


Introduction
Italian ryegrass [Lolium perenne L. ssp. multiflorum (Lam.) Husnot] [2n = 2x = 14 or 2n = 4x = 28] is a widely grown cool-season forage species (Guan et al. Reference Guan, Yuyama, Stewart, Ding, Xu, Kiyoshi and Cai2017), and it is considered the most nutritious forage, often exceeding 70% digestible dry matter and 18% to 20% crude protein (https://www.ryegrass.com/forage.html; Amaral et al. Reference Amaral, Kozloski, Santos, Castagnino, Fluck, Farenzena, Alves and Mesquita2011; Ohshima et al. Reference Ohshima, Nagatomo, Kubota, Tano, Okajima and Kayama1988; Özelçam et al. Reference Özelçam, Kırkpınar and Tan2015). However, due to its high adaptability, broad genetic diversity, and prolific seed production, which together drive its rapid invasion and spread, L. perenne ssp. multiflorum has become a problematic weed in winter crops, especially wheat (Triticum aestivum L.), despite its widespread use as a turf and cover crop species (Maity et al. Reference Maity, Singh, Jessup and Bagavathiannan2021b; Tucker et al. Reference Tucker, Morgan, Senseman, Miller and Baumann2006; Webster and Nichols Reference Webster and Nichols2012). It is usually a diploid (2n = 2x = 14) and is an obligate outcrossing species due to its self-incompatibility and spontaneous, natural hybridization with perennial ryegrass (Lolium perenne L.) and other related species, such as fescues (Festuca spp.) (Guan et al. Reference Guan, Yuyama, Stewart, Ding, Xu, Kiyoshi and Cai2017; Rosell Reference Rosell1967). Consequently, it shows high levels of diversity for adaptive traits such as growth habit, plant height, tillering habit, regrowth rate, leaf width, plant texture, awns, seed shattering, and seed dormancy across geographic locations (Maity et al. Reference Maity, Singh, Jessup and Bagavathiannan2021b, Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c). The greater diversity not only complicates its taxonomic identification in field conditions but also increases its potential to invade new areas and its vulnerability to rapid herbicide-resistance evolution (Singh et al. Reference Singh, Maity, Abugho, Swart and Bagavathiannan2020).
Some critical parameters to consider when the primary use of Lolium spp. is hay or silage include tillering habit, high biomass production, faster regrowth rate following cutting, and high nutritive value (Fu et al. Reference Fu, Song, Zhao and Jameson2018; Knorst et al. Reference Knorst, Yates, Byrne, Asp, Widmer, Studer and Kölliker2018). Many of these traits are considered equally important for high adaptability when Lolium spp. grow as a weed in agricultural systems (Dekker Reference Dekker2011). For example, the seed shattering tendency of Lolium spp. enriches the weed seedbank, ensuring infestation in subsequent years (Fu et al. Reference Fu, Song, Zhao and Jameson2018; Maity et al. Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c; Walsh et al. Reference Walsh, Broster, Schwartz-Lazaro, Norsworthy, Davis, Tidemann, Beckie, Lyon, Soni, Neve and Bagavathiannan2018). Likewise, high tiller production in Lolium ssp. is associated with high biomass production in pasture types (Nair Reference Nair2004), while it can pose severe competitive effects on crops when present as a weed (Peeper and Wiese Reference Peeper, Wiese and Donald1990). In addition to physiological and agronomic studies, molecular dissection of these adaptive traits using advanced tools may elucidate their genetic control and aid plant breeding and weed management efforts (Fu et al. Reference Fu, Song, Zhao and Jameson2018).
The adaptive traits critical for weed persistence in agricultural landscapes have not received the same attention as yield traits in crops (Stewart, Reference Stewart and Mercer2006). For example, the propensity for seed shattering is retained in most forage and weedy species or biotypes within a species, compared with cultivated types; however, efforts to understand the underlying mechanisms are limited (Fu et al. Reference Fu, Song, Zhao and Jameson2018). Although, specific gene(s), such as Sh1, have been identified as being responsible for seed shattering in many important cereal crops (Dong and Wang Reference Dong and Wang2015; Konishi et al. Reference Konishi, Izawa, Lin, Ebana, Fukuta, Sasaki and Yano2006; Lin et al. Reference Lin, Li, Shannon, Yeh, Wang, Bai, Peng, Jiarui, Trick, Clemente, Doebely, Schnable, Tuinistra, Tesso, White and Yu2012; Maity et al. Reference Maity, Lamichaney, Joshi, Bajwa, Subramanian, Walsh and Bagavathiannan2021a), it was not until 2018 that Fu et al. (Reference Fu, Song, Zhao and Jameson2018) confirmed the role of LpqSH1 and LpSH1 in Lolium perenne seed shattering by transcriptome analysis. Such genetic knowledge is necessary to understand seed shattering and other weedy traits in L. perenne ssp. multiflorum, such as tiller production, regrowth potential, stress tolerance, and fecundity, which could also be useful for breeding improved forage varieties.
