


- DNP
dinitrophenol
- UCP1
uncoupling protein 1
Since the identification of leptin as a signalling hormone from the adipose organ, the adipostat model for body-weight control has been the basis for analysis in the field (Fig. 1). The adipostat hypothesis basically states that any alterations in the total amount of energy reserves (as signalled to the brain via leptin) will be compensated by alterations either in food intake or in thermogenesis so that the body will revert to the ‘desired’ body weight(Reference Kennedy1). This hypothesis is inherently pessimistic in relation to any attempts at weight loss, because weight loss would then only be successful if the adipostat system could be manipulated into believing that energy reserves are higher than ‘desired’ or the adipostat could be reprogrammed. Thus, attempts to alter body weight through altered thermogenesis would be unsuccessful, as the individual would compensate by increasing food intake in order to restore body energy reserves.
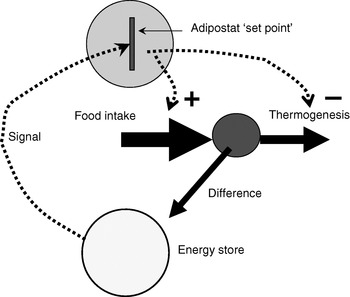
Fig. 1. The adipostat hypothesis for body-weight control. The scheme depicts a situation in which the ‘signal’ (normally considered to be leptin) indicates a level of body energy reserves lower than those required by the adipostat ‘set point’, which results in regulation of food intake (positively; ![]() ) and thermogenesis (negatively;
) and thermogenesis (negatively; ![]() ) to restore the desired level.
) to restore the desired level.
However, observations are summarized here that relate to different forms of thermogenesis; observations that may be said to override a rigid concept of the adipostat and thus represent a more optimistic view of the ability to affect body weight by increasing energy utilization.
Proof of principle: the effects of chemical uncouplers
The mitochondria of any organ of the body (except brown adipose tissue) are innately energy sparing through the ‘coupling’ of O2 consumption (i.e. thermogenesis) to ATP synthesis, only allowing the combustion of substrate when it is necessary, i.e. when ADP is available to be phosphorylated (Fig. 2(A)). As heat is released when ATP is hydrolysed, thermogenesis can in principle occur either when ATP utilization is increased (e.g. in muscular work) or when protons are allowed to re-enter the mitochondria without ATP synthesis taking place, i.e. in ‘uncoupled’ mitochondria (Fig. 2(B and C)).
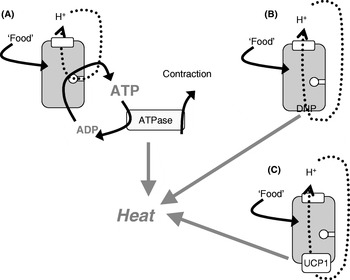
Fig. 2. Mitochondrial coupling and chemical and physiological uncouplers. (A) ‘Normal’ mitochondria in which protons from the respiratory chain are only allowed into the mitochondria in order to drive ATP synthesis; when the ATP is later hydrolysed, thermogenesis occurs (as in exercise or shivering thermogenesis). The mitochondria may become ‘uncoupled’ through an artificial uncoupler (dinitrophenol (DNP); B) or through the physiological uncoupler (uncoupling protein 1 (UCP1); C).
Conceptually, the issue of whether increased thermogenesis would lead to weight loss without compensation by increased energy intake can most clearly be investigated by using chemical uncouplers to increase the proton permeability of mitochondria and thus allow the mitochondria to burn off energy, even under conditions in which ATP is not utilized. Classically, the uncoupler used has been dinitrophenol (DNP; Fig. 2(B)), which can be administered orally and will affect all mitochondria in the body.
Administration of DNP to birds leads to an increase in metabolism and a shift in substrate use, from mainly carbohydrate to more lipid combustion(Reference Dominguez, Menkel and Fairbrother2); furthermore, birds lose body fat while body protein is spared(Reference Toyomizu, Okamoto and Tanaka3), i.e. exactly the type of weight loss that is of interest in the human setting. Thus, the birds do not compensate for the increased energy utilization by increasing food intake. Similarly, in mice, chronic treatment with DNP leads to body-weight loss in the form of fat, sparing proteins, with no food intake compensation(Reference Caldeira da Silva, Cerqueira and Barbosa4).
