


INTRODUCTION
The leishmaniases, including the cutaneous and visceral forms (CL and VL) of the disease, are amongst the most devastating infectious diseases of our time. The most recent estimates report about 0·2–0·4 million VL cases and 0·7–1·2 million CL cases each year (Alvar et al. Reference Alvar, Velez, Bern, Herrero, Desjeux, Cano, Jannin and Den Boer2012). Leishmaniasis management includes multiple difficulties, such as vector and animal reservoir control, but probably the most challenging of them all is treatment.
Over 20 species within the genus are implicated as the aetiological agents of the human leishmaniases in a spectrum of clinical manifestations. The visceral presentation is the most severe form and leads to death almost invariably if untreated. The aetiological agents for VL are Leishmania donovani and Leishmania infantum (in the Old World) and L. infantum chagasi (in the New World). The cutaneous or tegumentary form can be categorized as localized, diffuse, disseminated and mucocutaneous. Apart from the localized form, which can heal spontaneously after a chronic progression, all the other forms are severe, mutilating and respond poorly to the available therapeutic options. The most common agents of CL in the Old World are Leishmania major and Leishmania tropica, but L. infantum and Leishmania aethiopica are also detected in cutaneous patients. In the Americas, several species of the subgenus Viannia are responsible for human disease, the most common of them being Leishmania braziliensis. In a more restricted geographical pattern, Leishmania guyanensis, Leishmania panamensis and Leishmania peruviana are also common, while Leishmania shawi, Leishmania naiffi, Leishmania lainsoni and Leishmania lindenbergi are diagnosed less often in the Amazon region. In the Americas, species of the Leishmania subgenus, such as L. amazonensis, L. mexicana and L. venezuelensis are also agents of CL. Post kalazar dermal leishmaniasis, a cutaneous presentation of L. donovani infection, often arises after treatment of the visceral disease, more frequently in East Africa and India.
According to the Global Burden of Disease Study 2013, from 2005 to 2013, the number of disability adjusted life years (DALYs) as a result of leishmaniasis increased 15% globally (Murray et al. Reference Murray, Foreman, Abbasoglu Ozgoren, Abd-Allah, Abera, Aboyans, Abraham, Abubakar, Abu-Raddad, Abu-Rmeileh, Achoki, Ackerman, Ademi, Adou, Adsuar, Afshin, Agardh, Alam, Alasfoor, Albittar, Alegretti, Alemu, Alfonso-Cristancho, Alhabib, Ali, Alla, Allebeck, Almazroa and Alsharif2016). When the clinical forms were considered, the rise in DALYs was 14·4% for VL and 175·1% for CL. In 33 out of 50 countries with the highest CL incidence, a rise in age-adjusted DALYs was observed between 1990 and 2013. The greatest increases were found in Sudan, Iraq, Gambia, Benin, Venezuela, Paraguay, Ecuador and Honduras (Karimkhani et al. Reference Karimkhani, Wanga, Coffeng, Naghavi, Dellavalle and Naghavi2016).
The factors behind this increase in incidence derive from climate change, dislocation of populations from endemic rural areas to new agricultural or urban zones (Gadisa et al. Reference Gadisa, Tsegaw, Abera, Elnaiem, Den Boer, Aseffa and Jorge2015), as well as the installation of conflict zones (Berry and Berrang-Ford, Reference Berry and Berrang-Ford2016).
Large epidemics of VL and CL have been registered in conflict areas such as Sudan, Iraq, Afghanistan and Syria (Kreutzer et al. Reference Kreutzer, Grogl, Neva, Fryauff, Magill and Aleman-Munoz1993; Hyams et al. Reference Hyams, Riddle, Trump and Graham2001; Collin et al. Reference Collin, Davidson, Ritmeijer, Keus, Melaku, Kipngetich and Davies2004; Berry and Berrang-Ford, Reference Berry and Berrang-Ford2016) not only resulting in large mortality rates, but also in the emergence of new foci of transmission (Alawieh et al. Reference Alawieh, Musharrafieh, Jaber, Berry, Ghosn and Bizri2014; Sharara and Kanj, Reference Sharara and Kanj2014; Inci et al. Reference Inci, Ozturk, Mulayim, Ozyurt, Alatas and Inci2015), sometimes with unexpected clinical presentations (Kreutzer et al. Reference Kreutzer, Grogl, Neva, Fryauff, Magill and Aleman-Munoz1993).
Geographical expansion outside of conflict zones has been registered as well. Factors most likely associated with these expansions are human migration and climate change.
LEISHMANIASIS TREATMENT
There are only a few drugs available for leishmaniasis treatment and most of them have been in use for quite some time (Table 1). Curiously, the approaches to leishmaniasis treatment are mostly very broad and not species-specific, in spite of all the clinical diversity and of the great deal of correlation between species-specific determinants of clinical disease patterns. The specific choice of drug or length of treatment generally does not take into account the particularities of the different aetiological agents, because there are no easily accessible methods for species-specific diagnosis, while the co-existence of different species in the same geographical area is common. Therefore, in general, researchers and clinicians have not been able to ascertain whether species-specific schemes of treatment are more appropriate than an overall strategy.
Table 1. Summary of current antileishmanial drugs
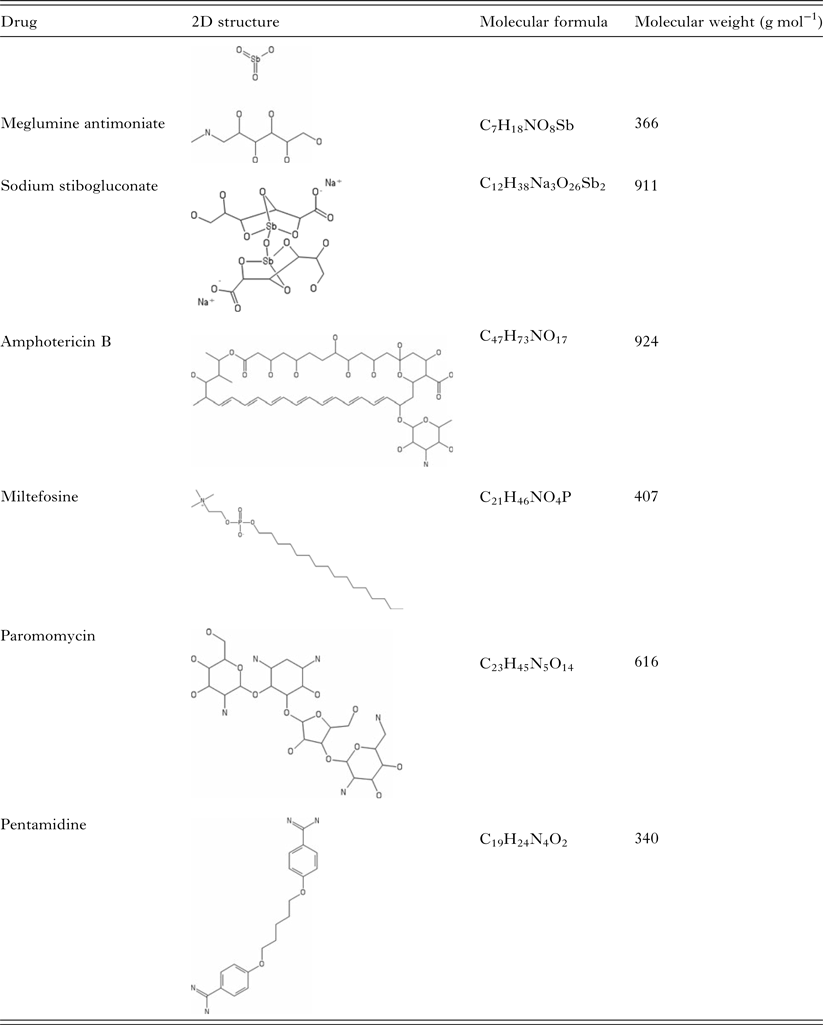
Pentavalent antimonials were the first class of drugs applied to leishmaniasis treatment. Together with amphotericin B, pentamidine, miltefosine and paromomycin, they constitute the resources available for leishmaniasis chemotherapy.
The initial observations that led to the global use of antimonials and amphotericin B as the mainstay of leishmaniasis therapy were made by Brazilians. Gaspar Vianna suggested trivalent antimonials could be useful to treat the first cases of CL diagnosed in São Paulo, in 1914 (Vianna, Reference Vianna1914). Pentavalent antimonials were developed during the 1920s, in India, and contributed to decrease considerably the treatment toxicity (Marsden, Reference Marsden1985). In the late 1950s, amphotericin B was described by Furtado and Lacaz as an alternative to the management of mucocutaneous leishmaniasis patients who frequently relapsed or did not respond to antimonial drugs (Sampaio et al. Reference Sampaio, Godoy, Paiva, Dillon and Da1960) and was soon applied to the treatment of severe VL (Prata, Reference Prata1963).
