


- CR
-
caloric restriction
- ETC
-
electron transport chain
- IUGR
-
in utero growth restricted
- ROS
-
reactive oxygen species
- SOD
-
superoxide dismutases
- SSB
-
single-strand break
- T2D
-
type 2 diabetes
- TOR
-
target of rapamycin
The global burden of ageing
Globally, the population is steadily ageing with individuals aged 60 years and older doubling since 1980 and this is forecast to reach 2 billion by 2050. This can be construed as an indicator of improved global health; however, an ageing population also brings increases in the prevalence of age-associated diseases. About 75 % of all deaths in the USA and other developed countries now result from age-related conditions such as cancer, diabetes, heart disease, stroke, neurodegeneration and dementia. Environmental factors during any stage of the life course can influence risk of these conditions. However, this review will focus on the importance of the environment during very early life on modulating risk of age-associated metabolic diseases, such as type 2 diabetes (T2D) and CVD.
Ageing and the thrifty phenotype hypothesis
Over 20 years ago, Hales and Barker proposed the thrifty phenotype hypothesis( Reference Hales and Barker 1 ) in which it was postulated that under conditions of suboptimal in utero nutrition, the fetus permanently alters its organ structure and adapts its metabolism to ensure immediate survival of the organism. This can occur through the ‘sparing’ of certain vital organs, especially the brain, at the expense of other organs, including the heart, pancreas, kidney and skeletal muscle (Fig. 1). This hypothesis was the result of striking epidemiological studies in which Barker and colleagues( Reference Barker, Winter and Osmond 2 ) and Hales et al. ( Reference Hales, Barker and Clark 3 ) determined the prevalence of CVD and T2D in 64-year-old men. It was found that those individuals who were born smaller and weighed significantly less at age 1 year, compared with those with a normal birth-weight, had the highest mortality rates from IHD( Reference Barker, Winter and Osmond 2 ), a higher incidence of T2D and abnormal glucose tolerance( Reference Hales, Barker and Clark 3 ). These findings have been robustly reproduced in many populations worldwide( Reference Phipps, Barker and Hales 4 , Reference Lithell, McKeigue and Berglund 5 ). Furthermore, studies in monozygotic twins discordant for T2D have strongly implicated the environment playing a pivotal role in mediating the associations between low birth-weight and subsequent development of disease in later life. These studies demonstrated that the diabetic twin had the lower birth-weight( Reference Pouslen, Vaag and Kyvik 6 – Reference Bo, Cavelli-Perin and Scaglione 8 ). Birth-weight has also been shown to be non-genetically linked with insulin sensitivity and glucose intolerance in elderly subjects( Reference Monrad, Grunnet and Rasmussen 9 , Reference Grunnett, Vielworth and Vaag 10 ), perhaps suggesting that the ageing process may be exacerbating the effects of low birth-weight. Studies of individuals exposed to famine while in utero, have also demonstrated very powerful direct evidence for the importance of maternal nutrition in early development. Ravelli et al. demonstrated that exposure to the Dutch famine of 1944–1945 significantly increased the risk of obesity( Reference Ravelli, Stein and Susser 11 ) and reduced glucose tolerance in later life( Reference Ravelli, van der Mullen and Michels 12 ).
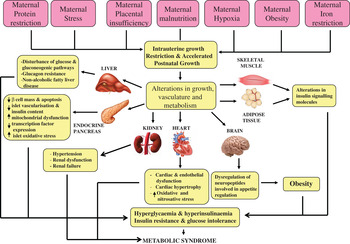
Fig. 1. (colour online) The thrifty phenotype hypothesis.
Accelerated postnatal growth
Although sparing of vital organs such as the brain, at the expense of other organs, is beneficial in continued conditions of poor postnatal nutrition, several epidemiological studies have shown that this is detrimental in postnatal conditions of adequate or overnutrition. A study of a cohort of South African children demonstrated that those who were born small, but who underwent rapid postnatal weight gain had the worst glucose tolerance at age 7 years( Reference Crowther, Cameron and Trusler 13 ). Moreover, Indian children with a low birth-weight, who underwent rapid postnatal catch-up growth, were insulin resistant and had increased cardiovascular risk factors at age 8 years( Reference Yajnik 14 ). In a Finnish cohort, males who were born small, but had higher than average body mass in childhood, had higher mortality from CHD, compared with low birth-weight men with average body masses( Reference Eriksson, Forsen and Tuomilehto 15 ). Moreover, early development of adiposity and insulin resistance was observed in children who were small for gestational age but rapidly gained weight postnatally( Reference Ibanez, Ong and Dunger 16 ).
