

Classically nutritional science has applied the principles of reductionism to investigate nutritional deficiencies, exploring complex systems by reducing them to the interactions of their component parts. This approach has been highly successful in identifying the roles of individual nutrients and has given us a clear description of the mechanisms underlying the major deficiency syndromes. However, in recent years the consumption of energy-dense but micronutrient-poor foodstuffs has created a situation where both over and undernutrition can exist simultaneously in a single human population(Reference Demaio and Branca1). Understanding the range of nutrient interactions caused by these imbalanced diets and the long-term consequences for health is an ongoing challenge for nutritional science.
The nutrient interactions associated with obesity are particularly important during pregnancy with nearly half of the women presenting at maternity clinics in developed countries such as Scotland classified as overweight or obese (Births in Scottish Hospitals (National Statistics Publication for Scotland) November 2016). Studies from both the developed and developing world show an inverse relationship between diet quality and BMI, particularly in pregnant women in poorer socioeconomic groups(Reference Laraia, Bodnar and Siega-Riz2, Reference Shin, Lee and Song3). This is because cheap and readily available foods tend to comprise energy-dense, micronutrient-poor products such as refined carbohydrates and oils(Reference Moubarac, Batal and Louzada4). Furthermore, an energy-dense diet increases the demand for micronutrient cofactors such as the vitamins in the B group, which are essential for energy metabolism. Imbalanced, energy-dense, micronutrient-poor diets, therefore, present with an array of nutrient interactions, all of which require adaptations in cellular metabolism and gene expression.
These interactions between metabolism and gene expression are especially important in the 9 months from conception to birth, as the human fetus grows and develops from a single cell to a recognisable infant. A plethora of changes transforms four totipotent stem cells in the preimplantation embryo into increasingly committed cell lineages in mesoderm, endoderm and ectoderm and finally into the terminally differentiated cell types in the organs. Growth and differentiation depend upon a continuous balanced supply of nutrients, derived in part from the maternal diet but also from nutrients released by mobilisation of the mother's tissues. Indeed this relationship continues into postnatal life, as the production of milk also depends on mobilisation of maternal reserves. Because of this interrelationship between maternal diet and reserves, seemingly, simple nutritional imbalances invoke surprisingly complex interactions between macro and micro-nutrients. Maternal metabolism is thus crucial in delivering a stable and balanced nutrient supply to the developing fetus.
Fetal development and long-term health
Over the past 20 years, it has become accepted that poor early development can have a profound effect on the risk of developing non-communicable diseases later in life. The original studies of the Boyd Orr Cohort(Reference Martin, Gunnell and Pemberton5) and subsequent studies by Barker and colleagues(Reference Barker and Osmond6–Reference Hales, Barker and Clark8) firmly established the relationship between poor growth (and by implication nutrition) in early life and an increased risk of non-communicable disease. Subsequently, these observations have been confirmed and extended in populations across the world. Extremes of birthweight, both above and below the mean, are associated with increased risk of the offspring developing a range of diseases including CVD(Reference Barker, Osmond and Winter7, Reference Barker, Osmond and Forsen9), metabolic disease(Reference Hales, Barker and Clark8, Reference Hales and Barker10), osteoporosis(Reference Cooper, Eriksson and Forsen11) and psychiatric disorders(Reference Thompson, Syddall and Rodin12, Reference Veena, Gale and Krishnaveni13). The concept of life-course nutrition encapsulates these ideas in a scheme where early fetal and neonatal development is plastic and influenced by environmental factors, principal amongst these being nutrition. Physiological changes introduced during fetal and neonatal life modulate the risk of developing the non-communicable disease later in life. Once an individual is set on a higher risk trajectory, interventions in adult life can only partly rescue or ameliorate the impact of early life insults(Reference Hanson and Gluckman14). Indeed, nutritional status may be important prior to conception as there is evidence that nutritional status affects follicle recruitment and oocyte maturation in the female and spermatogenesis in the male, modifying the metabolic competence of the gametes and influencing the subsequent development of the conceptus(Reference Fleming, Watkins and Velazquez15).