Numerous efforts have been made to understand the genetic control of adaptive traits in L. perenne ssp. multiflorum and related species using biparental mapping, with a primary focus on identifying quantitative trait loci (QTL) (Shinozuka et al. Reference Shinozuka, Cogan, Spangenberg and Forster2012). To date, high-throughput genotyping platforms such as the Lolium/Festuca DArT array (Kopecky et al. Reference Kopecky, Bartos, Lukaszewski, Baird, Černoch, Kölliker, Rognli, Blois, Caig, Lübberstedt, Studer, Shaw, Doležel and Kilian2009), the Lolium Infinium SNP-genotyping array (Blackmore et al. Reference Blackmore, Thomas, McMahon, Powell and Hegarty2015), and Genotyping-By-Sequencing (GBS) (Ashraf et al. Reference Ashraf, Byrne, Fé, Czaban, Asp, Pedersen, Lenk, Roulund, Didion, Jensen, Jensen and Janss2016; Pembleton et al. Reference Pembleton, Drayton, Bain, Baillie, Inch, Spangenberg, Wang, Forster and Cogan2016; Velmurugan et al. Reference Velmurugan, Mollison, Barth, Marshall, Milne, Creevey, Lynch, Meally, McCabe and Milbourne2016), have been used for genome-wide association studies (GWAS) to identify marker–trait associations, candidate genes, and genomic selection applications in L. perenne (Byrne et al. Reference Byrne, Nagy, Pfeifer, Armstead, Swain, Studer, Mayer, Campbell, Czaban, Hentrup, Panitz, Bendixen, Hedegaard, Caccamo and Asp2015; Velmurugan et al. Reference Velmurugan, Mollison, Barth, Marshall, Milne, Creevey, Lynch, Meally, McCabe and Milbourne2016). Association analysis involves two key approaches: candidate gene analysis and GWAS. Candidate gene analysis focuses on variations within specific genes linked to traits, while GWAS examines markers across the genome. GWAS is advantageous due to its better suitability for natural populations, which offer high genetic diversity and broader allelic exploration without the need for mapping populations (Buckler and Thornsberry Reference Buckler and Thornsberry2002; Byrne et al. Reference Byrne, Nagy, Pfeifer, Armstead, Swain, Studer, Mayer, Campbell, Czaban, Hentrup, Panitz, Bendixen, Hedegaard, Caccamo and Asp2015; Czaban et al. Reference Czaban, Sharma, Byrne, Spannagl, Mayer and Asp2015; Harper et al. Reference Harper, De Vega, Swain, Heavens, Gasior, Thomas, Evans, Lovatt, Lister, Thorogood, Skøt, Hegarty, Blackmore, Kudrna and Byrne2019; Mascher et al. Reference Mascher, Gundlach, Himmelbach, Beier, Twardziok, Wicker, Radchuk, Dockter, Hedley, Russell, Bayer, Ramsay, Liu, Haberer and Zhang2017; Salamini et al. Reference Salamini, Ozkan, Brandolini, Schafer-Pregl and Martin2002; Stranger et al. Reference Stranger, Stahl and Raj2011; Velmurugan et al. Reference Velmurugan, Mollison, Barth, Marshall, Milne, Creevey, Lynch, Meally, McCabe and Milbourne2016; Zhu et al. Reference Zhu, Gore, Buckler and Yu2008). However, population structure factors can cause spurious associations. Mixed linear models incorporating kinship matrices effectively address these issues, improving the reliability of GWAS (Flint-Garcia et al. Reference Flint-Garcia, Thornsberry and Buckler2003; Kang et al. Reference Kang, Zaitlen, Wade, Kirby, Heckerman, Daly and Eskin2008; Wang et al. Reference Wang, Jiang, Jia, Leach, Cockram, Waugh, Ramsay, Thomas and Luo2011).
The recent publication of two initial genome assemblies for L. perenne (Byrne et al. Reference Byrne, Nagy, Pfeifer, Armstead, Swain, Studer, Mayer, Campbell, Czaban, Hentrup, Panitz, Bendixen, Hedegaard, Caccamo and Asp2015; Velmurugan et al. Reference Velmurugan, Mollison, Barth, Marshall, Milne, Creevey, Lynch, Meally, McCabe and Milbourne2016), a high-quality reference genome assembly for common barley (Hordeum vulgare L.) (Mascher et al. Reference Mascher, Gundlach, Himmelbach, Beier, Twardziok, Wicker, Radchuk, Dockter, Hedley, Russell, Bayer, Ramsay, Liu, Haberer and Zhang2017), and the first assembly of the gene space of L. perenne ssp. multiflorum, which shows high similarity to L. perenne and is supported by previously published rice (Oryza sativa L.) and false brome (Brachypodium spp.) genomes (Harper et al. Reference Harper, De Vega, Swain, Heavens, Gasior, Thomas, Evans, Lovatt, Lister, Thorogood, Skøt, Hegarty, Blackmore, Kudrna and Byrne2019), is a notable accomplishment. Czaban et al. (Reference Czaban, Sharma, Byrne, Spannagl, Mayer and Asp2015) reported a high genome sequence conservation within the Lolium/Festuca species complex due to their ancestral relationship (Bulinska-Radomska and Lester Reference Bulinska-Radomska and Lester1988; Charmet et al. Reference Charmet, Ravel and Balfourier1997). Moreover, physical mapping techniques such as chromosome conformation capture sequencing help resolve ambiguities in sequence-based assemblies, easing the construction and validation of sequence scaffolds in genome assemblies (Mascher et al. Reference Mascher, Gundlach, Himmelbach, Beier, Twardziok, Wicker, Radchuk, Dockter, Hedley, Russell, Bayer, Ramsay, Liu, Haberer and Zhang2017). Recently, Frei et al. (Reference Frei, Veekman, Grogg, Stoffel-Studer, Morishima, Shimizu-Inatsugi, Yates, Shimizu, Frey, Studer and Copetti2021) developed the first high-quality haploid reference assembly for L. perenne using the Oxford Nanopore Technologies platform, and Chen et al. (Reference Chen, Kölliker, Mascher, Copetti, Himmelbach, Stein and Studer2024) developed an improved chromosome-level genome assembly of L. perenne ssp. multiflorum using collinearity with H. vulgare, employing high-throughput chromosome conformation capture.