Utilisation of animal models in developmental programming
These observations from studies in human subjects have been supported by numerous studies in animal models. These have been carried out in a range of species including non-human primates, sheep and rodents. These have all been important in providing proof of principle that the early environment can influence long-term health and have provided insight into underlying mechanisms. For obvious practical reasons, the majority of studies that have addressed the impact of the early environment on ageing parameters have been carried out in rodents. Conditions that have been associated with early nutrition are also those with an ageing-associated aetiology; therefore, this review will focus upon models of developmental programming and their associations with the ageing process.
Maternal low-protein restriction
The maternal low-protein rat model is one of the most extensively studied of all programming models and was established by Snoeck et al. ( Reference Snoeck, Remacle and Reusens 17 ). This involved the administration of a low (8 % protein) or a ‘normal’ (20 %) protein diet to pregnant rats. These diets had similar fat content and were made isoenergetic by the addition of carbohydrates to the low-protein diet. The low-protein fed rat offspring were in utero growth restricted (IUGR) with significantly reduced birth-weights compared with control-fed offspring; however the placental weights were similar between groups( Reference Snoeck, Remacle and Reusens 17 ). The IUGR rat offspring had severe perturbations in pancreatic islets, including reduced pancreatic islet cell size and proliferation, diminished β-cell mass, and decreased islet vascularisation, and increased islet apoptosis( Reference Dahri, Snoeck and Reusens-Billen 18 , Reference Petrik, Reusens and Arany 19 ). These alterations were programmed in utero as they were irreversible after the islets were removed from the disturbed metabolic environment( Reference Dahri, Snoeck and Reusens-Billen 18 , Reference Cherif, Reusens and Dahri 20 ). Strong justification for the use of this model to accurately dissect the mechanistic changes in human studies has been demonstrated. Offspring of protein-restricted rat dams had alterations in key insulin signalling molecules in skeletal muscle and adipose tissue, which were strikingly similar in specificity and magnitude to those observed in skeletal muscle and adipose tissue from low birth-weight men( Reference Ozanne, Jensen and Tingey 21 , Reference Ozanne, Olsen and Hansen 22 ). Male rat offspring exposed to the low-protein diet in utero demonstrate an age-dependent loss of glucose tolerance from insulin-sensitive and glucose-tolerant phenotype in early life( Reference Petry, Ozanne and Wang 23 ), through insulin resistance during middle age( Reference Shepherd, Crowther and Desai 24 ) and to the development of frank diabetes in old age( Reference Petry, Dorling and Pawlak 25 ). This age-associated loss of insulin sensitivity and glucose tolerance is also observed in human populations( Reference Chen, Bergman and Pacini 26 ). Therefore, it is certainly feasible that accelerated cellular ageing may be a potential underlying mechanism of developmental programming. Maternal protein restriction has also been demonstrated to lead hypertension and age-associated deterioration of renal function in offspring( Reference Nwagwu, Cook and Langley-Evans 27 ). This was associated with reduced renal mitochondrial respiration rate( Reference Engeham, Mdaki and Jewell 28 ) and impairment of recovery of rat hearts subjected to ischaemia/reperfusion injury( Reference Elmes, Gardner and Langley-Evans 29 ).
Cellular senescence and developmental programming
In 1961, Hayflick first demonstrated that all somatic cells have a finite division potential (between 40 and 60 population doublings in normal human fetal cells) before they cease division and enter a cell senescent phase( Reference Hayflick and Moorhead 30 ). A potential explanation for this replicative senescent phenotype of somatic cells may be derived from ‘the free radical theory of ageing’ hypothesis; established by Harman in 1956( Reference Harman 31 ), which suggested that free radicals accumulate during the ageing process and cause damage to cellular macromolecules including DNA, proteins and lipids, mediating the development of various pathologies, which could result in cellular senescence and organismal ageing. This has been supported by several studies( Reference Stadtman 32 , Reference Sohal, Agarwal and Dubey 33 ). However, it is known that free radicals such as superoxide (•O2 −) and reactive nitrogen species (ROS) including nitric oxide are formed in physiological as well as pathological processes (reviewed in ( Reference Bergendi, Benes and Durackova 34 , Reference Valko, Rhodes and Moncol 35 )); therefore it is only when excess •O2 − is produced, that damage to DNA, proteins and lipids through oxidation occurs (reviewed in( Reference Valko, Leibfritz and Moncol 36 )). Excess •O2 − can also combine with excess nitric oxide• and form peroxynitrite, which can damage proteins through nitrotyrosination and increase DNA single-strand break (SSB) damage( Reference Salgo, Bermudez and Squadrito 37 ) and damage lipids via peroxidation. Therefore, unsurprisingly, oxidative and nitrosative stress have been implicated in various pathological diseases, all of which have an age-related aetiology, including CVD( Reference Kukreja and Hess 38 , Reference Csiszar, Pacher and Kaley 39 ), cancer( Reference Valko, Izakovic and Mazur 40 ), neurological disorders( Reference Jenner 41 , Reference Sayre, Smith and Perry 42 ) and diabetes( Reference Rosca, Mustata and Kinter 43 ). Consequently, organisms have evolved to develop cellular defence mechanisms in order to maintain redox homoeostasis; so that levels are sufficient for physiological, beneficial cell functions, but can prevent high pathophysiological levels, which are associated with age-associated pathology.