Maternal metabolism
Pregnancy can be divided into two periods, an initial anabolic phase followed by a second catabolic phase(Reference Herrera16). The anabolic phase covers the first two-thirds of pregnancy. In this period, fetal tissues are a very small part of the mother's body and the available nutrients are directed towards the growth of key maternal organs such as liver and adipose tissue. The nutrient reserves stored in the anabolic phase can then be mobilised during the catabolic phase of gestation (from about 20 weeks onwards in human subjects), supplementing nutrients from the diet. For example, a large increase in lipolysis in adipose tissue late in gestation provides fatty acids for the fetus(Reference Herrera, Amusquivar and López-Soldado17) and also for the mammary gland in readiness for lactation(Reference Vernon and Pond18). These maternal reserves of protein and lipids are particularly important in supporting fetal metabolism in cases where the maternal diet is limited or imbalanced.
Maternal micronutrient reserves are also mobilised to meet the increased demands of pregnancy and in many cases are preferentially allocated to the fetus(Reference Costello and Osrin19, Reference Devakumar, Fall and Sachdev20). If these stores are exhausted during gestation, the deficit continues into lactation, affecting the metabolism of both the mother and her infant. For example, pregnancy has been estimated to require approximately 740 mg additional iron(Reference Fisher and Nemeth21). However, since an ideal diet only provides approximately 5 mg iron daily, a woman must enter pregnancy with iron stores of at least 300 mg if she is to have sufficient iron to avoid becoming deficient by the time the child is born(Reference Bothwell22). Many women, and particularly those in developing countries, have insufficient reserves and become iron deficient by the time the baby is born. Fetal and postnatal iron deficiency results in a range of adverse consequences for mother and infant including low birth weight, impaired cognitive development and poor immune function(Reference Lozoff23). Iron is a key component of enzymes of oxidative metabolism and as the electron-carrying capacity of iron-dependent enzymes is reduced by iron deficiency(Reference Evans and Mackler24, Reference Mackler, Grace and Finch25), glucose is increasingly used as a metabolic substrate in place of lipids(Reference Davis, Rendina and Peterson26, Reference Borel, Beard and Farrell27). This is also the case in pregnant animals as an increase in the incorporation of glucose into polar lipids suggest a shift from fat to glucose as the preferred substrate for oxidative metabolism(Reference Hay, McArdle and Hayes28). As pregnancy progresses there are more pronounced changes in lipid metabolism ultimately resulting in marked changes in both amounts and fatty acid composition of the milk(Reference O'Connor, Picciano and Sherman29).
A similar pattern is seen with other essential vitamins or minerals, all of which have important roles as cofactors or prosthetic groups in enzymes of major metabolic pathways. In developing countries, and to a lesser extent in developed countries, multiple micronutrient deficiencies are present, creating a complex array of potential nutrient interactions that are only now beginning to be studied with advanced metabolomics approaches. These complex interactions also make it difficult to evaluate the outcome of interventions. Frequently supplements with one or two components prove to be relatively ineffective when compared with multiple micronutrient supplements(Reference Ramakrishnan, Goldenberg and Allen30).
Consequences of altered fetal growth and development
Animal models have been extremely important both in exploring nutrient interactions in pregnancy and in understanding the underlying mechanisms. Protein metabolism during pregnancy provides a good example of the complex interactions between nutrients during the anabolic and catabolic phases of pregnancy. One of the most widely studied animal models is the rat low-protein model(Reference Langley, Browne and Jackson31). In this deceptively simple model, laboratory rats are fed a reduced protein diet for the duration of gestation and lactation and the offspring are compared with animals fed a control diet containing the recommended amount of protein. In most cases, the low protein diet contains 8 or 9 % protein (w/w) and is compared with a control diet containing 18–20 % w/w protein, which is the amount recommended by the National Research Council and American Institute for Nutrition for reproduction in the rat(Reference Reeves, Nielsen and Fahey32). Because of the bi-phasic nature of pregnancy, protein restriction has a limited effect on growth in the early stages of fetal life(Reference Rees, Hay and Buchan33). The effects of a modest protein restriction on growth only become more noticeable during the lactation phase(Reference Guzmán, Cabrera and Cárdenas34) when the mobilisation of maternal reserves is no longer able to compensate for the reduced protein supply(Reference Naismith and Robinson35, Reference Pine, Jessop and Allan36).