Indeed, the largest-scale ‘experiments’ involving mammals in relation to the effect of chemically-induced thermogenesis were those performed on human subjects in the USA in the 1930s in which >100 000 individuals consumed DNP as a slimming agent. Acutely, DNP leads to increased thermogenesis(Reference Cutting, Mehrtens and Tainter5) and is associated with continuous weight loss. In formal studies on approximately 100 subjects there was no dietary intervention, but with a mean dose of 300 mg DNP/d the subjects demonstrated a body-weight decrease of 0·5–1 kg/week, mainly through loss of fat from the hips and abdomen. The subjects experienced a feeling of warmth, as expected, but they did not become hyperthermic at these doses (moderately-increased thermogenesis does not normally lead to an increased body temperature because the body temperature ‘set point’ is not altered, so the excess heat will be dissipated). After >100 000 individuals had taken DNP in the 1930s it was noted that there were side effects, i.e. cataracts, skin rashes and loss of taste (but not the type of detrimental effects a bioenergetisist would expect, i.e. brain malfunction, muscle weakness etc.). Thus, DNP was prohibited in 1935; however, it is still used by some individuals as a ‘fat burner’ during defattening in body builders, and some reported fatalities seem to have been a result of overdoses, leading to hyperthermia. Whether the use of chemical uncouplers is a reasonable way to combat obesity is still an open question, but the DNP story clearly provides proof of principle. Furthermore, in human subjects increased thermogenesis does not necessarily lead to compensation with an increased food intake, i.e. the adipostat hypothesis does not accurately predict the actual outcome.
Exercise is thermogenic and leads to weight loss
The practical point of using an uncoupling drug against obesity is of course that a constant increased thermogenesis is obtained in the absence of any constant effort on the part of the subject involved. Another, but more cumbersome, way to increase thermogenesis is obviously through exercise. Indeed, in rodents voluntary or forced exercise (simple provision of a running wheel or forced running in running wheels) is normally associated with weight loss, and here again it is therefore evident that the increased energy utilized is not fully compensated by increased food intake. However, some experiments may be interpreted to indicate that in rodents running is associated with an endorphin-releasing effect that results in reduced food intake, in which case the adipostat hypothesis would not necessarily be invalidated(Reference Boveris and Navarro6).
The same interpretation may be made about exercise in human subjects. The general contention is that it has a weight-reducing effect, but in addition to possible endorphin effects it is difficult to ascertain whether subjects undertaking exercise programmes do so with the intention of losing weight and thus may not respond fully to the signals from the adipostat to increase food intake. Nonetheless, it has proved difficult to involve the general population in exercise, and its effect as a general remedy against obesity has therefore been limited.
Cold is thermogenic and leads to weight loss
Mammals are euthermic animals and if they are placed under conditions in which they lose heat to the environment they will increase their metabolism proportionally to exactly counteract the heat loss. However, in order to compensate over a prolonged period they will also have to eat more, which is exactly the response. Fig. 3 illustrates this process, in which (in a mouse) metabolism is increased in proportion to decreased ambient temperature, i.e. thermoregulatory thermogenesis is activated(Reference Golozoubova, Gullberg and Matthias7). However, this process also requires the mouse to eat more in proportion to the extra heat lost. Thus, living at lower temperatures allows (and even necessitates) increased food intake (more meals), which is in principle in agreement with the adipostat hypothesis.

Fig. 3. Effect of cold on (A) metabolism (adapted from Golozoubova et al.(Reference Golozoubova, Gullberg and Matthias7)) and (B) food intake in mice (V Golozoubova, B Cannon and J Nedergaard, unpublished results). For metabolism (A) values are means with their standard errors represented by vertical bars.
In an extension of this rationale, it follows that increased heat loss (and compensatory heat production) will counteract the effect of increased energy intake. There are several good illustrations of this weight-reducing effect of increased thermogenesis. Thus, mice fed a high-fat diet at a thermoneutral environmental temperature become obese on the diet (in contravention of the adipostat hypothesis), but when offered the same diet at a lower ambient temperature they do not become obese (the extra energy is released as heat to maintain body temperature)(Reference Rippe, Berger and Boiers8, Reference Nikonova, Koza and Mendoza9) (Fig. 4). It is notable that this effect is seen dramatically with a high-fat diet but is almost absent with a normal chow diet. The reasons for this response are unknown, but it should be noted that the lifestyle of present-day man is much more similar to that of a mouse at thermoneutrality eating a high-fat diet than to any of the other conditions.