Antioxidant defence is a major mechanism by which cells can regulate redox homoeostasis. Cellular antioxidants can be either enzymatic or non-enzymatic. Enzymatic antioxidants include superoxide dismutases (SOD), which are the first line of cellular defence against excessive O2 • and are responsible for the catalysis of the surfeit O2 • into H2O2 and O2. There are three major isoforms of SOD known to exist in eukaryotic cells and are named according to their cell localisations. Copper–zinc SOD (SOD1) is localised to the cytoplasm, manganese SOD (SOD2) is expressed within mitochondria, and extracellular SOD (SOD3) is found within the extracellular matrix. Although H2O2 is less volatile than O2, it has the potential to react with transition metals (such as Fe2+ or Cu+) to generate the highly destructive hydroxyl radical (OH•)( Reference Leonard, Harris and Shi 44 , Reference Valko, Morris and Cronin 45 ). Therefore, a further group of antioxidant enzymes, the peroxidases, including peroxiredoxins, catalase, glutathione peroxidases and glutathione reductase convert H2O2 into H2O and O2. Non-enzymatic endogenous antioxidants include ascorbate (vitamin C) and ubiquinol (coenzyme Q).
Studies utilising the maternal low-protein restriction of Snoeck et al. ( Reference Snoeck, Remacle and Reusens 17 ) have demonstrated that pancreatic islets from older rat offspring (age 15 months) had an accelerated cellular ageing phenotype, with evidence of decreased antioxidant defence capacity, increased fibrosis and oxidative stress, compared with age-matched control counterparts( Reference Tarry-Adkins, Chen and Jones 46 ). Moreover, a maternal protein restriction model in goats (40 % protein restriction) resulted in reductions of SOD in the offspring( Reference He, Sun and Tan 47 ).
Mechanisms of ageing and developmental programming
Telomeres are repeating guanine-rich nucleotide DNA sequences, which prevent chromosomal ends from being recognised as double-strand DNA breaks and prevent chromosomal deterioration or fusion with neighbouring chromosomes( Reference Blackburn 48 ). They are also particularly susceptible to oxidative damage due to their guanine–cytosine-rich sequences( Reference Oikawa and Kawanishi 49 , Reference Kawanishi and Oikawa 50 ). Telomeres shorten in eukaryotic somatic cells after each cellular division (between 20 and 200 base-pairs per division in human cells), due to the ‘end replication problem’( Reference Olivnikov 51 ) and this erosion is an integral part of the ageing process( Reference Armanios 52 , Reference Greider and Blackburn 53 ). Oxidative stress mediated damage to telomeric DNA is another major mechanism for telomere attrition( Reference Richter and von Zglinicki 54 , Reference von Zglinicki 55 ) and may contribute to the development of replicative senescence, as well as mediating oxidative damage induced by cellular senescence. DNA SSB damage has been reported to be a major mechanism of telomere shortening( Reference von Zglinicki, Pilger and Sitte 56 ) and oxidative stress can increase frequency of this damage( Reference Honda, Hjelmeland and Handa 57 ). Interestingly, telomeric DNA is deficient in the ability to repair DNA SSB, in contrast to the majority of genomic DNA( Reference Petersen, Saretzki and von Zglinicki 58 ). When telomeres reach a critically short length, they become dysfunctional and undergo a conformational change, resembling double-stranded breaks, which causes cells to enter irreversible G0/G1 growth arrest (senescence) or apoptosis( Reference Harley, Fuchter and Greider 59 ). This senescence is triggered by activation of the p53/p21/p19 and p16INK/retinoblastoma pathways( Reference Stein, Drullinger and Soulard 60 ).
Differences in the telomere length have been implicated in developmental programming. When low-protein-fed offspring were suckled by mothers fed a 20 % ‘normal’ protein diet, these ‘recuperated’ animals underwent rapid postnatal growth, had reduced longevity( Reference Jennings, Ozanne and Hales 61 ) and demonstrated accelerated telomere shortening in aorta( Reference Tarry-Adkins, Martin-Gronert and Chen 62 ), pancreatic islets( Reference Tarry-Adkins, Chen and Smith 63 ) and renal( Reference Jennings, Ozanne and Hales 61 ) tissues. This was accompanied by a reduction in SOD2 and increased p21 and p16INK in the pancreatic islets( Reference Tarry-Adkins, Chen and Smith 63 ). Moreover, evidence of accelerated cellular ageing is present in cardiac tissue from recuperated rats from weaning (age 22 d). This included evidence of increased oxidative stress, alterations in antioxidant defence capacity, increased DNA SSB damage, and increased DNA damage repair enzymes( Reference Tarry-Adkins, Martin-Gronert and Fernandez-Twinn 64 ). Increased frequency of DNA SSB damage was also observed in renal tissue from age 12-month recuperated animals compared with controls, which was associated with mitochondrial dysfunction( Reference Shelley, Tarry-Adkins and Martin-Gronert 65 ).