Whilst the requirements for individual amino acids have not been established in human subjects, estimates extrapolated from animal studies suggest the demand for individual amino acids changes with both time and the availability of other nutrients(Reference Kwon, Spencer and Bazer37). Plasma amino acid concentrations in the maternal circulation of the female rat reflect the two phases of protein metabolism in pregnancy; typically, concentrations fall in the first stages of gestation and then increase between days 19 and 21 (Fig. 1). Dietary protein restriction has very modest effects on the free amino acid concentrations because amino acids entering the peripheral circulation are constantly buffered by amino acids released by the degradation of existing tissue proteins(Reference Elango and Ball38) and only a limited number of amino acids are differentially affected, mostly decreasing. The only notable exception is threonine, one of the indispensable amino acids(Reference Rees, Hay and Antipatis39). The placenta also responds to a reduced dietary supply of protein and this helps to stabilise amino acid concentrations in the fetal circulation, which are usually higher than in the maternal circulation; however, the profile tends to follow the pattern in the maternal circulation(Reference Rees, Hay and Buchan33, Reference Gaccioli, Lager and Powell40).

Fig. 1. The biphasic nature of amino acid metabolism in the rat during gestation. Concentrations of glycine (top) leucine (middle) and threonine (bottom) in the maternal plasma of rats fed a diet containing high (18 % w/w) protein (open circles dotted line) and low (9 % w/w) protein (closed circles solid line). Adapted from Rees et al.(Reference Rees, Hay and Buchan33, Reference Rees, Hay and Antipatis39).
However, amino acids are more than just the monomers for protein synthesis; they are also important substrates for intermediary metabolism, creating a range of potential nutrient interactions in the low protein model. Animal studies suggest that a low protein diet produces changes in intermediary metabolism, which affect development throughout gestation, even in the pre-, and peri-conception phases of life before the conceptus has become implanted(Reference Kwong, Miller and Wilkins41). Changes in intermediary metabolism possibly linked to the oxidation of excess amino acids alters the available concentrations of other important metabolic fuels. For example, concentrations of TAG in the maternal circulation of animals fed a low protein diet are higher than in animals fed sufficient protein and this is likely to increase the supply of fatty acids to fetal tissues and influence their development(Reference McNeil, Maloney and Hay42).
Interactions between macro-nutrients in the maternal diet and the effect on the postnatal phenotype of the offspring
In adult life, the offspring of female rats fed a low protein diet exhibit changes in insulin action and in time, these develop into diabetic-like symptoms(Reference Harding, Jaquiery and Hernandez43). This phenotype, reflected in the area under the insulin release curve when animals are given a glucose challenge(Reference Rees, Hay and Cruickshank44), is the result of reduced growth and development of the pancreatic β-cells(Reference Reusens, Theys and Dumortier45) and a decrease in the insulin sensitivity of the tissues(Reference Martin-Gronert and Ozanne46). However, the extent of this programming is dependent on factors other than just the protein content of the maternal diet(Reference Langley-Evans47). The type of lipid and hence the fatty acid composition of the diet modifies the final outcome such that the offspring of rats fed a low protein diet prepared with maize oil, low in n-3 fatty acids, release less insulin in the glucose tolerance test compared with the offspring of animals fed a diet prepared with soya oil(Reference Maloney, Lilley and Czopek48, Reference Mennitti, Oliveira and Morais49).