For decades, treatment schemes with pentavalent antimonials were used as the first choice to treat VL all over the world, in spite of the parenteral route of administration, high toxicity and cost. However, an increase in the number of therapeutic failures was noted from the 1980s in India and doses required to lead to clinical cure had to be slowly increased (Thakur, Reference Thakur1984; Thakur et al. Reference Thakur, Kumar, Singh, Sharma, Prasad, Singh, Dhawan and Achari1984). WHO recommendations of pentavalent antimonial doses of 10 mg SbV kg−1 day−1 for 20 days in 1984 (WHO, 1984) were raised to 20 mg kg−1 day−1 and the period of administration to 28–30 days. The latest WHO recommendations have demoted the use of pentavalent antimonials as single drugs to second or lower choices for the treatment of VL in all endemic regions, making liposomal amphotericin B the first recommended drug in most areas (WHO, 2010). However, due to the constraints on liposomal amphotericin use, mostly related to price and availability, pentavalent antimonials are still largely used as the first option for VL in some endemic countries.
Amphotericin B deoxycholate and liposomal amphotericin B have been in use for the treatment of VL in the presence of concomitant illnesses or risk factors such as age extremes. Amphotericin B deoxycholate is highly toxic and has to be administered by slow intravenous infusion over 4 to 6 h, requiring hospitalization. Lipid formulations of amphotericin B display similar efficacy with significantly lower toxicity (Szoka et al. Reference Szoka, Milholland and Barza1987; Botero Aguirre and Restrepo Hamid, Reference Botero Aguirre and Restrepo Hamid2015). These formulations are now recommended as the first choice drug, with a good level of evidence for efficacy (Balasegaram et al. Reference Balasegaram, Ritmeijer, Lima, Burza, Ortiz Genovese, Milani, Gaspani, Potet and Chappuis2012). However, its cost is still prohibitive in several endemic regions.
Another drug available for VL treatment is miltefosine (hexadecylphosphocholine), a phosphatidylcholine analogue, initially developed as an antineoplastic drug, that showed good activity against L. donovani (Croft et al. Reference Croft, Neal, Pendergast and Chan1987; Kuhlencord et al. Reference Kuhlencord, Maniera, Eibl and Unger1992) and was shown to be very effective for the treatment of VL in India, country that was the first to approve its clinical use in 2002 (Sundar et al. Reference Sundar, Jha, Thakur, Engel, Sindermann, Fischer, Junge, Bryceson and Berman2002). The main drawbacks of miltefosine are its teratogenic potential and the long half-life that can lead to the selection of drug-resistant lines (Dorlo et al. Reference Dorlo, Van Thiel, Huitema, Keizer, De Vries, Beijnen and De Vries2008). However, it benefits from being administered orally. Side-effects are mainly gastrointestinal and, although severe in some patients, they are generally considered manageable. Efficacy of miltefosine for the treatment of L. donovani infections in Africa is still under study (Omollo et al. Reference Omollo, Alexander, Edwards, Khalil, Younis, Abuzaid, Wasunna, Njoroge, Kinoti, Kirigi, Dorlo, Ellis, Balasegaram and Musa2011), but seems to be low in populations with a high prevalence of HIV co-infection (Ritmeijer et al. Reference Ritmeijer, Veeken, Melaku, Leal, Amsalu, Seaman and Davidson2001). Miltefosine was used for some years as the first line therapy for VL in parts of India, but the emergence of a relevant relapse rate has moved miltefosine down the line as an option mainly for combination therapy (Sundar et al. Reference Sundar, Singh, Rai, Prajapati, Singh, Ostyn, Boelaert, Dujardin and Chakravarty2012; Rijal et al. Reference Rijal, Ostyn, Uranw, Rai, Bhattarai, Dorlo, Beijnen, Vanaerschot, Decuypere, Dhakal, Das, Karki, Singh, Boelaert and Dujardin2013).
The antileishmanial activity of paromomycin, an aminoglycoside antibiotic, was first described in the 1960s (Neal et al. Reference Neal, Gonzalez, Pinart, Rengifo-Pardo, Macaya, Alvar, Tweed, Gonzalez, Pinart, Reveiz and Alvar1968). It was proposed as a topical agent for CL treatment (El-On et al. Reference El-On, Livshin, Paz and Weinrauch1985). Paromomycin used by the parenteral route was later suggested as an alternative for VL (Chunge et al. Reference Chunge, Owate, Pamba and Donno1990). Clinical trials carried on in India indicated that paromomycin was effective for the treatment of VL (Sundar et al. Reference Sundar, Jha, Thakur, Sinha and Bhattacharya2007). However, the same response was not found in East Africa, where trials demonstrated geographical heterogeneity and lower cure rates after treatment with paromomycin than with antimonials (Hailu et al. Reference Hailu, Musa, Wasunna, Balasegaram, Yifru, Mengistu, Hurissa, Hailu, Weldegebreal, Tesfaye, Makonnen, Khalil, Ahmed, Fadlalla, El-Hassan, Raheem, Mueller, Koummuki, Rashid, Mbui, Mucee, Njoroge, Manduku, Musibi, Mutuma, Kirui, Lodenyo, Mutea, Kirigi and Edwards2010; Musa et al. Reference Musa, Younis, Fadlalla, Royce, Balasegaram, Wasunna, Hailu, Edwards, Omollo, Mudawi, Kokwaro, El-Hassan and Khalil2010, Reference Musa, Khalil, Hailu, Olobo, Balasegaram, Omollo, Edwards, Rashid, Mbui, Musa, Abuzaid, Ahmed, Fadlalla, El-Hassan, Mueller, Mucee, Njoroge, Manduku, Mutuma, Apadet, Lodenyo, Mutea, Kirigi, Yifru, Mengistu, Hurissa, Hailu, Weldegebreal, Tafes and Mekonnen2012). On the other hand, shorter courses of the association of paromomycin and antimonial were shown to be as effective as the long course of antimonial alone (Musa et al. Reference Musa, Khalil, Hailu, Olobo, Balasegaram, Omollo, Edwards, Rashid, Mbui, Musa, Abuzaid, Ahmed, Fadlalla, El-Hassan, Mueller, Mucee, Njoroge, Manduku, Mutuma, Apadet, Lodenyo, Mutea, Kirigi, Yifru, Mengistu, Hurissa, Hailu, Weldegebreal, Tafes and Mekonnen2012; Atia et al. Reference Atia, Mumina, Tayler-Smith, Boulle, Alcoba, Elhag, Alnour, Shah, Chappuis, Van Griensven and Zachariah2015), representing an advance in terms of therapeutic options. This scheme of association is now part of the WHO recommendations in these areas.
Historically, CL treatment has also made use of pentavalent antimonials as a first choice drug (Gonzalez et al. Reference Gonzalez, Pinart, Reveiz and Alvar2008, Reference Gonzalez, Pinart, Rengifo-Pardo, Macaya, Alvar, Tweed, Gonzalez, Pinart, Reveiz and Alvar2009). The toxicity and parenteral routes of administration, together with the duration of treatment stimulated the use of alternative routes and drugs to treat the cutaneous form of the disease. For example, in benign cases (one or few small lesions in non-critical body areas) caused by L. major, local options such as topical paromomycin, termotherapy, cryotherapy or intralesional antimonials have been applied.
The heterogeneity of clinical pictures, disease progression, complications and response to treatment of the various aetiological agents of CL makes the choice of therapy much less homogeneous than for VL. Another complicating factor is that most clinical trials conducted so far on the treatment of CL and mucocutaneous leishmaniasis, including studies on antimonials as well as other agents, were not well designed and reported, leading to inconclusive results (Gonzalez et al. Reference Gonzalez, Pinart, Reveiz and Alvar2008, Reference Gonzalez, Pinart, Rengifo-Pardo, Macaya, Alvar, Tweed, Gonzalez, Pinart, Reveiz and Alvar2009).