Rapid advances in low-cost next-generation sequencing approaches have broadened the scope and accuracy of GWAS in several species (López de Heredia et al. Reference López de Heredia, Mora-Márquez, Goicoechea, Guillardín-Calvo, Simeone and Soto2020). Among the NGS genotyping approaches, double-digest restriction site–associated DNA sequencing (ddRADseq) is a relatively inexpensive approach that has eased SNP discovery and genotyping by not requiring a reference genome and providing a variable range of genome coverage by selecting appropriate restriction enzymes in various non-model plant species for genomic studies (Lang et al. Reference Lang, Weiß, Kersten, Latorre, Nagel, Nickel, Meyer and Burbano2020; Reitzel et al. Reference Reitzel, Herrera, Layden, Martindale and Shank2013). In this method, for sequencing a large number of samples in a single lane—that is, multiplexing—genomic DNA is digested with two restriction enzymes and then barcoded adapters are ligated onto the digested ends, followed by their PCR amplification (Elshire et al. Reference Elshire, Glaubitz, Sun, Poland, Kawamoto and Buckler2011; Scheben et al. Reference Scheben, Batley and Edwards2017). Because of these advantages, the present study aimed at using the ddRADseq approach for assessing genetic diversity and identifying the genomic region(s) associated with tiller production, regrowth rate, and seed shattering, which are considered critical for weed adaptation and persistence, by using GWAS in L. perenne ssp. multiflorum.
Materials and Methods
Plant Materials and Phenotyping
For this study, about 50 mature spikes each for 56 wild L. perenne ssp. multiflorum populations from wheat fields and field edges in the Texas Blacklands region during 2017, 25 half-sibs from the L. perenne ssp. multiflorum breeding program at the Texas A&M University Overton station, and nine known reference samples of L. perenne ssp. multiflorum (4), L. perenne (3), rigid ryegrass (Lolium rigidum Gaudin) (1), and poison ryegrass (Lolium temulentum L.) (1) from the Germplasm Resources Information Network (GRIN) of the USDA-Agricultural Research Service (USDA-ARS Plant Germplasm Introduction and Testing Research Unit, Pullman, WA, https://npgsweb.ars-grin.gov/) were collected/obtained (Table 1). Seeds of all Lolium ssp. populations were planted in pots (14-cm diameter and 12-cm height) filled with a potting soil mixture (LC1 Potting Mix, Sun Gro® Horticulture, Agawam, MA, USA) in a greenhouse with 15 replications per population during fall 2017. The experiment was conducted in the Norman Borlaug Greenhouse Research Facility, Texas A&M University (TAMU), College Station, maintained at 26/22 C day/night temperatures and a 10-h photoperiod. Three adaptive traits (regrowth rate, tiller production, and seed shattering) were phenotyped following Maity et al. (Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c), as described in Table 2. Because the plants within a population showed high phenotypic diversity, each of the 15 plants was sampled for leaf tissue to extract DNA.
Details of Lolium ssp. plant materials used for the study.

a Half-sibs and cultivars were obtained from the Texas A&M University Overton Station, TX.
b Reference samples were obtained from the Germplasm Resources Information Network (GRIN) of the USDA-Agricultural Research Service (USDA-ARS Plant Germplasm Introduction and Testing Research Unit, Pullman, WA, https://npgsweb.ars-grin.gov/).
Methodology used for assessing seed shattering, tiller production, and regrowth rate, and phenotyping results in Lolium multiflorum ssp. multiflorum.

a See Maity et al. (Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c) for details.
