.5, (
$\bar{2}$02); 2.348, 38.5, (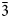 $\bar{3}$51); 2.174, 42.0, (261). Potassic-magnesio-arfvedsonite is the product of strongly peralkaline and potassic (perpotassic) magma compositions. Trace-element analysis shows that this amphibole did not exert significant control on trace-element distribution in the crystallising peralkaline magma.
$\bar{3}$51); 2.174, 42.0, (261). Potassic-magnesio-arfvedsonite is the product of strongly peralkaline and potassic (perpotassic) magma compositions. Trace-element analysis shows that this amphibole did not exert significant control on trace-element distribution in the crystallising peralkaline magma.
Discreditation of the pyroxenoid mineral name ‘marshallsussmanite’ with a reinstatement of the name schizolite, NaCaMnSi3O8(OH)
-
- Published online by Cambridge University Press:
- 22 April 2019, pp. 473-478
-
- Article
- Export citation
NEWSLETTER 49
IMA Commission on New Minerals, Nomenclature and Classification (CNMNC)
New minerals and nomenclature modifications approved in 2019
-
- Published online by Cambridge University Press:
- 24 May 2019, pp. 479-483
-
- Article
-
- You have access
- HTML
- Export citation
Front Cover (OFC, IFC) and matter
MGM volume 83 issue 3 Cover and Front matter
-
- Published online by Cambridge University Press:
- 09 July 2019, pp. f1-f2
-
- Article
-
- You have access
- Export citation
Back Cover (IBC, OBC) and matter
MGM volume 83 issue 3 Cover and Back matter
-
- Published online by Cambridge University Press:
- 09 July 2019, pp. b1-b2
-
- Article
-
- You have access
- Export citation