While topical agents are applied to L. major infections, New World CL is generally more severe with the potential of metastasis or progression to severe forms (diffuse or disseminated as well as mucocutaneous leishmaniasis) (Jirmanus et al. Reference Jirmanus, Glesby, Guimaraes, Lago, Rosa, Machado and Carvalho2012). In addition, the spontaneous cure rate is very low in Americas (Cota et al. Reference Cota, De Sousa, Fereguetti, Saleme, Alvarisa and Rabello2016). For these reasons, exclusive local treatment is generally not recommended and systemic drugs are employed with a great degree of geographical variation, including drugs with very specific applications, such as ketoconazole (for L. mexicana) (WHO, 2010). The systemic drugs applicable are pentavalent antimonials, pentamidine, amphotericin B and miltefosine. Notwithstanding, antimonials are still the first line treatment for CL in most leishmaniasis-endemic countries. Meglumine antimoniate is still used as first line option to treat CL in Brazil, for example, where even with more extensive schemes of antimonial treatment, recent studies have demonstrated a failure rate of over 40% in patients with tegumentary leishmaniasis in different geographic areas (Machado et al. Reference Machado, Ampuero, Guimaraes, Villasboas, Rocha, Schriefer, Sousa, Talhari, Penna and Carvalho2010; Chrusciak-Talhari et al. Reference Chrusciak-Talhari, Dietze, Chrusciak Talhari, Da Silva, Gadelha Yamashita, De Oliveira Penna, Lima Machado and Talhari2011; Neves et al. Reference Neves, Talhari, Gadelha, Silva, Guerra, Ferreira and Talhari2011).
The use of amphotericin B in CL is generally reserved as a second or third option after treatment failure, mainly due to toxicity.
Pentamidine was synthesized at the end of the 1930s, as part of a diamidine group and as a synthalin analogue that showed activity against Trypanosoma. The demonstration of activity against Leishmania led to its use for the treatment of VL in India in the early 1940s (Bray et al. Reference Bray, Barrett, Ward and de Koning2003). The high frequency of toxic effects, as for example, cardiotoxicity and metabolic disturbances, made pentamidine a forgotten option until the emergence of resistance against antimonials. However, the efficacy in the treatment of VL patients was unsatisfactory and a high number of relapses was noted (Jha et al. Reference Jha, Singh and Jha1991), suggesting that resistance against pentamidine could be easily selected. Pentamidine has found its niche in the treatment of infections in areas of South America where L. guyanensis is more prevalent (Van Der Meide et al. Reference Van Der Meide, Sabajo, Jensema, Peekel, Faber, Schallig and Fat2009).
The application of miltefosine in the treatment of CL is not yet completely established. The susceptibility of cutaneous Leishmania species to miltefosine is lower as compared with the susceptibility of L. donovani (Escobar et al. Reference Escobar, Matu, Marques and Croft2002). Clinical trials performed in Colombia and Nicaragua showed that miltefosine's efficacy was variable, and seemed to correlate not only with the Leishmania species, but also with geographical heterogeneity within the same species (Soto and Berman, Reference Soto and Berman2006). The drug is approved for the treatment of CL in Colombia. It has been tried in Brazil in two studies that showed efficacy of about 70% for miltefosine in CL patients (Machado et al. Reference Machado, Ampuero, Guimaraes, Villasboas, Rocha, Schriefer, Sousa, Talhari, Penna and Carvalho2010; Chrusciak-Talhari et al. Reference Chrusciak-Talhari, Dietze, Chrusciak Talhari, Da Silva, Gadelha Yamashita, De Oliveira Penna, Lima Machado and Talhari2011). Miltefosine was also shown to be non-inferior to pentavalent antimonial for the treatment of CL caused by L. major in Iran (Mohebali et al. Reference Mohebali, Fotouhi, Hooshmand, Zarei, Akhoundi, Rahnema, Razaghian, Kabir and Nadim2007).
Paromomycin was initially reported as a potential topical drug in animal models (El-On et al. Reference El-On, Jacobs, Witztum and Greenblatt1984) and tested for CL treatment as a topical agent with variable results (Kim et al. Reference Kim, Chung, Bleys and Ghohestani2009). Topical treatment of Old World CL with paromomycin was generally more effective than placebo and equivalent to intralesional pentavalent antimonials (El-On et al. Reference El-On, Halevy, Grunwald and Weinrauch1992; Ben Salah et al. Reference Ben Salah, Ben Messaoud, Guedri, Zaatour, Ben Alaya, Bettaieb, Gharbi, Belhadj Hamida, Boukthir, Chlif, Abdelhamid, El Ahmadi, Louzir, Mokni, Morizot, Buffet, Smith, Kopydlowski, Kreishman-Deitrick, Smith, Nielsen, Ullman, Norwood, Thorne, Mccarthy, Adams, Rice, Tang, Berman and Ransom2013). The association of paromomycin with methylbenzethonium chloride led to an increase in side-effects. Against New World species, topical paromomycin was inferior to pentavalent antimonials (Soto et al. Reference Soto, Fuya, Herrera and Berman1998; Armijos et al. Reference Armijos, Weigel, Calvopina, Mancheno and Rodriguez2004).
All therapeutic alternatives mentioned above present considerable toxicity, high cost and, with the exception of miltefosine, have to be administered by the parenteral route. Furthermore, the risk or demonstration of parasite resistance has become a major concern.
DRUG RESISTANCE
Drug resistance is a phenotype of decreased susceptibility to a given drug acquired after drug selection. In the absence of previous exposure to drugs, natural variation in drug susceptibility does occur amongst species and clinical isolates of Leishmania. Reports of proven parasite resistance originated mainly from areas of anthroponotic transmission, such as the Indian subcontinent (Sundar, Reference Sundar2001). In endemic regions of zoonotic transmission, there is no definite demonstration of resistance yet. However, the recent disease urbanization may eventually change this scenario, leading to the emergence of drug resistant parasites in these regions (Harhay et al. Reference Harhay, Olliaro, Costa and Costa2011). Another serious concern is the increased number of HIV–leishmaniasis co-infected patients. In general, these patients respond poorly to the treatment and exhibit high relapse rates, representing a potential source for the emergence of drug-resistant parasites (Molina et al. Reference Molina, Gradoni and Alvar2003).
A progressive decrease in the efficacy of pentavalent antimonials was described initially in VL patients. In the Northeast of India, where 85–95% of the VL patients were cured after treatment with antimony in 1981, therapeutic failures reached 60–70% by 1995 (Olliaro et al. Reference Olliaro, Guerin, Gerstl, Haaskjold, Rottingen and Sundar2005). Acquired resistance was confirmed in clinical isolates of L. donovani from this endemic region that were shown to be 3-fold less susceptible in vitro than isolates from patients who did respond to chemotherapy (Lira et al. Reference Lira, Sundar, Makharia, Kenney, Gam, Saraiva and Sacks1999). Currently, the drug is not in use in the district of Bihar, India and in parts of Nepal and Bangladesh, due to the spread of antimony resistance. The contamination of drinking water with arsenic in Bihar has been implicated as an additional factor associated with the development of antimonial resistance (Perry et al. Reference Perry, Wyllie, Prajapati, Menten, Raab, Feldmann, Chakraborti, Sundar, Boelaert, Picado and Fairlamb2015).
In other VL endemic regions, lack of response to antimony does occur but detailed mapping of therapeutic failures is missing. Leishmania infantum isolates from dogs treated with antimony were shown to present decreased susceptibility to antimonials (Gramiccia et al. Reference Gramiccia, Gradoni and Orsini1992). On the other hand, variability in clinical response can also be explained by intrinsic differences in the sensitivity of Leishmania species and strains to antimonials (Berman et al. Reference Berman, Chulay, Hendricks and Oster1982; Navin et al. Reference Navin, Arana, Arana, Berman and Chajon1992).
Studies performed in the laboratory have demonstrated that the parasite is easily selected in vitro for antimony resistance by drug pressure (Papadopoulou et al. Reference Papadopoulou, Roy, Dey, Rosen and Ouellette1994; Haimeur et al. Reference Haimeur, Brochu, Genest, Papadopoulou and Ouellette2000; Do Monte-Neto et al. Reference Do Monte-Neto, Coelho, Raymond, Legare, Corbeil, Melo, Frezard and Ouellette2011; Monte-Neto et al. Reference Monte-Neto, Laffitte, Leprohon, Reis, Frezard and Ouellette2015). The main drug resistance mechanisms have been correlated with reduction of drug concentration within the parasite, either by a decreased uptake mediated by the aquaglyceroporin AQP1, the primary route of antimony entry (Gourbal et al. Reference Gourbal, Sonuc, Bhattacharjee, Legare, Sundar, Ouellette, Rosen and Mukhopadhyay2004) or by increased efflux of the drug mediated by the ABC transporter ABCC3 (also known as MRPA) (Legare et al. Reference Legare, Richard, Mukhopadhyay, Stierhof, Rosen, Haimeur, Papadopoulou and Ouellette2001). Antimony resistant lines also present increased levels of thiols (cysteine, glutathione and trypanothione) due to overexpression/amplification of genes involved in the synthesis of gluthathione and polyamines, the components of trypanothione, the main intracellular thiol in Leishmania (Mukhopadhyay et al. Reference Mukhopadhyay, Dey, Xu, Gage, Lightbody, Ouellette and Rosen1996; Grondin et al. Reference Grondin, Haimeur, Mukhopadhyay, Rosen and Ouellette1997; Legare et al. Reference Legare, Papadopoulou, Roy, Mukhopadhyay, Haimeur, Dey, Grondin, Brochu, Rosen and Ouellette1997; Guimond et al. Reference Guimond, Trudel, Brochu, Marquis, El Fadili, Peytavi, Briand, Richard, Messier, Papadopoulou, Corbeil, Bergeron, Legare and Ouellette2003; Do Monte-Neto et al. Reference Do Monte-Neto, Coelho, Raymond, Legare, Corbeil, Melo, Frezard and Ouellette2011). Interestingly, several of the mechanisms associated with antimony resistance in vitro were found in L. donovani clinical isolates. For these parasites, susceptibility in vitro of intracellular amastigotes correlated well with the clinical response (Mukherjee et al. Reference Mukherjee, Padmanabhan, Singh, Roy, Girard, Chatterjee, Ouellette and Madhubala2007).