This and other similar results suggest that changes in maternal metabolism, modifying the supply of macronutrients to the developing fetal organs, may be an important part of the programming mechanism. Increases in maternal plasma TAG concentrations are a feature of a number of different models of fetal programming in the rat, including low protein, high fat(Reference Williams, Seki and Vuguin50) and folate deficient diets(Reference McNeil, Hay and Rucklidge51) as well as iron deficiency(Reference Hay, McArdle and Hayes28), suggesting an important role for lipid metabolism in programming. A proposed mechanism envisages the setting of metabolic set points during development in response to the available nutrient supply(Reference Plagemann52). For example, the regulation of blood glucose by insulin is a closed loop biological control system, maintaining plasma concentrations between the tightly constrained maximum and minimum values. The set points at which insulin or glucagon are released in response to changes in the prevailing concentrations of glucose are learned during development. If the prevailing glucose concentrations are higher than average during development, then a correspondingly higher set point may be established during fetal development affecting both insulin release and tissue insulin sensitivity.
Nutrient interactions caused by maternal obesity during pregnancy
Whilst many prospective studies in human populations or animal models have focused on fetal growth restriction caused by nutrient deficiency, maternal obesity caused by the oversupply of energy is now perceived to be a greater problem. Increasing numbers of women are now presenting at antenatal clinics with a higher than normal BMI. Obesity in pregnancy carries a number of risks for the mother with higher rates of preeclampsia and premature birth, while the child is at increased risk of macrosomia and gross malformations such as neural tube defects(Reference Poston, Caleyachetty and Cnattingius53). There is also evidence for long-term changes in both metabolism and behaviour in the offspring of obese mothers(Reference Mikkelsen, Hohwü and Olsen54).
Obesity in human populations is not the simple product of a high-energy diet, the simultaneous presence of a double-burden of obesity and malnutrition has been described in populations worldwide(Reference Wahlqvist, Krawetz and Rizzo55). There is an inverse association between BMI and diet quality(Reference Laraia, Bodnar and Siega-Riz2, Reference Murakami and Livingstone56) and this translates into an inverse association between BMI and micronutrient status(Reference Kimmons, Blanck and Tohill57, Reference Sen, Iyer and Meydani58). Deficiencies in the morbidly obese include vitamins B1, B12, folic acid, Vitamin A, Vitamin D and minerals including iron and zinc(Reference Kaidar-Person, Person and Szomstein59). The double burden is a product of socioeconomic factors that promote the consumption of a diet containing large quantities of refined fat or carbohydrate products. Whilst these low-cost foods are plentiful and have a high-energy content, processing tends to deplete them of key vitamins. For example, milling wheat and other grains remove the vitamin-rich aleurone layer, containing approximately 80 % of total niacin together with considerable quantities of other B vitamins, betaine and choline(Reference Likes, Madl and Zeisel60, Reference Brouns, Hemery and Price61). At present little is known about the range of complex nutrient interactions involving vitamins, minerals and amino acids involved in the metabolism of excessive amounts of energy. Incomplete or diverted biochemical reactions could lead to toxicity such as damage caused by the accumulation of NEFA(Reference Brookheart, Michel and Schaffer62). As the prevalence of obesity increases, we cannot consider the consequences of excess fat consumption in isolation. The influence of marginal micronutrient deficiency must also be taken into account, even in wealthy Western countries.
Folic acid, the B vitamins and choline in pregnancy
Central in the metabolism of lipids is the B group of vitamins, which include folic acid. There is a direct relationship between maternal folate status and birthweight, illustrating the importance of folic acid for fetal development(Reference Relton, Pearce and Parker63, Reference van Uitert and Steegers-Theunissen64). Folate derivatives are also important for specific processes in development. It is well known that folic acid supplements reduce the risk of neural tube defects in pregnancy, although questions remain over the precise mechanism. Recent evidence suggests that an improvement in the formate supply and a corresponding increase in nucleotide biosynthesis may be an important factor(Reference Kim, Lei and Guo65). Formate, produced in the mitochondria is transported into the cytoplasm to provide formyl tetrahydrofolate groups necessary for a range of reactions some of which may be essential for neural tube development(Reference Brosnan and Brosnan66).