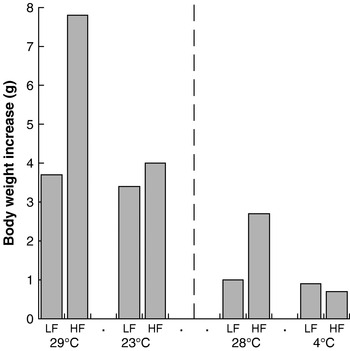
Fig. 4. Cold protects against diet-induced obesity in mice. Data are shown for low-fat (LF)- and high-fat (HF)-fed NMRI mice kept at 29°C and 23°C, representing the increase in body weight over 25 d. (Adapted from Rippe et al.(Reference Rippe, Berger and Boiers8).) Data are also shown for similarly-fed C57BL/6J mice kept at 28°C and 4°C, representing the increase in body weight over 1 week. (Adapted from Nikonova et al.(Reference Nikonova, Koza and Mendoza9).)
Correspondingly, the response to a given diet at a given ambient temperature will depend on how the animal experiences that temperature, i.e. how well insulated it is. Thus, any mouse mutation that has decreased insulation as (one of) its phenotypes will demonstrate an apparent resistance to diet-induced obesity (when maintained at normal animal-house temperatures). Such resistance is thus simply a result of the increased thermogenesis necessary to counteract the increased heat loss, and this thermogenesis results in weight regulation. Most heat is clearly lost from naked mice, but many mutations or genetic manipulations that result in skin phenotypes will show the same effect, e.g. the Elovl3 KO mouse(Reference Westerberg, Månsson and Golozoubova10), the SCD1 KO mouse(Reference Lee, Dobrzyn and Dobrzyn11, Reference Sampath, Flowers and Liu12) and probably several other mutations. In all these mutations thermogenesis demonstrates its weight-reducing potential and no other metabolic alteration need be invoked to explain the weight-reducing effect of the genetic manipulation.
Man is poorly insulated against heat loss and may in this context be considered a tropical animal. Indeed, exposure of naked man to cold results in large increases in heat demand, as demonstrated in classical experiments(Reference Erikson, Krog and Andersen13) (Fig. 5).

Fig. 5. Cold-induced thermogenesis in man; the effect of ambient temperature on metabolic rate in naked adult male subjects. Values are means for five subjects; the BMR of each subject is set to 100. The subjects increased their metabolism by cycling sufficiently on an ergometer to avoid goose pimples or shivering. (Adapted after Erikson et al.(Reference Erikson, Krog and Andersen13).)
The sources of cold-induced thermogenesis
Extra heat can be generated in three different ways, one of which is by exercise (as was the case in the experiment illustrated in Fig. 5, in which in order to maintain body temperature the subjects cycled more the colder it became). If no exercise is performed the brain will activate muscles and shivering will set in. Thus, in man, living at lowered ambient temperatures will increase thermogenesis dramatically and similarly to what is the case in mice (Fig. 3). If the extra energy lost is fully compensated by increased food uptake (as with the mouse; Fig. 3), the result will not be a decrease in body weight.
There is, however, a third way to increase thermogenesis in the cold: non-shivering thermogenesis. Although the anatomical site has been discussed for many years, and is still being discussed, it is our contention that non-shivering thermogenesis solely derives from the activity of brown adipose tissue and uncoupling protein 1 (UCP1) in that tissue (Fig. 2(C))(Reference Nedergaard, Golozoubova and Matthias14). In mammals that remain in the cold for a prolonged period brown adipose tissue will be recruited and the animals will no longer have to shiver to produce heat(Reference Golozoubova, Hohtola and Matthias15). Thus, brown-fat-derived thermogenesis is indeed a more comfortable type of thermogenesis than shivering. However, it is important to realize that the heat loss, and therefore the amount of thermogenesis needed to counteract the heat loss, is exactly the same irrespective of whether the heat derives from shivering or non-shivering thermogenesis(Reference Golozoubova, Hohtola and Matthias15). Thus, although some literature may imply that brown fat is needed for the weight-reducing effect of cold, this is not the case; the only issue is whether or not food intake will compensate for the energy lost.