Resistance to antimony can also arise from inhibition of drug reduction or inactivation of the active drug form (Croft et al. Reference Croft, Sundar and Fairlamb2006). Pentavalent antimony is a prodrug that requires conversion to trivalent antimony to be active against the parasite. Two enzymes mediate this enzymatic reduction: a thiol-dependent reductase (TDR1) and an arsenate reductase (ACR2) (Denton et al. Reference Denton, Mcgregor and Coombs2004; Zhou et al. Reference Zhou, Messier, Ouellette, Rosen and Mukhopadhyay2004). The conversion to the active form is mediated by the host and by intracellular amastigotes, but not by promastigotes (Roberts and Rainey, Reference Roberts and Rainey1993; Shaked-Mishan et al. Reference Shaked-Mishan, Ulrich, Ephros and Zilberstein2001).
In addition, other molecular markers have been associated with antimony resistance by functional cloning, such as ABC transporters, phosphatases and hypothetical proteins (Choudhury et al. Reference Choudhury, Zander, Kube, Reinhardt and Clos2008; Manzano et al. Reference Manzano, Garcia-Hernandez, Castanys and Gamarro2013; Nuhs et al. Reference Nuhs, Schafer, Zander, Trube, Tejera Nevado, Schmidt, Arevalo, Adaui, Maes, Dujardin and Clos2014; Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016; Perea et al. Reference Perea, Manzano, Castanys and Gamarro2016). However, their role in clinical resistance and treatment failure is still unknown.
Amphotericin is a polyene antibiotic that targets the main membrane sterol of the parasite, ergosterol. The drug has been in use, although not as a first-line option in the treatment of leishmaniasis for decades, and, until recently, without indication that clinical resistance was emerging, even when patients were exposed to multiple courses of treatment (Lachaud et al. Reference Lachaud, Bourgeois, Plourde, Leprohon, Bastien and Ouellette2009). However, lack of response to treatment with amphotericin B has now been reported in India, where this drug has become the first-line option in areas where refractoriness to antimony is widespread (Purkait et al. Reference Purkait, Kumar, Nandi, Sardar, Das, Kumar, Pandey, Ravidas, Kumar, De, Singh and Das2012, Reference Purkait, Singh, Wasnik, Das, Kumar, Paine, Dikhit, Singh, Sardar, Ghosh and Das2015).
Leishmania is not easily selected in vitro by amphotericin pressure. However, stepwise drug increase was applied successfully to select Leishmania tarentolae and L. donovani amphotericin resistant lines (Mbongo et al. Reference Mbongo, Loiseau, Billion and Robert-Gero1998; Singh et al. Reference Singh, Papadopoulou and Ouellette2001). These parasites exhibited gene amplification (Singh et al. Reference Singh, Papadopoulou and Ouellette2001) and changes in drug-binding affinity to the plasma membrane as a result of a modified sterol composition (Mbongo et al. Reference Mbongo, Loiseau, Billion and Robert-Gero1998). Recently, a screening method termed Cos-Seq was described employing functional cloning coupled to next-generation sequencing that allows the identification of potential drug targets and drug resistance genes (Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016). This strategy led to the identification of a hypothetical protein that contains transmembrane domains and a secretory signal peptide responsible for mediating amphotericin B resistance in promastigotes (Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016).
After its approval for VL treatment in India (Sundar et al. Reference Sundar, Jha, Thakur, Engel, Sindermann, Fischer, Junge, Bryceson and Berman2002) miltefosine replaced antimonials and was used as first-line therapy in northeastern India. However, an increase in the number of relapses was noted after a few years (Sundar et al. Reference Sundar, Singh, Rai, Prajapati, Singh, Ostyn, Boelaert, Dujardin and Chakravarty2012; Rijal et al. Reference Rijal, Ostyn, Uranw, Rai, Bhattarai, Dorlo, Beijnen, Vanaerschot, Decuypere, Dhakal, Das, Karki, Singh, Boelaert and Dujardin2013) affecting as many as 20% of patients infected with L. donovani 6–12 months after the end of treatment. The parasites isolated from relapsing patients did not present decreased drug susceptibility in vitro (Prajapati et al. Reference Prajapati, Sharma, Rai, Ostyn, Salotra, Vanaerschot, Dujardin and Sundar2013; Rijal et al. Reference Rijal, Ostyn, Uranw, Rai, Bhattarai, Dorlo, Beijnen, Vanaerschot, Decuypere, Dhakal, Das, Karki, Singh, Boelaert and Dujardin2013), suggesting that the reduced clinical efficacy was related to other factors such as selection of parasites with increased virulence/infectivity or lower exposure to the drug due to heterogeneous pharmacokinetics (Rai et al. Reference Rai, Cuypers, Bhattarai, Uranw, Berg, Ostyn, Dujardin, Rijal and Vanaerschot2013; Dorlo et al. Reference Dorlo, Rijal, Ostyn, De Vries, Singh, Bhattarai, Uranw, Dujardin, Boelaert, Beijnen and Huitema2014).
On the other hand, parasite resistance to miltefosine has been described in clinical isolates of L. infantum from a HIV-coinfected patient (Cojean et al. Reference Cojean, Houze, Haouchine, Huteau, Lariven, Hubert, Michard, Bories, Pratlong, Le Bras, Loiseau and Matheron2012) and L. panamensis, both rescued after treatment (Obonaga et al. Reference Obonaga, Fernandez, Valderrama, Rubiano, Castro Mdel, Barrera, Gomez and Gore Saravia2014).
Miltefosine interferes in cell membrane composition by inhibiting phospholipid metabolism and affecting phosphatidylcholine and phosphatidylethanolamine synthesis due to a decrease in intracellular choline (Rakotomanga et al. Reference Rakotomanga, Blanc, Gaudin, Chaminade and Loiseau2007). Parasites treated with miltefosine also present a significant reduction in mitochondrial membrane potential and in cytochrome-c oxidase activity (Santa-Rita et al. Reference Santa-Rita, Henriques-Pons, Barbosa and De Castro2004; Luque-Ortega and Rivas, Reference Luque-Ortega and Rivas2007). The drug binds to the outer leaflet of the plasma membrane and is internalized by the endocytic pathway or by a flippase activity mediated by the miltefosine transporter (MT) and its non-catalytic subunit Ros3. This MT–Ros3 complex is responsible for phosphocoline accumulation in an ATP-dependent process (Perez-Victoria et al. Reference Perez-Victoria, Gamarro, Ouellette and Castanys2003, Reference Perez-Victoria, Sanchez-Canete, Seifert, Croft, Sundar, Castanys and Gamarro2006b; Coelho et al. Reference Coelho, Trinconi, Costa and Uliana2014). Miltefosine is eliminated by exocytosis or by a floppase activity that may be mediated by members of ABC transporter subfamilies ABCB and ABCG (Perez-Victoria et al. Reference Perez-Victoria, Perez-Victoria, Parodi-Talice, Jimenez, Ravelo, Castanys and Gamarro2001; Castanys-Munoz et al. Reference Castanys-Munoz, Alder-Baerens, Pomorski, Gamarro and Castanys2007, Reference Castanys-Munoz, Perez-Victoria, Gamarro and Castanys2008).