Fetal cell proliferation requires the production of large quantities of DNA and membrane lipids as well as the synthesis of new protein. All of these processes are dependent on folate-mediated C1 metabolism and derivatives of folic acid are important intermediates in the methionine cycle, which is in turn linked to lipid and nucleotide metabolism (Fig. 2). The methionine cycle maintains intracellular pools of S-adenosyl methionine, the donor of methyl groups in more than hundred essential metabolic reactions. In addition to methionine, the methionine cycle also depends on a variety of other nutrients, particularly the amino acids glycine-serine and threonine as well as micronutrients such as the B vitamins(Reference Rees67).
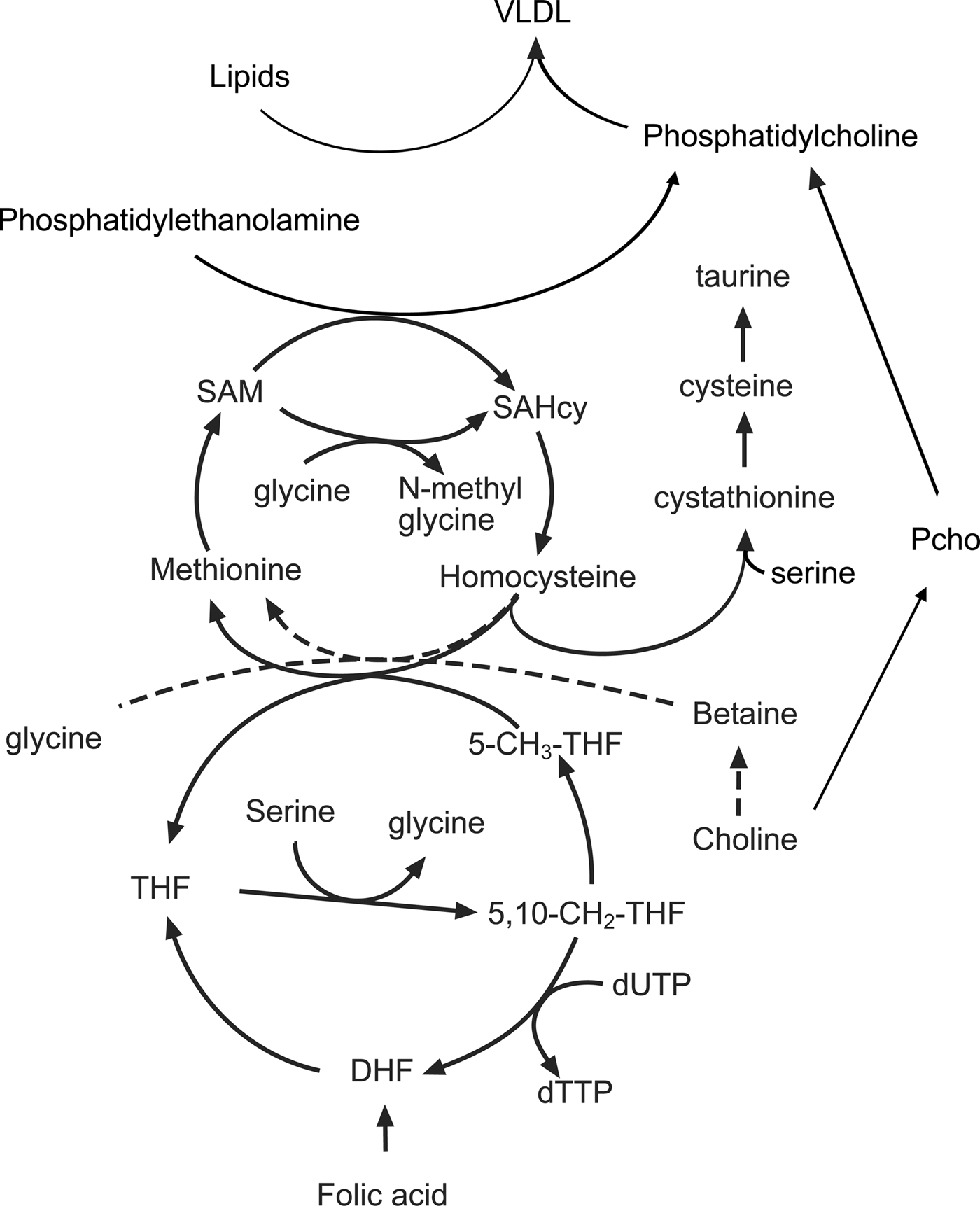
Fig. 2. The interactions between amino acids, choline, the methionine cycle and lipid metabolism in the liver. 5-CH3-THF, 5 methyl tetrahydrofolate; 5,10, CH2 THF, 5,10 methylene tetrahydrofolate; DHF, dihydrofolate; Pcho, phosphocholine; SAM, S-adenosyl methionine; SAHcy, S-adenosyl homocysteine; THF, tetrahydrofolate.