DNA Extraction and ddRADseq Analysis
Genomic DNA was extracted from 100 mg of young leaf tissue using the modified CTAB protocol (Murray and Thompson Reference Murray and Thompson1980) in the AgriGenomics Laboratory, TAMU. DNA quality was assessed by 1% agarose gel electrophoresis, and the concentration was quantified using a Nanodrop 1000 UV-VIS Spectrophotometer (Thermo Fisher, Waltham, MA, USA). The DNA samples were diluted to a final concentration of 50 ng µl−1 and submitted to the Texas A&M Genomics and Bioinformatics Unit of AgriLife Research (TXGEN; https://www.txgen.tamu.edu/) in College Station, for ddRADseq (Peterson et al. Reference Peterson, Weber, Kay, Fisher and Hoekstra2012) using the NovaSeq 6000 S4-X2 platform (Illumina, San Diego, CA, USA; paired-end 150 bp) with some modifications. Restriction enzymes, PstI (a rare cutter) and MluCI (a frequent cutter) were used to prepare the libraries. These two restriction enzymes were selected based on a simulation study and a size range selected for ddRADseq.
The sequenced reads were initially processed using the synteny-based draft assembly of the L. perenne genome (only scaffolds without the chromosome information) (ASM173568v1; Byrne et al. Reference Byrne, Nagy, Pfeifer, Armstead, Swain, Studer, Mayer, Campbell, Czaban, Hentrup, Panitz, Bendixen, Hedegaard, Caccamo and Asp2015), as outlined in Supplementary Figure S1. However, final filtering (with missing data < 0.5 and Minor Allele Frequency > 0.02) yielded only 221 SNPs. Hence, the de novo assembly was performed using STACKs software. For the de novo analysis, the raw sequence data were initially evaluated for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/ projects/fastqc/). Reads were subsequently de-multiplexed and trimmed to 120 bp with process_radtags in the STACKs software (Catchen et al. Reference Catchen, Hohenlohe, Bassham, Amores and Cresko2013; https://catchenlab.life.illinois.edu/stacks/), with no mismatches permitted in the barcode sequences. Loci were then assembled de novo, and SNPs were identified using the de novo map pipeline in STACKs, without relying on a reference genome.
De novo loci assembly in STACKs is controlled by three key parameters that determine the trade-off between the number of loci, genotyping error rates, and the extent of missing data. The first two parameters influence locus construction at the individual level: –m defines the minimum number of raw reads required to form a stack (allele) within an individual, and –M specifies the maximum number of mismatches permitted between alleles within an individual when merging them into a locus. A high value of –m may result in allele dropout, whereas a value that is too low can lead to the generation of false alleles. A higher –M value can lead to false heterozygous polymorphisms (by merging non-homozygous loci), whereas a low value can result in false homozygous loci (by splitting a single locus into two). The third parameter, –n, is the number of mismatches permitted between stacks while constructing the population catalog.
Parameter selection is study specific, as it depends on both the biological characteristics of the target species (e.g., inherent level of polymorphism and ploidy) and the experimental design (e.g., choice of restriction enzyme, number of multiplexed samples, and sequencing depth). The parameter set of m = 5, M = 2, and n = 1 was used to minimize errors, based on previous studies such as Paris et al. (Reference Paris, Stevens and Catchen2017) and Mastretta-Yanes et al. (Reference Mastretta-Yanes, Arrigo, Alvarez, Jorgensen, Piñero and Emerson2015). Following the initial SNP calling, additional filtering was applied to exclude loci with excessive missing data and to remove individuals with low coverage. Individual SNPs with more than 50% missing data within each population group were removed, resulting in 3,766 total SNPs. After removal of monomorphic markers, 3,079 SNPs remained for GWAS analysis. De novo assembly generates a reference lacking chromosome information. Many GWAS software packages (like TASSEL) require a chromosome number for each marker to process the data. Because the association analysis was performed at the single-locus level, a hypothetical chromosome (chromosome 1 in this study) was assigned to each marker to ensure that the software treated the dataset as a single linkage group.
The SNP data and global positioning system coordinates were also used to investigate whether genetic differentiation increases with increasing geographic distance between the wild accessions, a pattern known as “isolation by distance” (IBD). The IBD analysis was performed using RStudio 2022.12.0 packages vegan (Oksanen et al. Reference Oksanen, Blanchet, Kindt, Legendre, Minchin, Ohara, Simpson, Solymos, Stevens, Wagner and Oksanen2013) and geosphere (Hijmans et al., Reference Hijmans, Williams, Vennes and Hijmans2021). A Mantel test with 999 permutations was conducted to assess the correlation between genetic and geographic distance matrices among the wild accessions.