Susceptibility to miltefosine in vitro is intrinsically variable in different species and clinical isolates of Leishmania (Yardley et al. Reference Yardley, Croft, De Doncker, Dujardin, Koirala, Rijal, Miranda, Llanos-Cuentas and Chappuis2005; Coelho et al. Reference Coelho, Trinconi, Costa and Uliana2014; Fernandez et al. Reference Fernandez, Diaz-Toro, Ovalle, Valderrama, Muvdi, Rodriguez, Gomez and Saravia2014; Obonaga et al. Reference Obonaga, Fernandez, Valderrama, Rubiano, Castro Mdel, Barrera, Gomez and Gore Saravia2014). This differential susceptibility among species and isolates may be explained by variations in the activity and substrate specificity of MT–Ros3 machinery, rate of cell division, biochemical targets, drug metabolism and biochemical composition of the plasma membrane (Perez-Victoria et al. Reference Perez-Victoria, Sanchez-Canete, Seifert, Croft, Sundar, Castanys and Gamarro2006b; Sanchez-Canete et al. Reference Sanchez-Canete, Carvalho, Perez-Victoria, Gamarro and Castanys2009). On the other hand, recent studies have shown an absence of correlation between miltefosine susceptibility in vitro and treatment outcome, indicating that this variation in vitro may not affect clinical efficacy (Rijal et al. Reference Rijal, Ostyn, Uranw, Rai, Bhattarai, Dorlo, Beijnen, Vanaerschot, Decuypere, Dhakal, Das, Karki, Singh, Boelaert and Dujardin2013; Hendrickx et al. Reference Hendrickx, Eberhardt, Mondelaers, Rijal, Bhattarai, Dujardin, Delputte, Cos and Maes2015).
Resistance to miltefosine is easily selected in vitro by stepwise increase in drug concentrations (Seifert et al. Reference Seifert, Matu, Javier Perez-Victoria, Castanys, Gamarro and Croft2003; Coelho et al. Reference Coelho, Boisvert, Mukherjee, Leprohon, Corbeil and Ouellette2012, Reference Coelho, Trinconi, Costa and Uliana2014; Obonaga et al. Reference Obonaga, Fernandez, Valderrama, Rubiano, Castro Mdel, Barrera, Gomez and Gore Saravia2014) or by chemical mutagenesis (Perez-Victoria et al. Reference Perez-Victoria, Gamarro, Ouellette and Castanys2003; Coelho et al. Reference Coelho, Trinconi, Senra, Yokoyama-Yasunaka and Uliana2015). In general, mechanisms of miltefosine resistance are associated with a defect in drug internalization mediated by the machinery MT–Ros3 (Perez-Victoria et al. Reference Perez-Victoria, Sanchez-Canete, Seifert, Croft, Sundar, Castanys and Gamarro2006b). Mutations in the MT and Ros3 genes have been found after selection with miltefosine, with a higher frequency of mutations in the MT gene (Perez-Victoria et al. Reference Perez-Victoria, Gamarro, Ouellette and Castanys2003, Reference Perez-Victoria, Sanchez-Canete, Castanys and Gamarro2006a; Coelho et al. Reference Coelho, Boisvert, Mukherjee, Leprohon, Corbeil and Ouellette2012, Reference Coelho, Trinconi, Costa and Uliana2014; Kulshrestha et al. Reference Kulshrestha, Sharma, Singh and Salotra2014; Mondelaers et al. Reference Mondelaers, Sanchez-Canete, Hendrickx, Eberhardt, Garcia-Hernandez, Lachaud, Cotton, Sanders, Cuypers, Imamura, Dujardin, Delputte, Cos, Caljon, Gamarro, Castanys and Maes2016; Shaw et al. Reference Shaw, Lonchamp, Downing, Imamura, Freeman, Cotton, Sanders, Blackburn, Dujardin, Rijal, Khanal, Illingworth, Coombs and Carter2016). Interestingly, the resistance phenotype mediated by MT inactivation persists in animal models of visceral and cutaneous disease (Seifert et al. Reference Seifert, Perez-Victoria, Stettler, Sanchez-Canete, Castanys, Gamarro and Croft2007; Coelho et al. Reference Coelho, Trinconi, Costa and Uliana2014), indicating that MT activity is essential for miltefosine efficacy in vivo.
Pentamidine was not used extensively for VL treatment, due to failure and toxicity, but it has its place in the treatment of CL. Parasites resistant to pentamidine are rapidly obtained in vitro for several Leishmania species (Ellenberger and Beverley, Reference Ellenberger and Beverley1989; Sereno and Lemesre, Reference Sereno and Lemesre1997; Basselin et al. Reference Basselin, Denise, Coombs and Barrett2002; Coelho et al. Reference Coelho, Gentil, Da Silveira and Cotrim2008). Pentamidine resistant lines display changes in intracellular concentrations of arginine and polyamines, reduction in pentamidine accumulation in the mitochondria and increased efflux of the drug (Basselin et al. Reference Basselin, Denise, Coombs and Barrett2002). It is possible that the ABC transporter PRP1, isolated previously by functional cloning, mediates this efflux (Coelho et al. Reference Coelho, Beverley and Cotrim2003). In addition to PRP1, functional cloning strategies using Cos-Seq identified a hypothetical protein that mediates low level of resistance to pentamidine in promastigotes (Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016). It would be interesting to evaluate whether parasites isolated from unresponsive cases are associated with overexpression of these genes.
Paromomycin is an aminoglycoside antibiotic that targets protein synthesis in the parasite (Maarouf et al. Reference Maarouf, Lawrence, Croft and Robert-Gero1995; Fernandez et al. Reference Fernandez, Malchiodi and Algranati2011). The drug binds to the ribosome, specifically to the ribosomal A-site (Shalev et al. Reference Shalev, Kondo, Kopelyanskiy, Jaffe, Adir and Baasov2013). Paromomycin also induces alterations in membrane fluidity and lipid metabolism and affects mitochondrial activity (Maarouf et al. Reference Maarouf, De Kouchkovsky, Brown, Petit and Robert-Gero1997). Resistant parasites can be selected in vitro and these lines show a decreased drug accumulation, although the protein/transporter involved is still unknown (Jhingran et al. Reference Jhingran, Chawla, Saxena, Barrett and Madhubala2009; Bhandari et al. Reference Bhandari, Sundar, Dujardin and Salotra2014). Mutations in the small subunit ribosomal RNA gene were found in paromomycin resistant mutants of Escherichia coli and Tetrahymena (Fong et al. Reference Fong, Chan, Rodriguez, Gately, Berman and Grogl1994), but not in Leishmania-resistant lines, suggesting that the mechanisms leading to paromomycin resistance are variable. So far, there is only one paromomycin resistance gene identified in Leishmania (Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016). It encodes a hypothetical protein that contains a leucine-rich repeat domain that also confers resistance to pentamidine (Gazanion et al. Reference Gazanion, Fernandez-Prada, Papadopoulou, Leprohon and Ouellette2016). The mechanism of multiple drug resistance mediated by this protein is still uncertain. Clinical resistance to paromomycin has not yet been reported, fact perhaps explained by the recent introduction of the drug for the chemotherapy of the disease.
Heterogeneity in paromomycin susceptibility has been observed in different species and clinical isolates of the parasite (Neal et al. Reference Neal, Allen, Mccoy, Olliaro and Croft1995; Kulshrestha et al. Reference Kulshrestha, Singh, Kumar, Negi and Salotra2011; Prajapati et al. Reference Prajapati, Mehrotra, Gautam, Rai and Sundar2012). Whether these differences detected in vitro affect treatment response in the field remains to be determined.
In fact, studies to determine the correlation between the various molecular markers of drug resistance described in vitro and treatment failure/clinical resistance need to be expanded. The problem of treatment failure is a complex one given that it can also be due to mishandling of antileishmanial drugs, individual variability in the susceptibility of Leishmania species and isolates, pharmacokinetics and drug–host immune response interaction (Croft et al. Reference Croft, Sundar and Fairlamb2006). An additional factor that may also affect clinical efficacy and lead to treatment failure is the presence of Leishmania RNA virus 1 (LRV-1) in parasites of the Viannia subgenus. Recent data indicated that the virus may subvert the host immune response and affect clinical response to drugs (Adaui et al. Reference Adaui, Lye, Akopyants, Zimic, Llanos-Cuentas, Garcia, Maes, De Doncker, Dobson, Arevalo, Dujardin and Beverley2015; Bourreau et al. Reference Bourreau, Ginouves, Prevot, Hartley, Gangneux, Robert-Gangneux, Dufour, Sainte-Marie, Bertolotti, Pratlong, Martin, Schutz, Couppie, Fasel and Ronet2016).
Given that resistance to all antileishmanial drugs has been detected, at least in the laboratory, there is now a consensus on the necessity of adopting combination therapy as the preferred treatment option, especially for the life-threatening VL (WHO, 2012).