A crucial intermediate in the cycle is the non-protein amino acid homocysteine, concentrations of which are increased when the rate of production exceeds the capacity of the reactions that convert it back to methionine (remethylation) or convert it to cysteine (transulphuration). The importance of C1 metabolism and the methionine cycle for fetal growth is illustrated by the inverse relationship between plasma homocysteine concentrations and birth weight(Reference Bergen, Schalekamp-Timmermans and Jaddoe68). Nutrient interactions have a bearing on the rate of homocysteine removal as both remethylation and transulphuration reactions depend on the availability of other amino acids and vitamin cofactors. The converse is also true in that amino acids and cofactors can become depleted depending on whether homocysteine is remethylated or removed by transulphuration. For example, both choline and derivatives of folic acid provide methyl groups that are used to convert homocysteine back to methionine. The flux through the remethylation reactions increases when methionine is limited and is dependent on the availability of choline, betaine and folic acid in the diet(Reference Wilson, van den Borne and Calder69). Reducing the supply of folic acid increases the utilisation of choline and reduces the use of serine, glycine and threonine. As a result, the phosphocholine pool is depleted and concentrations of serine, glycine and threonine increase in the maternal plasma(Reference Maloney, Hay and Rees70). Restricting the amount of methionine in the diet also reduces concentrations of taurine, another non-protein amino acid implicated in fetal programming(Reference Boujendar, Arany and Hill71, Reference Boujendar, Reusens and Merezak72). If additional methionine is provided in the diet then these folate and choline dependent changes in methionine cycling are lost showing that surplus methionine can be used as a source of methyl groups. Homocysteine can be converted by the transulphuration pathway to cysteine, via the intermediate cystathionine in a reaction dependent upon vitamin B6, pyridoxal phosphate and the amino acid serine(Reference Gregory, DeRatt and Rios-Avila73). This pathway is an important route for the direct use of methionine as a methyl donor and is essential for reproduction as animals lacking the gene coding for cystathionine-β-synthase are infertile as the conceptus fails to implant(Reference Nuño-Ayala, Guillén and Arnal74).
Measurements of methionine turnover using stable isotopes show that in the rat both maternal and fetal livers have active methionine cycles. There is a gradual decline in the remethylation of methionine in the maternal liver as gestation progresses. However, at the same time, methionine is actively produced from homocysteine in the fetal liver, with homocysteine taken up from the maternal circulation and converted to methionine by the fetal tissues(Reference Wilson, Holtrop and Calder75). When animals are fed a diet low in folic acid and choline there is a decrease in the remethylation of homocysteine in the maternal liver; however, to compensate for this there is an increase in the rate of methionine cycling in the fetal liver. In part, this reflects the preferential partitioning of the available folic acid to the fetal tissues but also suggests that there is an increase in the utilisation of methyl groups in fetal tissues, perhaps to produce products normally produced in the maternal compartment.