Ozanne and Hales have also demonstrated that offspring of control (20 %) protein-fed dams that were suckled by low (8 %) protein-fed dams, until weaning, had increased longevity in both rats( Reference Jennings, Ozanne and Hales 61 ) and mice( Reference Ozanne and Hales 66 ), compared with animals fed a control (20 %) protein diet during both gestation and lactation. These mice were also protected against longevity reduction when fed an obesogenic diet after weaning( Reference Ozanne and Hales 66 ). These data may suggest that the mild stress of reduced protein intake during the suckling postnatal period is eliciting a protective effect of lifespan extension, which is in keeping with a recent ageing theory; ‘the hormesis hypothesis of ageing’, first highlighted by Minius in 2000( Reference Minois 67 ). This suggests that the exposure of an organism to a mild stress (which in larger doses would be detrimental) can improve the functional ability of organisms. Much support for this theory has been gained from studies conducted in a variety of animal models including yeast, flies, worms and rodents, which have demonstrated that a mild stress, such as caloric restriction (CR) can increase longevity (reviewed in( Reference Le Bourg 68 )). Indeed, CR is the most potent and reproducible environmental variable capable of extending lifespan.
Uterine placental ligation
Uterine placental ligation( Reference Simmons, Gounis and Bangalore 69 , Reference Ogata, Bussey and Finley 70 ) is an elegant model of placental insufficiency which results in an IUGR phenotype. This model does not completely restrict blood supply to the fetus, but reduces it adequately to reflect human uteroplacental insufficiency, which can be caused by preeclampsia, maternal smoking and abnormalities in placental development. These neonatal IUGR rats demonstrated reduced glucose, insulin, insulin-like growth factor 1, amino acid and oxygen concentrations( Reference Simmons, Gounis and Bangalore 69 – Reference Unterman, Lascon and Gotway 71 ). These IUGR animals later developed age-associated diabetes( Reference Simmons, Templeton and Gertz 72 ). Other studies demonstrated that bilateral uterine vessel ligation resulted in IUGR rat offspring with increased arterial vascular stiffness and selective endothelial uterine artery dysfunction( Reference Maccuza, Wlodek and Dragomir 73 ) and caused nephron deficits and modest renal insufficiency( Reference Moritz, Maccuza and Siebel 74 ).
Placental insufficiency in both animal models of uterine placental ligation( Reference Heltemes, Gingery and Soldner 75 ) and in human studies( Reference Karowicz-Bilinska, Suzin and Sieroszewski 76 – Reference Wang and Walsh 79 ) is strongly associated with the development of oxidative stress, and it is thought that the mitochondria plays an important role in mediating this effect. Mitochondria are a major source of ROS generation in cells and are generated from the mitochondrial electron transport chain (ETC). This couples electron transfer from an electron donor (such as NADH) to an electron acceptor (O2), with consequential transfer of protons across the membrane and production of an electrochemical proton gradient, which can drive synthesis of ATP. However, during this process, electrons can leak out of the complexes of the ETC and can combine with O2 to form the free radical •O2 −. In 1980, Miquel proposed the ‘mitochondrial theory of free radicals in ageing’, in which he suggested that mitochondrially generated ROS can cause accumulation of somatic mutations to the mitochondrial genome and result in cellular senescence and apoptosis in post-mitotic cells( Reference Miquel, Economos and Fleming 80 ). This establishes a ‘vicious circle’ of mitochondrial DNA damage, altered oxidative phosphorylation and overproduction of ROS. Others have supported this theory, whereby mitochondria isolated from old animals produced more ROS compared with their younger counterparts( Reference Sohal and Sohal 81 ). Furthermore, accumulation of mitochondrial DNA damage( Reference Wei and Lee 82 , Reference Pang, Ma and Wei 83 ) and reduction of mitochondrial respiratory chain function in human tissue( Reference Yen, Chen and King 84 , Reference Trounce, Byrne and Marzuki 85 ) and that from rats and dogs( Reference Sugiyama, Takasawa and Hayakawa 86 ) have been demonstrated with increased age.