COMBINATION THERAPY
Combination therapy consists of using two or more drugs with synergistic or additive effects and distinct mechanisms of action with the aim of increasing the spectrum of activity and therapeutic efficacy. Ideally, combination therapy will reduce the overall dose of drugs and treatment duration, resulting in lower toxicity and higher compliance. It can also reduce costs and therefore the burden on the health system (Van Griensven et al. Reference Van Griensven, Balasegaram, Meheus, Alvar, Lynen and Boelaert2010; WHO, 2010). Combination therapy can also help delaying the selection of drug-resistant parasites, prolonging the effective life of available medicines, as has been reported in diseases such as tuberculosis, malaria and AIDS (Vandamme et al. Reference Vandamme, Van Laethem and De Clercq1999; Kremsner and Krishna, Reference Kremsner and Krishna2004; Van Griensven et al. Reference Van Griensven, Balasegaram, Meheus, Alvar, Lynen and Boelaert2010; Mitchison and Davies, Reference Mitchison and Davies2012).
The implementation of combination therapy for leishmaniasis requires the determination of the best combination schemes and their efficacy in clinical settings. Efficacy studies reported or in progress have employed combinations of drugs already available – pentavalent antimonials, amphotericin B, miltefosine and paromomycin – and were performed mainly with VL patients in India and Africa.
Ongoing initiatives have focused mainly on two combination schemes: pentavalent antimonial associated with paromomycin and amphotericin B plus miltefosine. The first strategy is being applied mainly in Africa, given the high incidence of antimonial resistance in Asia.
Initial clinical trials testing the interaction between pentavalent antimonial and paromomycin date from the early 1990s, were performed in Kenya, India and Sudan, and demonstrated an enhanced overall efficacy and/or reduced treatment duration for the combination as compared with antimonial alone (Chunge et al. Reference Chunge, Owate, Pamba and Donno1990; Thakur et al. Reference Thakur, Olliaro, Gothoskar, Bhowmick, Choudhury, Prasad, Kumar and Verma1992; Seaman et al. Reference Seaman, Pryce, Sondorp, Moody, Bryceson and Davidson1993). A combination of sodium stibogluconate combined with paromomycin for 17 days was shown to be effective in southern Sudan with higher survival and initial cure rates in patients treated with the combination than with sodium stibogluconate alone (Melaku et al. Reference Melaku, Collin, Keus, Gatluak, Ritmeijer and Davidson2007). A phase 3 trial in Ethiopia, Kenya, Sudan and Uganda confirmed that the use of sodium stibogluconate combined with paromomycin for 17 days was safe and non-inferior to the standard 30-day antimonial treatment (Musa et al. Reference Musa, Khalil, Hailu, Olobo, Balasegaram, Omollo, Edwards, Rashid, Mbui, Musa, Abuzaid, Ahmed, Fadlalla, El-Hassan, Mueller, Mucee, Njoroge, Manduku, Mutuma, Apadet, Lodenyo, Mutea, Kirigi, Yifru, Mengistu, Hurissa, Hailu, Weldegebreal, Tafes and Mekonnen2012).
Based on these data, WHO recommendations were changed in 2010 to advocate the use of sodium stibogluconate and paromomycin combination as the first-line treatment in East Africa (Ethiopia, Eritrea, Kenya, Somalia, Sudan and Uganda) and Yemen (WHO, 2010). A recent assessment of this treatment scheme indicated a cure rate of 86% at 6 months (Atia et al. Reference Atia, Mumina, Tayler-Smith, Boulle, Alcoba, Elhag, Alnour, Shah, Chappuis, Van Griensven and Zachariah2015). However, it has also made clear that, although representing an improvement over past options given the reduction in hospitalization time, it still lacks in comprehensiveness: a considerable number of patients are excluded from this scheme because of underlying conditions, such as HIV co-infection, pregnancy, renal failure or due to disease severity, highlighting the necessity of the continued search for new drugs and treatment regimens to improve the handling of VL.
The interaction of amphotericin B and miltefosine has been investigated mainly in Indian VL patients. Phase 2 and 3 studies, performed in Bihar, India, have evaluated combinations of a single dose of liposomal amphotericin B followed by miltefosine given for 7, 10 or 14 days (Sundar et al. Reference Sundar, Rai, Chakravarty, Agarwal, Agrawal, Vaillant, Olliaro and Murray2008). Other combination alternatives were also evaluated, such as single-dose liposomal amphotericin B plus a 10-day course of intramuscular paromomycin and the combination of miltefosine and paromomycin for 10 days (Sundar et al. Reference Sundar, Sinha, Rai, Verma, Nawin, Alam, Chakravarty, Vaillant, Verma, Pandey, Kumari, Lal, Arora, Sharma, Ellis, Strub-Wourgaft, Balasegaram, Olliaro, Das and Modabber2011). All tested combinations were well tolerated and showed high efficacy with cure rates at 6–9 months follow-up higher than 95% and were therefore non-inferior to the standard treatment with amphotericin B in that endemic area.
A short course of amphotericin B combined with miltefosine is an attractive option for Indian VL due to the ease of administration, efficacy and tolerance. The reduction in time and amount of drug administered contribute to reduced toxicity and cost of therapy, besides protecting miltefosine from selection of drug resistant parasites.
Combination therapy is also strongly recommended for HIV/Leishmania co-infected patients (WHO, 2010). Visceral leishmaniasis in HIV co-infected patients represents a tremendous treatment challenge since patients that do respond to monotherapy present a high relapse rate. Furthermore, given the seriousness of the co-morbidities, high mortality rates and increased antileishmanial drug toxicity are observed (Alvar et al. Reference Alvar, Aparicio, Aseffa, Den Boer, Canavate, Dedet, Gradoni, Ter Horst, Lopez-Velez and Moreno2008). Antiretroviral treatment is a crucial measure associated with antileishmanial therapy. Protease inhibitors available for second-line treatment of HIV-positive patients have demonstrated antileishmanial activity in vitro (Kumar et al. Reference Kumar, Lodge, Trudel, Ouellet, Ouellette and Tremblay2010; Valdivieso et al. Reference Valdivieso, Rangel, Moreno, Saugar, Canavate, Alvar and Dagger2010; Santos et al. Reference Santos, Vitorio, Branquinha, Pedroso E Silva, Santos and D'avila-Levy2013; Van Griensven et al. Reference Van Griensven, Diro, Lopez-Velez, Boelaert, Lynen, Zijlstra, Dujardin and Hailu2013) and, pending clinical studies, might become valuable partners in combination therapy against leishmaniasis in these patients.
Unfortunately, there are still no evidence-based guidelines for the use of drug combinations in the treatment of these patients. A retrospective analysis of the treatment of HIV–Leishmania co-infected patients with a combination of liposomal amphotericin and miltefosine revealed the safety and efficacy of this scheme (Mahajan et al. Reference Mahajan, Das, Isaakidis, Sunyoto, Sagili, Lima, Mitra, Kumar, Pandey, Van Geertruyden, Boelaert and Burza2015). A lower relapse rate was detected in these patients in comparison with previous data available for monotherapy with liposomal amphotericin (Burza et al. Reference Burza, Mahajan, Sinha, Van Griensven, Pandey, Lima, Sanz, Sunyoto, Kumar, Mitra, Kumar, Verma and Das2014). A phase 3 clinical trial aimed at evaluating the safety and efficacy of liposomal amphotericin and miltefosine combination in co-infected patients is ongoing in Ethiopia.
Secondary prophylaxis is adopted in some countries after primary treatment for leishmaniasis in AIDS patients, at least until the immune status of the patient can be considered minimally reconstituted. Even with ongoing prophylaxis, relapse rates can be very high (Diro et al. Reference Diro, Ritmeijer, Boelaert, Alves, Mohammed, Abongomera, Ravinetto, De Crop, Fikre, Adera, Colebunders, Van Loen, Menten, Lynen, Hailu and Van Griensven2015). There are no data available on the use of drug combinations for secondary prophylaxis.
Current knowledge on combination therapy for CL is even more limited. The use of pentavalent antimonial combined with pentoxifylline, an inhibitor of tumour necrosis factor alpha, has proven effective against aggressive forms, such as mucocutaneous leishmaniasis (Lessa et al. Reference Lessa, Machado, Lima, Cruz, Bacellar, Guerreiro and Carvalho2001; Machado et al. Reference Machado, Lessa, Lessa, Guimaraes, Bang, Ho and Carvalho2007). This combination scheme was also effective in CL patients in Iran (Sadeghian and Nilforoushzadeh, Reference Sadeghian and Nilforoushzadeh2006).
For severe forms of CL, the use of topical or local treatment is not advisable. However, the combination of a topical agent to a systemic drug may represent an interesting option and should be further investigated.