GWAS
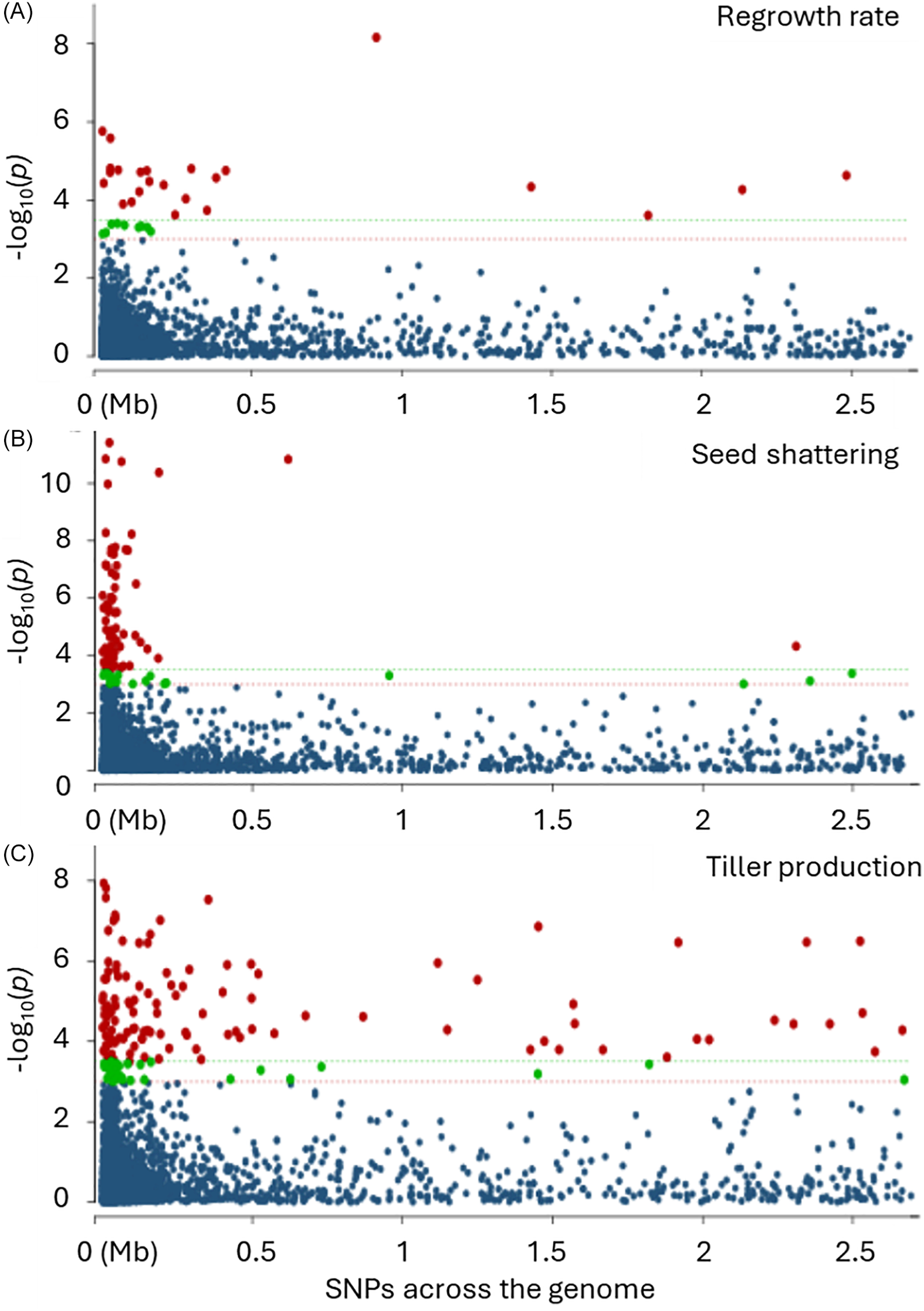
The principal component analysis (PCA) was performed in RStudio 2022.12.0, using the R function prcomp() and the libraries factoextra and FactoMiner for visualization and analysis. SNP data imputation was done in TASSEL v. 5.0 using LD KNNI (Money et al. Reference Money, Gardner, Migicovsky, Schwaninger, Zhong and Myles2015). GWAS was performed using a generalized linear model and a kinship matrix in TASSEL v. 5.0 (Trait Analysis by Association, Evolution and Linkage; Bradbury et al. Reference Bradbury, Zhang, Kroon, Casstevens, Ramdoss and Buckler2007) to identify the SNPs associated with tiller production, regrowth rate, and seed shattering. Because the analysis was performed by assigning all markers to a single chromosome, the markers on a Manhattan plot appeared as a single cluster rather than multiple chromosomes. For declaring significant marker–trait association by the GWAS, a P-value threshold of 3.27 × 10−6 (Bonferroni’s correction: calculated by dividing a standard significance level [alpha, in this case 0.05] by the number of tests performed) and a false discovery rate (FDR) of P < 0.01 were set. The Bonferroni multiple test correction was done using the formula [−log10 (0.01/effective number of SNPs)] to identify the highly significant SNPs (Wu et al. Reference Wu, Feng, Lian, Teng, Wei, Yu, Xie, Yan, Fan, Li, Ma, Liu, Yu, Wang and Zhou2015). Finally, the genomic regions flanking the highly significant SNPs (P < 0.00001) for the number of tillers per plant (#25), regrowth rate (#19), and seed shattering (#25) were examined to identify potential candidate genes.