Simmons and colleagues have demonstrated an impairment of oxidative phosphorylation in both skeletal muscle( Reference Selak, Storey and Peterside 87 ) and liver mitochondria( Reference Peterside, Selak and Simmons 88 ) of IUGR rats, as well as a progressive accumulation of ROS generation and mitochondrial DNA mutations, and a progressive decline in activities of complex I and III of the ETC in the pancreatic islets of IUGR rats( Reference Simmons, Suponitsky-Kroyter and Selak 89 ). This may be associated with the low oxygen levels observed in IUGR fetuses, as it has been shown that hypoxia decreases the activity of ETC complexes and increases ROS production( Reference Esposti and McLennan 90 , Reference Chandel, Budinger and Schumaker 91 ). Taken together this may suggest that insulin-sensitive tissues of IUGR rats exhibit mitochondrial dysfunction, which may lead to accelerated cellular ageing of these tissues. Several studies have also utilised ovine models of placental insufficiency to induce IUGR in offspring. This model bears striking similarities to the human situation, with decreased fetal and placental weights, reduced fetal oxygen concentrations, decreased placental oxygen transfer rates, decreased fetal glucose and insulin concentrations, decreased glucose-stimulated insulin secretion and diminished β-cell mass (reviewed in( Reference Barry, Rozance and Anthony 92 )).
Maternal hypoxia
Maternal hypoxia has been studied in a number of human populations. It has been demonstrated that babies born to mothers living at high altitude are also significantly smaller than those born at normal altitude, and that this IUGR was associated with fetal hypoglycaemia and hypoinsulinaemia( Reference Zamudo, Torricos and Fik 93 , Reference Kingdom and Kaufman 94 ). Placental insufficiency is thought to contribute to this, through impaired placental invasion of maternal blood vessels or through poor placental vascular development( Reference Zamudo, Torricos and Fik 93 , Reference Kingdom and Kaufman 94 ). Animal models of maternal hypoxia recapitulate these findings, demonstrating that IUGR can result from maternal hypoxia( Reference De Grauw, Myers and Scott 95 , Reference Jacobs, Robinson and Owens 96 ). Giussani et al. initially, using a chick embryo model( Reference Giussani, Salinas and Villena 97 ) and subsequently a rat model( Reference Herrera, Camm and Cross 98 ) demonstrated that this IUGR was due to the hypoxia per se, and not maternal malnutrition, as severe hypoxia can result in reduced maternal food intake. Maternal hypoxia has also been shown to programme cardiac dysfunction in these rat and chick embryo models( Reference Giussani, Camm and Nui 99 , Reference Rouwet, Tintu and Schellings 100 ), including promotion of fetal cardiac overload, leading to ventricular and aortic wall thickening( Reference Camm, Hansell and Kane 101 ) and endothelial dysfunction( Reference Morton, Rueda-Clausen and Davidge 102 ). As observed in the model of protein restriction and placental insufficiency, evidence of oxidative stress has been demonstrated in this model. This has been reported in the placenta (increased levels of 4-hydroxynonenal and heat-shock protein 70)( Reference Richter, Camm and Modi 103 ), and in the heart (increased nitrotyrosine staining and heat-shock protein 70)( Reference Morton, Rueda-Clausen and Davidge 102 ) of rats and in sheep( Reference Thakor, Richer and Kane 104 ). Furthermore, the administration of the antioxidants allopurinol( Reference Thakor, Richer and Kane 104 ) and vitamin C( Reference Herrara, Kane and Hansell 105 ) reversed these phenotypes in sheep. Therefore, it is plausible that hypoxia accelerates ageing in the placenta and cardiovascular systems, potentially leading to eventual cellular senescence, apoptosis and organ dysfunction.
Maternal stress
Severe maternal stress can induce IUGR in offspring, through various pathways including transplacental transport of the stress hormone glucocorticoid, via activation of the hypothalamic–pituitary–adrenal axis. It has also been shown that babies with a low birth-weight have higher plasma cortisol levels throughout life, which also indicates hypothalamic–pituitary–adrenal-axis programming. Indeed, severe maternal stress can deleteriously affect placental physiology, including alterations in blood flow and changes in metabolism, which impact upon oxygen and glucose availability. This has been demonstrated in both human studies( Reference French, Hagan and Evans 106 – Reference Reinisch, Simon and Karow 108 ), and animal models, including rats( Reference Nyirenda, Lindsay and Kenyon 109 ) and sheep( Reference Sloboda, Newnham and Challis 110 ), where these IUGR offspring demonstrated abnormal glucose tolerance( Reference Nyirenda, Lindsay and Kenyon 109 , Reference Sloboda, Newnham and Challis 110 ). Prenatal exposure to rats with glucocorticoids has also been shown to elevate plasma glucose, insulin and hepatic phosphoenolpyruvate carboxykinase in the next generation( Reference Drake, Walker and Seckl 111 ). Inhibition of 11β-hydroxysteroid (the fetoplacental barrier to maternal glucocorticoids) can also reduce birth-weight, increase the hypothalamic–pituitary–adrenal-axis activity and anxiety-related behaviours, and cause hyperglycaemia, hyperinsulinaemia and hypertension in the offspring (reviewed in( Reference Seckl and Holmes 112 )).