DRUG SCREENING
The need for new chemotherapeutic alternatives for leishmaniasis is evident. The Target Product Profile (TPP) proposed by DNDi (http://www.dndi.org/diseases-projects/leishmaniasis) includes consideration of: (a) activity against all Leishmania species, (b) efficacy against VL and CL, (c) efficacy with courses of treatment shorter than 14 days, (d) oral drug with single daily dose preferred, (e) safer than available treatment, (f) low cost per treatment and (g) stability under conditions that can be achieved in target regions. It is also highly desirable that efficacy is maintained in immunosuppressed patients. Furthermore, it is desirable that new drugs are compatible for combination therapy.
Drug discovery projects have been undertaken by many laboratories using classical drug-to-target or target-to-drug strategies. Drug discovery processes and challenges applicable to infectious diseases have been recently reviewed (Lechartier et al. Reference Lechartier, Rybniker, Zumla and Cole2014; Manjunatha et al. Reference Manjunatha, Smith, Lechartier, Rybniker, Zumla and Cole2015) and challenges specific to trypanosomatids have been also described in detail (Don et al. Reference Don, Ioset, Reguera, Calvo-Alvarez, Alvarez-Velilla and Balana-Fouce2014; Reguera et al. Reference Reguera, Calvo-Alvarez, Alvarez-Velilla and Balana-Fouce2014).
Screening of chemical libraries for the specific target inhibition or phenotypic outcomes, as well as drug repurposing have all been employed for leishmaniasis. Unbiased screening of chemical libraries makes use of phenotypic evaluations and has the potential of identifying new entities directed at novel targets (Annang et al. Reference Annang, Perez-Moreno, Garcia-Hernandez, Cordon-Obras, Martin, Tormo, Rodriguez, De Pedro, Gomez-Perez, Valente, Reyes, Genilloud, Vicente, Castanys, Ruiz-Perez, Navarro, Gamarro and Gonzalez-Pacanowska2015; Kaiser et al. Reference Kaiser, Maes, Tadoori, Spangenberg and Ioset2015a; Pena et al. Reference Pena, Pilar Manzano, Cantizani, Kessler, Alonso-Padilla, Bardera, Alvarez, Colmenarejo, Cotillo, Roquero, De Dios-Anton, Barroso, Rodriguez, Gray, Navarro, Kumar, Sherstnev, Drewry, Brown, Fiandor and Julio Martin2015; Khare et al. Reference Khare, Nagle, Biggart, Lai, Liang, Davis, Barnes, Mathison, Myburgh, Gao, Gillespie, Liu, Tan, Stinson, Rivera, Ballard, Yeh, Groessl, Federe, Koh, Venable, Bursulaya, Shapiro, Mishra, Spraggon, Brock, Mottram, Buckner, Rao and Wen2016; Khraiwesh et al. Reference Khraiwesh, Leed, Roncal, Johnson, Sciotti, Smith, Read, Paris, Hudson, Hickman and Grogl2016). The opposite strategy elects a target that has been deemed of importance for the parasite and ideally is absent in the host. Examples of targets that have been used for drug screening purposes are thypanothione reductase (Richardson et al. Reference Richardson, Nett, Jones, Abdille, Gilbert and Fairlamb2009; Pandey et al. Reference Pandey, Sharma, Bhatt, Sundar and Prajapati2015), N-myristoyltransferase (Bell et al. Reference Bell, Mills, Williams, Brannigan, Wilkinson, Parkinson, Leatherbarrow, Tate, Holder and Smith2012; Rackham et al. Reference Rackham, Yu, Brannigan, Heal, Paape, Barker, Wilkinson, Smith, Leatherbarrow and Tate2015), inositol phosphorylceramide synthase (Mina et al. Reference Mina, Mosely, Ali, Shams-Eldin, Schwarz, Steel and Denny2010), dipeptidylcarboxypeptidase (Gangwar et al. Reference Gangwar, Baig, Shah, Biswas, Batra, Siddiqi and Goyal2012), among others. Target-based drug screening has the disadvantage of not always translating into parasite killing or in vivo activity, as has been extensively demonstrated by target-based efforts. Therefore, a trend towards high-throughput phenotypic screening has been noted in the last years. Also, the introduction of high-content screening platforms emerged as a promising approach, allowing large libraries to be tested against Leishmania intracellular amastigotes (Siqueira-Neto et al. Reference Siqueira-Neto, Moon, Jang, Yang, Lee, Moon, Chatelain, Genovesio, Cechetto and Freitas-Junior2012; Forestier et al. Reference Forestier, Spath, Prina, Dasari, Don, Ioset, Reguera, Calvo-Alvarez, Alvarez-Velilla and Balana-Fouce2015). This approach has several advantages including the differentiation between activity against the parasite or the host cell, and the possibility of identification of singular cell events that can guide the research on the drug's mode of action.
Another aspect that has been advanced into earlier stages of drug screening is the evaluation of pharmacokinetics and pharmacodynamics (Lin and Lu, Reference Lin and Lu1997). Adequate properties are fundamental criteria if a positive hit is to achieve the status of a drug candidate. Bioavailability, drug metabolism, activity of metabolites and drug–drug interactions should be evaluated as soon as possible in the drug discovery process (Riley et al. Reference Riley, Kenna, Li, Liu, Chang, Groessl, Zimmerman, He, Isbell, Tuntland, Shearer, Smith, Diaz, Asher, Ramirez, Lin and Lu2004; Shearer et al. Reference Shearer, Smith, Diaz, Asher, Ramirez, Riley, Kenna, Li, Liu, Chang, Groessl, Zimmerman, He, Isbell and Tuntland2005; Li et al. Reference Li, Liu, Chang, Groessl, Zimmerman, He, Isbell, Tuntland, Lin, Lu, Shearer, Smith, Diaz, Asher and Ramirez2013).
The use of amphotericin B and miltefosine for leishmaniasis treatment are examples of successful repurposing. Other active drugs that were or are used for human or canine leishmaniasis, such as various azoles, pentoxyfilline and allopurinol were all repurposed from other indications. Drug screening based on drug repurposing is still an active area of research in leishmaniasis. Examples include the demonstration of antileishmanial activity of protein kinase inhibitors (Sanderson et al. Reference Sanderson, Yardley and Croft2014), phosphoinositide-3-kinases (Diaz-Gonzalez et al. Reference Diaz-Gonzalez, Kuhlmann, Galan-Rodriguez, Madeira Da Silva, Saldivia, Karver, Rodriguez, Beverley, Navarro and Pollastri2011) and of the anti-Mycobacterium drugs clofazimine and delamanid (Evans et al. Reference Evans, Croft, Peters and Neal1989; Kaiser et al. Reference Kaiser, Maser, Tadoori, Ioset and Brun2015b; Patterson et al. Reference Patterson, Wyllie, Norval, Stojanovski, Simeons, Auer, Osuna-Cabello, Read and Fairlamb2016). Another example is the antileishmanial activity of the group of molecules named SERM (selective oestrogen receptor modulators), of which the best-studied example is tamoxifen.
Tamoxifen is uniformly active against all Leishmania species tested so far (Miguel et al. Reference Miguel, Yokoyama-Yasunaka, Andreoli, Mortara and Uliana2007, Reference Miguel, Zauli-Nascimento, Yokoyama-Yasunaka, Katz, Barbieri and Uliana2009) and against clinical isolates from CL and VL patients (Miguel et al. Reference Miguel, Zauli-Nascimento, Yokoyama-Yasunaka, Pereira, Jeronimo, Ribeiro-Dias, Dorta and Uliana2011). The mechanisms of the leishmanicidal activity have not been fully elucidated but do not depend on interaction with oestrogen receptors (Bonano et al. Reference Bonano, Yokoyama-Yasunaka, Miguel, Jones, Dodge and Uliana2014). Amongst other effects, the drug reduces the acidification of the parasitophorous vacuoles that normally harbour a hydrolytic acid environment (Miguel et al. Reference Miguel, Yokoyama-Yasunaka, Andreoli, Mortara and Uliana2007).
Tamoxifen is used continuously for 5 years in breast cancer patients, with minimal side-effects in the short term (Gajdos and Jordan, Reference Gajdos and Jordan2002), and therefore should be safe for leishmaniasis treatment. Interestingly, all attempts to generate parasites resistant to tamoxifen in vitro were unsuccessful (Coelho et al. Reference Coelho, Trinconi, Senra, Yokoyama-Yasunaka and Uliana2015), indicating that this might be an interesting molecule to use in combination therapy.
In vitro drug screening, whatever the strategy used, will produce candidates to new drugs that need to be tested in vivo, in suitable animal models.
ANIMAL MODELS
The definition of a relevant animal model should be based on the purpose of the test. In the case of animal models for drug development, it is to be expected that they should mimic natural infection and exhibit pathology, immune response and clinical evolution comparable with the disease in humans. Ideally, drug pharmacokinetics and pharmacodynamics in the model should also be comparable with humans. The animals should also be easy to keep and handle and preferably not very expensive. One has to take into consideration that this is a chronic disease and long periods will be required to allow response to be noticed and confirmed.