Impact of limited C1 metabolism on the metabolism of the mother
One of the most obvious features of pregnant rats fed a methyl-deficient diet is an accumulation of lipid in the maternal liver late in gestation(Reference McNeil, Hay and Rucklidge51). The liver has an important role in this period, processing lipids in the diet alongside those mobilised from the adipose tissue in the catabolic phase of pregnancy. Despite the increased flow of TAG through the hepatocytes, intracellular concentrations are normally low as a lipid is rapidly exported as VLDL, which consist of a lipid core surrounded by an outer layer of phospholipids and lipoproteins. The predominant phospholipid in VLDL is phosphatidylcholine (PC) and this must be produced by the liver to maintain high rates of VLDL secretion. However, diets low in choline and associated methyl donors restrict PC synthesis, and lipid that cannot be exported accumulates as droplets within the hepatocytes. The liver produces PC (Fig. 2) either through the cytidine diphosphocholine pathway (also known as the Kennedy pathway) using choline as a precursor or by the conversion of phosphatidylethanolamine to PC by the enzyme phosphatidylethanolamine methyl transferase(Reference Vance76, Reference Cole, Vance and Vance77). Since the phosphatidylethanolamine methyl transferase pathway requires three methyl groups from S-adenosyl methionine for the synthesis of a single PC molecule, it creates a substantial demand for methyl groups(Reference Stead, Brosnan and Brosnan78). It becomes apparent from the complex interrelationships that changes in the supply of amino acids and choline can interact with maternal micronutrient status to produce a range of different outcomes. Further complexity is introduced by increases in the flow of lipids through the liver, a product of both excess in the mother's diet and mobilisation of excess body fat.
Analysis of fetal tissues suggests that methyl deficiency produces changes in the fetal liver(Reference McNeil, Hay and Rucklidge79, Reference Pooya, Blaise and Garcia80) heart(Reference Garcia, Gueant-Rodriguez and Pooya81), gut(Reference Bossenmeyer-Pourié, Blaise and Pourié82) and brain(Reference El Hajj Chehadeh, Dreumont and Willekens83, Reference Geoffroy, Kerek and Pourié84). Measurements of methionine flux show that methyl metabolism is protected in the late gestation fetus(Reference Wilson, Holtrop and Calder75) suggesting that it is the deficiency or excess of the products of folate-dependent metabolism that influences the regulation of gene expression(Reference Garcia, Gueant-Rodriguez and Pooya81).
Fetal metabolism: the totipotent stem cells of the preimplantation embryo
The process of tissue differentiation is accompanied by widespread changes in gene expression. Actively transcribed genes are located in euchromatin, which are loosely packaged areas of chromatin accessible for transcription. Genes not required are tightly packaged into heterochromatin, which is less accessible. These changes in chromatin structure require extensive covalent modification of both DNA and the proteins that surround it(Reference Reid, Dai and Locasale85). Prominent changes include the addition of methyl groups to DNA (DNA methylation) and histone proteins (histone methylation) and the acetylation of histone proteins (histone acetylation). Euchromatin is characterised by the presence of low levels of DNA methylation and hyperacetylated histones whereas heterochromatin has high levels of DNA methylation and hypoacetylation of histones.
It is becoming apparent that the metabolic state of the cell can modify the epigenetic regulation of gene expression during differentiation through changes in the availability of cofactors required for the modification of DNA and histones(Reference Shyh-Chang and Daley George86, Reference Dahan, Lu and Nguyen87). The most widely studied, thanks to the availability of effective tools to precisely measure changes in the modification of individual bases, are the epigenetic marks created by DNA methylation. Changes in DNA methylation and gene expression in the offspring of animals fed a methyl-deficient diet were first shown in the classical studies of the Agouti mouse(Reference Waterland and Jirtle88). Subsequent studies of human populations have identified a relationship between methyl donors in the maternal diet and DNA methylation in the offspring(Reference Dominguez-Salas, Moore and Cole89). The advent of high throughput methods for assessing DNA methylation has made it possible to explore the relationships between maternal nutrition and fetal epigenotype and a range of population-wide studies are currently in progress(Reference Bianco-Miotto, Craig and Gasser90, Reference Kooijman, Kruithof and van Duijn91).