Analyzing SNP Sequences Using BLASTN
The nucleotide sequences (135 to 150 bp) flanking the highly significant SNPs associated with the studied adaptive traits were retrieved and blasted in the NCBI database (https://www.ncbi.nlm.nih.gov/) to identify potential candidate genes from the published reports. Sequence matches from BLAST analysis of SNPs provide valuable insight into a marker’s genomic location, potential function, and evolutionary relationships. These matches can also help determine whether an SNP resides within a known gene, a regulatory region, or an intergenic region. As genome assemblies and associated gene sequences for Lolium spp. were not readily available in the NCBI database until recently, only incomplete, fragmented assemblies were available for the grasses of the Lolium and Festuca species complex. At first, the flanking nucleotide sequences were analyzed using the BLASTN database v. 2.17.0+ (standard nucleotide BLAST) without any filters (Altschul et al. Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997). Only the hits with >85% sequence identity in L. perenne ssp. multiflorum and relevant species were considered.
In some cases, the BLASTN search results did not provide any chromosome-specific information for L. perenne ssp. multiflorum. Following this, the BLASTN analysis was repeated using the L. perenne reference genome (L. perenne assembly MPB_Lper_Kyuss_1697; GCA_019359855.1). This analysis yielded chromosome-level information for most SNPs, except in two cases in which the gene of interest aligned with sequences from H. vulgare. These two SNPs were assigned to chromosome numbers based on the H. vulgare genome, given its high genomic similarity with the Lolium genome (Chen et al. Reference Chen, Kölliker, Mascher, Copetti, Himmelbach, Stein and Studer2024; Frei et al. Reference Frei, Veekman, Grogg, Stoffel-Studer, Morishima, Shimizu-Inatsugi, Yates, Shimizu, Frey, Studer and Copetti2021). Finally, BLASTN analysis was conducted using all Lolium species. Consequently, L. perenne, L. rigidum, and H. vulgare were considered in case of regrowth rate, but Tausch’s goatgrass (Aegilops tauschii Coss.), rivet wheat (Triticum turgidum L.; syn.: Triticum dicoccoides Koern. ex Schweinf.), L. multiflorum, L. perenne, L. rigidum, and wild barley [Hordeum vulgare ssp. spontaneum (K. Koch) Asch. & Graebn.] were considered for tiller count, whereas L. perenne, L. rigidum, and H. vulgare ssp. vulgare were considered for seed shattering based on sequence identity in BLASTN searches.
Results and Discussion
Phenotypic Data
All three traits considered in this study were highly variable among the populations (Table 2). The regrowth rate varied significantly across the populations, from very slow to very fast, with most having a fast regrowth rate. The number of tillers per plant ranged from 1 to 31, with an average of nine. Seed shattering (%) was highly variable among populations (0% to 100%), with 18% average shattering.
Population Structure Analysis
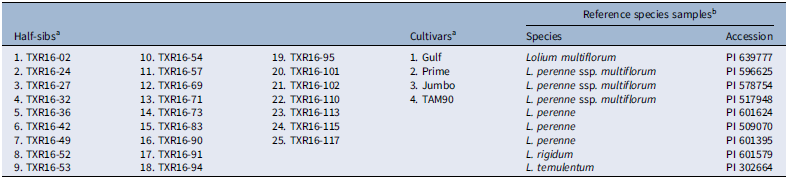
A total of 3,766 SNP markers were identified after removing SNPs with greater than 50% missing data, of which 3,079 SNPs were used to estimate marker-trait associations after removing monomorphic SNPs. The principal components (PCs) were obtained in the TASSEL program, explaining 5.4% and 3.4% of the variation for the first two PCs, respectively. Three distinct clusters were identified from the PCA plots (Figure 1). The first cluster represented the cultivars and reference samples of the four species received from the USDA-GRIN, that is, L. perenne ssp. multiflorum, L. perenne, L. rigidum, and L. temulentum. The half-sibs, that is, the breeding lines, along with a few wild populations, were present in the second cluster. This may possibly be attributed to the founder effect in recurrent selective breeding programs leading to reduced genetic diversity (Tamura et al. Reference Tamura, Arakawa, Kiyoshi and Yonemaru2022). The presence of a few wild populations in this cluster is likely due to repeated natural outcrossing between cultivated and wild types in the region, leading to introgression of alleles from cultivated to wild types and/or ferality of the cultivated types (Balfourier et al. Reference Balfourier, Imbert and Charmet2000; Maity et al. Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c). The third cluster consisted of only wild populations (Figure 1).
Principal component analysis (PCA) plots showing the clusters and the percentage variation explained by the first two components for all the lines, including wild populations, cultivars, half-sibs, and USDA-GRIN reference samples. Populations are indicated by different colors and letters: C, cultivars; H, half-sibs; W, wild; and USDA references: M, Lolium perenne ssp. multiflorum; P, Lolium perenne; R, Lolium rigidum; and T, Lolium temulentum.