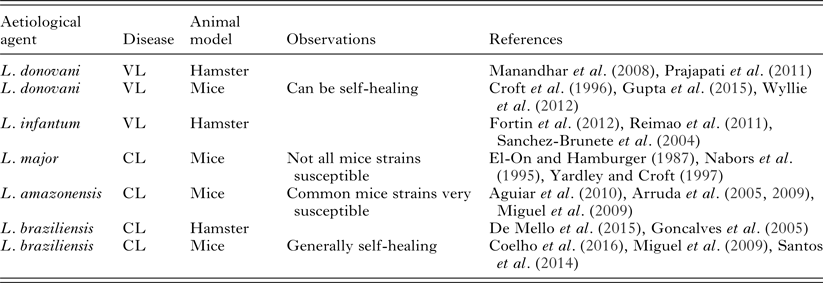
The establishment of a good experimental model for leishmaniasis will depend on fixing quite a number of variables. The multiplicity of leishmaniasis clinical presentations presents the first challenge. Animal models of localized CL and of VL have been described (Table 2). However, we have not yet found good models for mucosal, diffuse or disseminated leishmaniasis.
Table 2. Commonly used leishmaniasis experimental models employed for in vivo testing
The experimental design of an in vivo efficacy test for a candidate drug has to start from the choice of parasite and host species (and therefore of clinical disease) that will be used as a model. Within the parasite species chosen, we should also define which strain will be used, since even within the same species, virulence and behaviour can be variable, as well as response to treatment. Strains of L. donovani from India and Africa are good examples of this heterogeneity (Kauffmann et al. Reference Kauffmann, Dumetz, Hendrickx, Muraille, Dujardin, Maes, Magez and De Trez2016). It is important to keep in mind that Leishmania parasites kept under in vitro conditions can lose their virulence.
As for the inoculum appropriate to mimic the natural infection, the ideal procedure would include sandfly transmission, given the impact of insect factors in the success of the infection (Sacks and Kamhawi, Reference Sacks and Kamhawi2001; Aslan et al. Reference Aslan, Dey, Meneses, Castrovinci, Jeronimo, Oliva, Fischer, Duncan, Nakhasi, Valenzuela and Kamhawi2013). However, that is not practical since phlebotomine colonies are extremely difficult to keep. Instead, one frequently settles for using metacyclic parasites, which are the infective form inoculated by the sandfly. Separation of metacyclic from procyclic promastigotes is well established for some species (Da Silva and Sacks, Reference Da Silva and Sacks1987) but, for most Leishmania species, the best that can be achieved is to enrich the population in metacyclics by density gradient (Spath and Beverley, Reference Spath and Beverley2001).
The number of parasites used in the inoculum has an impact on the development of disease. Generally but within limits, the higher the number of parasites used in the inoculum, the shorter is the incubation period. Higher numbers in the inoculum may also be associated with a more disseminated pattern of disease, with visceralization of cutaneous species (Ribeiro-Romao et al. Reference Ribeiro-Romao, Moreira, Osorio, Cysne-Finkelstein, Gomes-Silva, Valverde, Pirmez, Da-Cruz and Pinto2014).
Also, parasites inoculated at different sites in the same host species may behave distinctly. For example, L. amazonensis inoculation in C57Bl/6 mice in the rump skin or in the footpad results in the development of lesions with different characteristics: while rump inoculation led to the development of a large ulcerated lesion, the same number of parasites inoculated in the footpad led to only minimal oedema (Felizardo et al. Reference Felizardo, Gaspar-Elsas, Lima and Abrahamsohn2012). For VL models, inoculation in the venous territory is preferable to establish infection successfully (Moreira et al. Reference Moreira, Vitoriano-Souza, Roatt, Vieira, Coura-Vital, Cardoso, Rezende, Ker, Giunchetti, Carneiro and Reis2016).
The time to start the treatment is the next question. Some investigators choose to perform proof-of-concept tests of drug efficacy treating inoculated animals immediately after (sometimes even prior to) parasite inoculation. These are very artificial conditions and would never be encountered in clinical settings. In our opinion, the infection has to be fully established before treatment is initiated, either with lesions clearly observable in the case of CL models, or at times when the infection has been demonstrated to be fully established in VL models. Lately, with the advent of parasites expressing reporter proteins, it is possible to make use of in vivo imaging to establish the parasite burden of subject animals before initiating treatment.
Dose, route and frequency of administration, as well as duration of treatment have to be carefully chosen. These decisions would be ideally made based on solubility, pharmacokinetics and pharmacodynamic properties of the test drug. Unfortunately, these data are not always available and are not easily obtained for a new compound. The next best thing, in our opinion, is to determine the maximal tolerated dose that can be used as a starting point in a proof-of-concept testing.
Parameters of efficacy have to be determined. The inoculation of some L. braziliensis strains into BALB/c mice is followed by the appearance of a lesion that heals spontaneously in some weeks (De Moura et al. Reference De Moura, Novais, Oliveira, Clarencio, Noronha, Barral, Brodskyn and De Oliveira2005). In these circumstances, the only possible measure of efficacy is to compare the development of lesions in treated and untreated animals at the point of maximal lesion size and parasite burden for control animals (Miguel et al. Reference Miguel, Zauli-Nascimento, Yokoyama-Yasunaka, Katz, Barbieri and Uliana2009). On the other hand, disease established by inoculation of L. infantum chagasi in hamsters develops into a chronic disease that ultimately leads to death (Reimao et al. Reference Reimao, Oliveira, Trinconi, Cotrim, Coelho and Uliana2015). In this model, a possible end-point could be survival. The method to evaluate efficacy is also variable depending on the model. Lesion size may be useful for CL evaluation, but it has to be taken carefully. For example, L. amazonensis lesions in BALB/c mice increase progressively in size until they ulcerate. From that point onwards, the size of the lesion, measured as footpad thickness, for example, decreases. That could be mistaken as a sign of drug-driven improvement if only the lesion size is considered.
The most strict criterion to determine drug efficacy is the evaluation of parasite burden. Classically, this was done in leishmaniasis by limiting dilution (Lima et al. Reference Lima, Bleyenberg and Titus1997), a cumbersome technique. More recently, real-time PCR is being employed as an alternative to limiting dilution (Nicolas et al. Reference Nicolas, Prina, Lang and Milon2002; Srivastava et al. Reference Srivastava, Sweat, Azizan, Vesely and Kyle2013). Both limiting dilution and qPCR require animal sacrifice since tissue is obtained and processed for parasite quantification. Bioimaging has brought several improvements to this scenario. Sensitive reporters have been applied successfully to Leishmania detection, allowing the determination of parasite burden before treatment is initiated and during the follow up of the same animals. Good correlations have been observed between bioimaging and the most traditional techniques (Lang et al. Reference Lang, Goyard, Lebastard and Milon2005; Michel et al. Reference Michel, Ferrua, Lang, Maddugoda, Munro, Pomares, Lemichez and Marty2011; Reimao et al. Reference Reimao, Trinconi, Yokoyama-Yasunaka, Miguel, Kalil and Uliana2013, Reference Reimao, Oliveira, Trinconi, Cotrim, Coelho and Uliana2015).
A sustained response to treatment is an important characteristic of a good candidate drug and that should be evaluated through a thorough post-treatment follow-up of treated animals.
CONCLUSIONS/FUTURE DIRECTIONS
Shortcomings in leishmaniasis chemotherapy are evident. Given the reduced clinical success rates, parasite resistance, toxicity and/or cost of current drugs, new effective alternatives are clearly necessary. Due to the wide variety of aetiological agents, pre-clinical studies should include animal models representative of the different forms of the disease. It is very important to make sound choices on experimental models to test drug candidates. General criteria for efficacy evaluation on pre-clinical studies should be agreed upon, so that studies performed in different laboratories can be compared. The development of methods for species-specific diagnosis easily performed and widely accessible would allow the investigation of better protocols to treat specific forms of CL. Combination therapy is clearly a necessity and extensive clinical tests of combination schemes should pave the way to fundamentally change leishmaniasis therapy.
ACKNOWLEDGEMENT
We wish to thank Dr. Jeffrey J. Shaw for support and countless fruitful discussions. We thank Jenicer Yokoyama-Yasunaka for the continuous support.
FINANCIAL SUPPORT
This work was supported by the São Paulo Research Foundation (FAPESP) (grant number 2015/09080-2); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant number 473343/2012-6). C. T. T. and A. C. C. were supported by FAPESP fellowships (2015/23832-7 and 2012/14629-5, respectively). S. R. B. U. is the recipient of a senior researcher scholarship from CNPq.