The mechanistic basis linking maternal stress to offspring metabolism is not clear; however oxidative stress is likely to be a major contributory factor. Entringer et al. demonstrated that severe stress in pregnant women was associated with shorter leucocyte telomere length in their children during young adulthood( Reference Entringer, Epel and Kumsta 113 ). Moreover, a recent study in chick embryos demonstrated that in utero exposure to glucocorticoids resulted in higher levels of ROS and increased leucocyte telomere shortening( Reference Haussmann, Longenecker and Marchetto 114 ). In addition, prenatal exposure to high levels of glucocorticoids in rats increased susceptibility of cerebellar granule cells to oxidative stress-induced cell death, increased mitochondrial dysfunction and reduced catalase expression( Reference Ahlbom, Goqvadze and Chen 115 ). These data suggest that severe maternal stress can accelerate cellular ageing. It is common clinical practice for pregnant women at risk of preterm labour to be prescribed with repeat courses of glucocorticoids to aid lung maturation of the premature baby, and indeed studies in sheep treated with glucocorticoids have reported reductions in oxidative stress in the lungs of pre-term lambs( Reference Walther, Jobe and Ikegami 116 , Reference Dani, Corsini and Burchielli 117 ).
Maternal caloric/nutrient restriction
Several models of maternal CR have been used in a variety of formats, ranging from models of moderate to severe restriction. Rat dams exposed to 50 % maternal CR from day 15 of pregnancy until weaning gave birth to offspring that were growth restricted, with a 50 % reduction in body and organ weights at weaning. These offspring also had a 70 % reduction in β-cell mass at age 21 d and decreased insulin content in adulthood( Reference Garofano, Czernichow and Breant 118 ). In this model, as in the model of maternal protein restriction, ageing plays a pivotal role in the development of glucose intolerance( Reference Garofano, Czernichow and Breant 119 ). Franco et al. also utilised a 50 % maternal CR model and demonstrated that both male and female offspring were hypertensive and had endothelial dysfunction( Reference Franco Mdo, Arruda and Fortes 120 ). They also demonstrated that enhanced oxidative stress was a potential mechanism for these observations( Reference Franco Mdo, Dantas and Akamine 121 ). Maternal dietary restriction, using a diet representative of that of Brazil, caused placental oxidative stress in rat dams, which later led to changes in kidney proximal tubule sodium ATPases in the offspring( Reference Vieira-Filho, Lara and Silva 122 ). In another rat model, 50 % maternal CR resulted in offspring that were growth restricted, however when suckled by ad libitum fed rat dams, these offspring underwent rapid postnatal growth, becoming heavier than the control offspring( Reference Desia, Gayle and Babu 123 ). A non-human primate model of maternal nutrient restriction (70 % of control food consumption) resulted in alterations in the renal transcriptome and kidney morphology of the offspring of nutrient restricted primates( Reference Cox, Nijland and Gilbert 124 ). Furthermore, the mammalian target of rapamycin (TOR) signalling pathway was found to be central to this phenotype( Reference Nijland, Schlabritz-Loutsevitch and Hubbard 125 ). Mammalian TOR is a serine/threonine kinase that regulates cell growth, cell proliferation and cell survival and it has recently been suggested that ageing can result from overactivation of TOR or mammalian TOR signalling pathways.
Maternal obesity and gestational diabetes
The deleterious effects of low birth-weight on long-term metabolic health are well established; however, it has become apparent that high birth-weight is also a clear indicator of increased risk of disease in later life. This relationship was initially found in epidemiological studies of Pima Indians( Reference Pettitt, Moll and Knowler 126 ) in which there is a very high prevalence of both T2D and maternal obesity, demonstrating that risk of developing T2D in later life is increased by both low and high birth-weight. This suggests that a U-shaped curve exists between birth-weight and risk of T2D development. A possible mechanism for the elevated risk of T2D in a high birth-weight population is the increased prevalence of gestational diabetes. Glucose can cross the placental barrier but maternal insulin cannot; therefore the fetus must regulate its own glucose homoeostasis by insulin production from fetal β-cells of pancreatic islets. In situations of maternal hyperglycaemia (which occurs in gestational diabetes), higher levels of fetal insulin are produced. Insulin is a potent growth factor in fetal life; therefore this results in gestational diabetic mothers giving birth to macrosomic offspring. Boney et al. demonstrated that macrosomic offspring of mothers with gestational diabetes were at increased risk of developing metabolic syndrome in childhood( Reference Boney, Verma and Tucker 127 ). Human studies have further characterised phenotypes of offspring born to gestationally diabetic mothers. These individuals were more obese and were hyperglycaemic compared with offspring of women who developed diabetes after pregnancy. They also had an increased propensity to develop diabetes in adulthood( Reference Boerschmann, Pfluger and Henneberger 128 ). More recently, studies of children exposed to maternal obesity and gestational diabetes in utero have shown higher incidence of insulin resistance( Reference Zielinsky and Piccoli 129 ), heart hypertrophy( Reference Zielinsky and Piccoli 129 ) and CVD in later life( Reference Krishnaveni, Venna and Hill 130 , Reference Lee, Jang and Park 131 ).