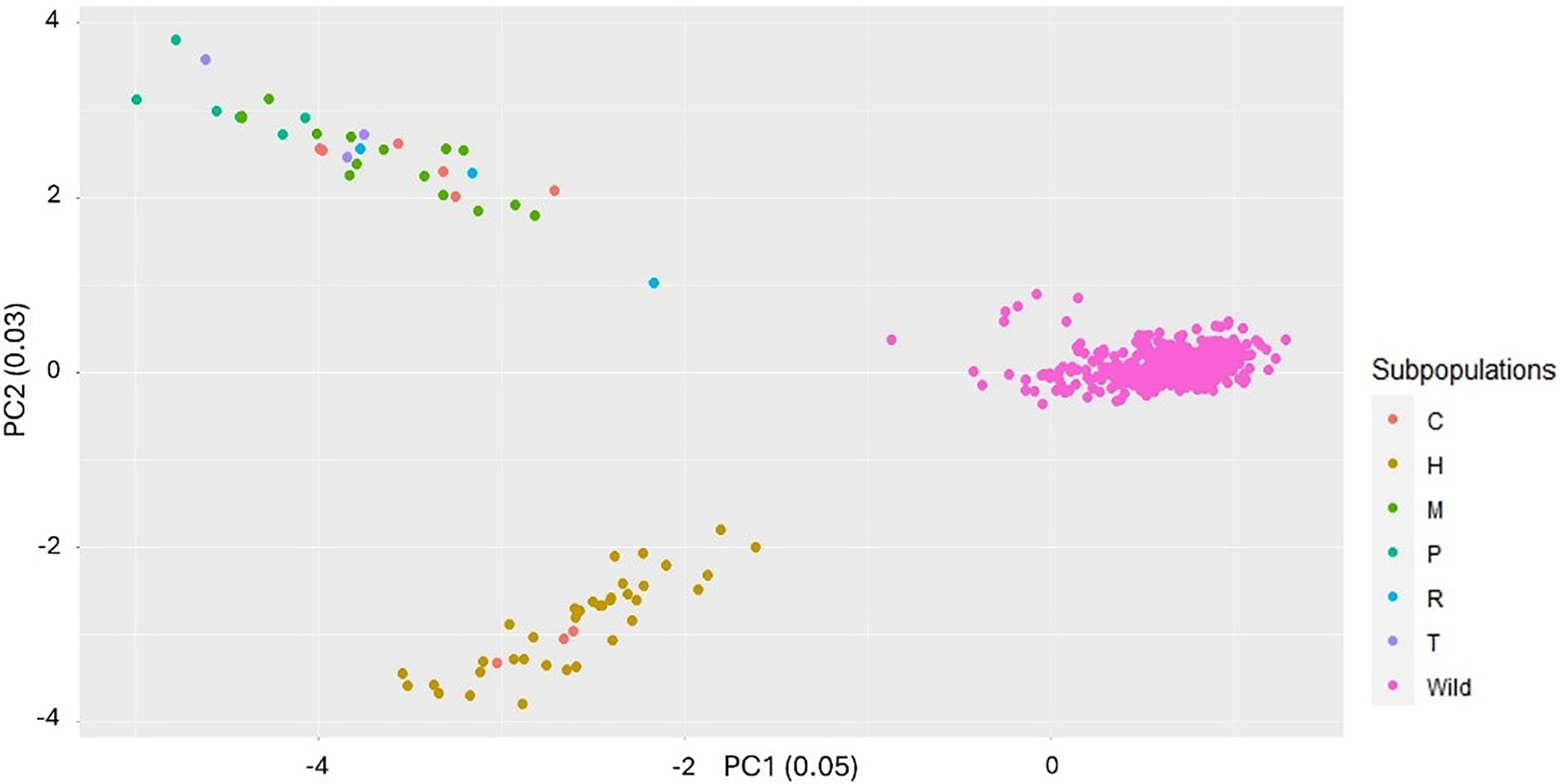
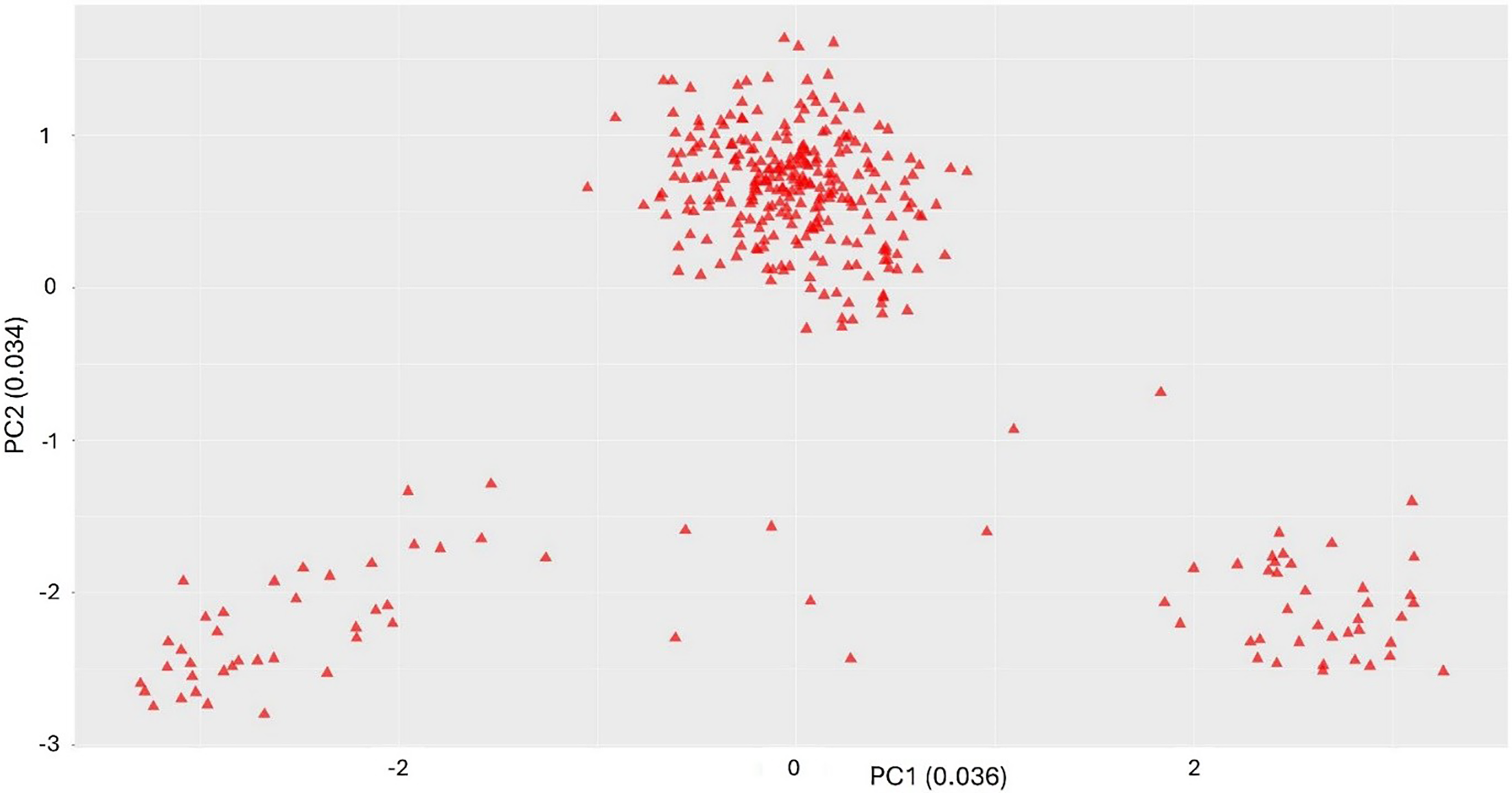
When wild populations were analyzed separately, they showed three distinct clusters, although the clustering pattern is unknown (Figure 2). The markers did not clearly differentiate the clusters, likely due to collapsed haplotype SNPs. A very weak but significantly positive correlation (r = 0.12, P < 0.0001) was observed between genetic and geographic distances (Supplementary Figure S2). This indicates that while some genetic differentiation is occurring, distance is not a primary factor structuring the populations. These results support the IBD model at the genetic level, despite evidence of long-range gene flow. The first two principal components, PC1 and PC2, explained 3.6% and 3.4% variance, respectively (Figure 2). When PCA analysis excluding wild species was performed, two clusters were identified (Figure 3). The first cluster comprised half-sibs and three cultivars, and the second cluster comprised USDA references and cultivars with no clear grouping among them. Several studies have clustered wild biotypes and/or landraces of L. perenne primarily based on morphological and/or genetic diversity (Bolaric et al. Reference Bolaric, Barth, Melchinger and Posselt2005a, Reference Bolaric, Barth, Melchinger and Posselt2005b, Reference Bolaric, Barth, Melchinger and Posselt2005c; Lopes et al. Reference Lopes, Reis, Barata and Nunes2009; Monestiez et al. Reference Monestiez, Goulard and Charmet1994; Oliveira et