The methylation of DNA is only part of the process of cell differentiation; the posttranslational modification of histones also plays a vital part in maintaining the pluripotent state in stem cells. Initially isolated from early embryos, embryonic stem cells are difficult to maintain in tissue culture, as they are prone to spontaneous differentiation. In a breakthrough study, threonine was identified as a critical component of the culture media(Reference Wang, Alexander and Wu92) as rodent cells grown in low threonine media rapidly lost the capacity to differentiate. Subsequent studies demonstrated that the catabolism of threonine produces both glycine and acetyl CoA; glycine is used to produce C1 folate derivatives and acetyl CoA is used for protein acetylation(Reference Shyh-Chang, Locasale and Lyssiotis93, Reference Shyh-Chang and Ng94). In a similar way, human stem cells use methionine as the principal source of methyl groups for histone methylation(Reference Shiraki, Shiraki and Tsuyama95). The reversal of epigenetic marking by the removal of methyl groups from histones and DNA is also sensitive to the availability of specific metabolites. Histone demethylases and ten-eleven translocation methylcytosine dioxygenases, which catalyse the first step of DNA demethylation require both the tri-carboxylic acid cycle intermediate α-ketoglutarate and ferric iron(Reference Shyh-Chang and Ng94, Reference Tatapudy, Aloisio and Barber96).
These changes in intermediary metabolism are linked to the function of the mitochondria during cell differentiation. Undifferentiated, or ‘naive’ embryonic stem cells, have relatively high levels of oxidative phosphorylation. When differentiation is initiated and cells enter the ‘primed’ state, oxidative phosphorylation becomes uncoupled and metabolism switches to glycolysis, using glucose for energy and increasing the production of acetate for the acetylation of histones(Reference Moussaieff, Kogan and Aberdam97). Energy production from oxidative phosphorylation then increases again as the cells become terminally differentiated. Mitochondria in oocytes and preimplantation embryos have an immature structure with underdeveloped cristae and low rates of oxidative phosphorylation, the classical mitochondrial morphology with well-developed cristae and an active oxidative phosphorylation only appears in terminally differentiated cells(Reference Prigione, Ruiz-Pérez and Bukowiecki98). The reprogramming of differentiated somatic cells into induced pluripotent stem cells reverses these changes and restores the glycolytic state. Changes in metabolism occur before changes in gene expression, suggesting that altered metabolism is required for the cell fate change rather than a consequence of it(Reference Tatapudy, Aloisio and Barber96).
Future perspectives
Optimal fetal development is important for long-term health and there is clear evidence that the supply of macro and micro-nutrients in the maternal diet interact to programme fetal development. There are a variety of potential mechanisms including endocrine programming, changes in organ development and epigenetic programming of gene expression. The rapidly developing field of stem cell metabolism is now opening a new window on the differentiation of cells during development. At present, these studies are limited to in vitro analysis of cells in culture but clearly show that several metabolic pathways have the potential to influence stem cell growth and differentiation through changes in intracellular pools of intermediary metabolites, such as α-ketoglutarate, S-adenosyl methionine, and acetyl-CoA(Reference Zhang, Zhao and Dahan99). It is possible that these changes can produce some profound differences in cellularity of the organs by preventing the premature differentiation of cells. It remains to be seen how these findings will translate into the whole body physiology of the developing fetus, however, it is easy to see potential links between metabolism and the energy-dense but micronutrient-poor diets consumed by many human subjects. Whilst it is possible that maternal metabolism is sufficiently well buffered to protect the developing stem cells there is good evidence from animal models to show that imbalanced diets alter the supply of glucose, amino acids and particularly lipids to the developing fetus. A number of new analytical methods are now needed to explore the relationship between metabolism and chromatin structure. By coupling these with insights into metabolism from the developing field of metabolomics, we may be in a good position to begin to document the complex nutrient interactions that influence chromatin structure, fetal development and long-term health.
Acknowledgements
The author would like to thank colleagues at the Rowett Institute and members of his laboratory, past and present, for facilitating the development of this research area.
Financial Support
Work in the author's laboratory was supported by The Scottish Government Strategic Research Programme.
Conflict of Interest
None.
Authorship
The author had sole responsibility for all aspects of preparation of the present paper.