The detrimental effects of overnutrition during fetal and early postnatal life have also been observed in animal models. Offspring of mice fed a highly palatable obesogenic diet before mating and during pregnancy and lactation were hyperphagic in early postnatal life, had increased adiposity, were hypertensive and insulin resistant, and were heavier in later life( Reference Samuelsson, Matthews and Argenton 132 , Reference Nivoit, Morens and Van Assche 133 ). In addition, evidence of mitochondrial dysfunction was observed in this model, with reductions in complex II–III linked activity of the ETC in skeletal muscle of male offspring exposed to maternal obesity( Reference Shelley, Martin-Gronert and Rowlerson 134 ). Our laboratory has demonstrated that maternal obesity in the mouse programmes cardiac hypertrophy in the offspring which was associated with hyperinsulinaemia, protein kinase B, extracellular signal-related kinase and mammalian TOR activation, which was independent of obesity in the offspring. These animals demonstrated evidence of increased oxidative stress with elevation in 4-hydroxynonenal levels and reduction in SOD2 protein expression( Reference Fernandez-Twinn, Blackmore and Siggens 135 ). It has also been shown that maternal obesity prior to conception is associated with altered mitochondria in mouse oocytes and zygotes, including increases in mitochondrial potential, mitochondrial DNA content and biogenesis; moreover ROS generation was also increased, again suggestive of increased oxidative stress and an increased ageing phenotype( Reference Igosheva, Abramov and Poston 136 ). In addition, rat offspring fed a ‘junk food’ diet during pregnancy and lactation have a greater preference for ‘junk food’ and increased obesity in adulthood( Reference Bayol, Farrington and Stickland 137 ). This adiposity was more pronounced in females( Reference Bayol, Simbi and Bertrand 138 ) and this diet has recently been shown to promote non-alcoholic fatty liver disease in the offspring( Reference Bayol, Simbi and Fowkes 139 ).
Maternal iron restriction
Iron deficiency is very common with 2 billion people affected globally and causes many abnormalities including long-term cognitive impairment (reviewed in ( Reference Beard 140 )). Epidemiological studies have shown that maternal iron deficiency is associated with increased incidence of IUGR( Reference Singla, Tyagi and Kumar 141 , Reference Lee, Kim and Kim 142 ). Large placental weights and a high ratio of placental weight to birth-weight are observed( Reference Godfrey, Redman and Barker 143 ). A potential mechanism for these altered whole body and organ growth trajectories may be the alterations of placental cytokine expression, which was observed in rat offspring born to iron-restricted mothers( Reference Gambling, Charinia and Hannah 144 ). Other laboratories have shown that maternal iron restriction causes long-term problems for the offspring, including hypertension( Reference Lewis, Forhead and Petry 145 , Reference Gambling, Dunford and Wallace 146 ), changes in renal morphology( Reference Lisle, Lewis and Petry 147 ) and changes in placental vascularisation( Reference Lewis, Doherty and James 148 ). So far, models of maternal iron restriction have not addressed the potential role of oxidative stress.
Developmental programming, epigenetics and oxidative stress
Epigenetics can be defined as any change in phenotype or gene expression caused by modifications (including DNA methylation or histone methylation, acetylation, phosphorylation and ubiquination), which is independent of changes in genotype. It is known that environmental cues can be ‘remembered’ during the lifespan and changes to the epigenetic landscape are associated with the ageing process. It is now emerging that epigenetic modification of transcription factors is a common underlying mechanism in many models of developmental programming. This includes epigenetic silencing of the pancreatic development gene Pdx1 in offspring of mothers with placental insufficiency( Reference Park, Stoffers and Nicholls 149 ). Moreover, rat offspring of a maternal low-protein diet have demonstrated epigenetic alterations in the PPAR-α( Reference Lillycrop, Phillips and Jackson 150 ). In addition, we have also demonstrated that in utero exposure to a low-protein diet can alter the dynamics of age-associated epigenetic changes at the hepatocyte nuclear factor 4-α locus( Reference Sandovici, Smith and Dekker-Nitert 151 ). Oxidative stress can induce epigenetic modifications, including DNA methylation and histone modification. DNA breaks caused by oxidant damage can provide access sites to DNA methyltransferases, which promote DNA methylation; moreover ROS can directly interact with histones resulting in disruption of normal gene expression. During oxidative stress, guanine residues are replaced with the oxygen radical adduct 8-hydroxyguanine and this can profoundly alter methylation status of adjacent cytosines and cause alteration of gene expression( Reference Cerda and Weitzman 152 ). Therefore, it is feasible that a molecular mechanism for the observed changes in epigenetic modification in several models of developmental programming may well be the development of oxidative stress.