It may also be implied that more brown fat in itself should have a weight-reducing effect, although such an effect has not been clearly demonstrated. Rather, an augmentation of the amount of brown adipose tissue and UCP1 in an animal through non-adrenergic mechanisms (that work directly on the brown-fat cells(Reference Petrovic, Shabalina and Timmons16)) does not in itself lead to weight reduction. Only if the tissue is also artificially adrenergically stimulated to activate UCP1 does the increased amount of brown-fat-derived thermogenesis lead to loss of body weight, but then again this is because there is no food intake compensation(Reference Sell, Berger and Samson17).
Metaboloregulatory thermogenesis leads to weight loss
For reasons that are not really fully understood mammals may demonstrate a type of thermogenesis, also brown fat derived, even in the absence of any need for thermoregulatory thermogenesis, i.e. under conditions in which no extra heat is needed to compensate heat loss in addition to that resulting from the general ongoing processes in the body (i.e. a type of thermogenesis that occurs at thermoneutrality). This type of thermogenesis has been implicated as being weight reducing for decades(Reference Trayhurn, Thurlby and James18–Reference Rothwell and Stock20) but only recently has a functional analysis become possible, i.e. that an inability to demonstrate metaboloregulatory thermogenesis results in an increased propensity for obesity, as will be discussed.
Absence of uncoupling protein 1-dependent thermogenesis induces obesity in mice
Extrapolation of the earlier summary of the weight-reducing effect of thermogenesis in the opposite direction would be that the absence of thermogenesis in itself should cause obesity, principally as illustrated in Fig. 6. Thus, if a proportion of the daily energy intake is normally combusted in brown adipose tissue, the absence of any thermogenic capacity in brown adipose tissue should lead to obesity, everything else being equal. It was therefore unexpected, indeed disappointing, when it was found that mice lacking thermogenic capacity in their brown adipose tissue, i.e. UCP1-ablated mice, did not spontaneously become obese(Reference Enerbäck, Jacobsson and Simpson21). It was even found that they were ‘protected’ against diet-induced obesity (and this latter phenomenon is still not understood)(Reference Liu, Rossmeisl and McClaine22).
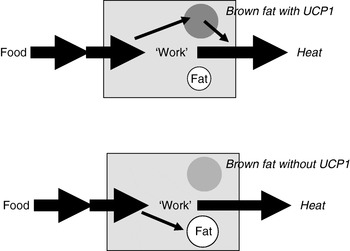
Fig. 6. Loss of thermogenesis should cause obesity. UCP1, uncoupling protein 1.
However, routine experiments with mice are performed at environmental temperatures of 18°C −22°C, which is cold for mice (see. Fig. 3), and thus the mice must constantly activate thermogenesis to combat cold; if there is any diet-, food- or meal-induced thermogenesis the heat from this process will therefore contribute to this cold-combating process.
The situation is quite different at a thermoneutral temperature, because no thermoregulatory thermogenesis is needed and only the effect of the metaboloregulatory thermogenesis will be evident. Indeed, in mice living at thermoneutrality obesity may be induced simply by ablation of UCP1, even when the mice are eating normal non-obesogenic diets; furthermore, the obesity-inducing effect of a high-fat diet is much augmented(Reference Feldmann, Golozoubova and Cannon23). Thus, at least in mice, the absence of UCP1-dependent thermogenesis is in itself obesity inducing. This finding demonstrates, for reasons that are still unclear, that even at thermoneutrality a proportion of energy intake is normally combusted without work being involved.
Does absence of uncoupling protein 1-dependent thermogenesis induce obesity in man?
Until recently, this question would have been considered irrelevant, because it was believed that adults do not possess active brown adipose tissue. However, developments in medical radiology (in connection with tumour localization and control of metastases) led during the early years of the present decade to the realization that a substantial proportion of adults possess active brown adipose tissue and that the activity of this tissue is under physiological control. This information was buried in radiological journals but was collated and presented as a coherent argument in 2007(Reference Nedergaard, Bengtsson and Cannon24). It is thus now possible to pose this question. However, several issues need clarification. First, how common is active brown adipose tissue in adults? Does its presence or absence correlate with leanness or obesity? If it does, is the correlation causative?