The potential of antioxidant therapy in models of developmental programming
A common mechanism for the observed phenotypic outcomes in most of these animal models of developmental programming is oxidative stress and this finding fully recapitulates epidemiological studies in which IUGR children demonstrate increased lipid peroxidation( Reference Franco Mdo, Dantas and Akamine 153 – Reference Hracsko, Orvos and Norvak 155 ), increased DNA damage( Reference Hracsko, Orvos and Norvak 155 ) and reduced antioxidant enzyme capacity( Reference Hracsko, Orvos and Norvak 155 ) compared with children of a normal birth-weight. Therefore, several animal models of developmental programming have focused upon using antioxidants as a therapeutic intervention in order to reverse the observed phenotypic changes. These included the reduction of adiposity and improvement of glucose tolerance resulting from the exposure to a high-fat diet, by maternal supplementation with high concentrations of vitamins A, C, E and selenium( Reference Sen and Simmons 156 ). Moreover, cardiac dysfunction in both rat( Reference Camm, Hansell and Kane 101 ) and sheep( Reference Thakor, Richer and Kane 104 ) models of acute hypoxia has been demonstrated to be improved by maternal supplementation with vitamin C. In addition, prevention of hypoxia-induced placental oxidative stress with vitamin C has also been observed in rats( Reference Richter, Camm and Modi 103 ). In a model of maternal protein restriction, hypertension, vascular dysfunction and microvascular rarefaction were prevented by antenatal treatment with the antioxidant Lazaroid( Reference Cambonie, Comte and Yzdorczyk 157 ) however, the physiological relevance of this antioxidant dose for translation into human studies is not known. Prenatal exposure to hypoxia in sheep also increased oxidative stress in the offspring, and maternal administration of an antioxidant; allopurinol reversed this phenotype( Reference Herrara, Kane and Hansell 105 , Reference Derks, Oudijk and Torrence 158 ). These studies show proof of principle that maternal antioxidants can prevent detrimental programming effects; however, the doses used to achieve these effects may not be able to be used safely in pregnant women. In addition, they focus on interventions to the mother.
Future perspectives
Many epidemiological studies and animal studies have demonstrated that growth and nutrition during early life development can influence the long-term physiology and health and as a result, the lifespan of the individual. However, the molecular mechanisms that underpin this phenomenon are only starting to be dissected. Studies in human populations, animal models and cell systems all seem to be pointing to the accumulation of oxidative stress and consequently accelerated cellular ageing, as an important underlying molecular mechanism (Fig. 2). Several developmental programming studies have demonstrated proof of principle that maternal antioxidant therapy may reverse some of deleterious effects of a suboptimal early life exposure. However, both animal( Reference Petry, Dorling and Pawlak 25 , Reference Nwagwu, Cook and Langley-Evans 27 ) and human studies( Reference Curhan, Willet and Rimm 159 , Reference Mi, Law and Zhang 160 ) demonstrate that evaluation of suboptimal in utero exposure may only be possible until later postnatal life and therefore further studies are required to address the potential beneficial effects of targeted postnatal antioxidant supplementation. This targeted intervention has the potential to combat the burden of common age-related diseases such as T2D, CVD and the metabolic syndrome that represent the major health-care issues of the 21st century.
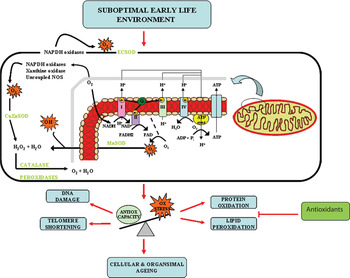
Fig. 2. (colour online) Oxidative stress (Ox stress) as an underlying mechanism of developmental programming and ageing.
Acknowledgements
The authors would like to thank James Adkins for IT support in the preparation and submission of this manuscript.
Financial support
This work was supported by The British Heart Foundation. S. E. O is a British Heart Foundation Senior Fellow and a member of the MRC Metabolic Diseases Unit.
Conflicts of interest
None.
Authorship
Both authors contributed equally to the writing of this manuscript.