The prevalence of brown adipose tissue has been reported in most retrospective analyses to be of the order of <10%. If present in so few individuals, the absence or presence of brown-fat-derived thermogenesis could hardly explain the substantial numbers of obese subjects. However, data from retrospective studies grossly underestimate the prevalence of brown adipose tissue because the tissue is visible only when functionally active. In contrast, recent studies where brown adipose tissue activity has been physiologically induced have clearly demonstrated that a high proportion of human subjects possess active brown adipose tissue(Reference van Marken Lichtenbelt, Vanhommerig and Smulders25, Reference Virtanen, Lidell and Orava26, Reference Saito, Okamatsu-Ogura and Matsushita27). Only a few studies demonstrate any correlation between the presence or absence of brown adipose tissue and obesity.
However, at least two studies in which a broader population basis was examined concur in showing that there is a clear correlation between the absence of brown adipose tissue and proneness to obesity(Reference Saito, Okamatsu-Ogura and Matsushita27, Reference Zingaretti, Crosta and Vitali28) (summarized in Fig. 7). This outcome thus implies that there could be a functional correlation in man just as there is in mouse, i.e. that the absence of brown-fat-derived thermogenesis could suffice to induce obesity.

Fig. 7. Correlation between the absence or presence of brown adipose tissue and obesity in human subjects. Individual observations for thirty-five Italians (•, ○)(Reference Zingaretti, Crosta and Vitali28) and nineteen Japanese (▪, □)(Reference Saito, Okamatsu-Ogura and Matsushita27) plotted as a function of BMI. (•, ▪), Presence of uncoupling protein 1 (UCP1) and active brown adipose tissue; (○, □), absence of UCP1 and active brown adipose tissue. For the Japanese subjects values <0·5 units of uptake was considered to be absence of brown adipose tissue. The BMI of the subjects without brown fat is shifted to the right. In the combined data the mean BMI of the subjects possessing brown adipose tissue was 21 kg/m2 whereas the mean BMI of the subjects without brown adipose tissue was 25 kg/m2 (P<0·004), which for a subject of height 1·70 m corresponds to a weight difference of approximately 15 kg.
There are indeed no studies in human subjects that unequivocally establish that the absence of brown-fat-derived thermogenesis is sufficient to cause obesity. However, some circumstantial data exist. In the distal enhancer of the human gene for UCP1 at position −3826, a single-nucleotide polymorphism has been detected and the alternative nucleotides A or G have been shown to be related to the level of UCP1 gene expression, with the A allele being associated with a higher extent of expression(Reference Esterbauer, Oberkofler and Liu29) (Fig. 8(A)). Notably, subjects with two A alleles show higher thermogenic responses to a high-fat meal(Reference Nagai, Sakane and Ueno30), and in a population study it was found that there is a greater risk of being in a higher quartile of BMI if a subject possesses the AG or GG alleles(Reference Sramkova, Krejbichova and Vcelak31) (Fig. 8(B)).

Fig. 8. Effects of the −3826 polymorphism in the human uncoupling protein 1 (UCP1) gene. (A) Levels of UCP1 mRNA in human intraperitoneal adipose tissue as a function of the three allele combinations at the locus. (Adapted from Esterbauer et al.(Reference Esterbauer, Oberkofler and Liu29).) (B) The distribution of the alleles A/A (▪) and A/G and G/G (□) in a population divided into quartiles according to BMI. (Adapted from Sramkova et al.(Reference Sramkova, Krejbichova and Vcelak31).)
Thus, developments within the last few years have unexpectedly indicated that the absence of brown-fat-derived thermogenesis may be sufficient to cause obesity in mice as well as in man.
Conclusions
Data have been presented that imply that the adipostat hypothesis does not adequately predict the outcome when thermogenesis is altered. Indeed, our conclusions are that decreased thermogenesis may in itself be causative of obesity; correspondingly, ‘induced’ thermogenesis counteracts obesity (even without dietary intervention). Thus, activation of thermogenesis is an anti-obesity tool irrespective of whether it is accomplished by artificial uncoupling, exercise, shivering, or recruitment and activation of brown adipose tissue. The latter would seem to be the more physiological, and the more comfortable, of these means of promoting thermogenesis.
Acknowledgements
Both authors contributed to the design and writing of this manuscript. Work from the authors' laboratory is supported by the Swedish Research Council, the Swedish Cancer Foundation and the European Union collaborative project ADAPT. The authors declare no conflict of interest.