al. Reference Oliveira, Lindner, Bregu, García and González1997), whereas Blackmore et al. (Reference Blackmore, Thomas, McMahon, Powell and Hegarty2015) grouped European ecotypes of L. perenne in relation to their geographic origin and distribution. Maity et al. (Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c) reported clustering of the L. perenne ssp. multiflorum populations from the same region based on their morphological trait diversity, both in vegetative and reproductive traits. However, Maity et al. (Reference Maity, Singh, Martins, Ferreira and Bagavathiannan2021c) did not confirm any specific pattern in L. perenne ssp. multiflorum, indicating the spontaneous allele exchange among the wild populations by natural outcrossing (Balfourier et al. Reference Balfourier, Imbert and Charmet2000).
Principal component analysis (PCA) plots showing the clusters and the percentage variation explained by the first two components for the Lolium perenne ssp. multiflorum wild populations.

Principal component analysis (PCA) plots showing the clusters and the percentage variation explained by the first two components for all the lines except wild populations. Populations are indicated by different colors and letters: Populations are indicated by different colors and letters: C, cultivars; H, half-sibs; and USDA-GRIN references: M, Lolium perenne ssp. multiflorum; P, Lolium perenne; R, Lolium rigidum; and T, Lolium temulentum.

Marker–Trait Associations and BLAST Sequence Matches
Tiller Production
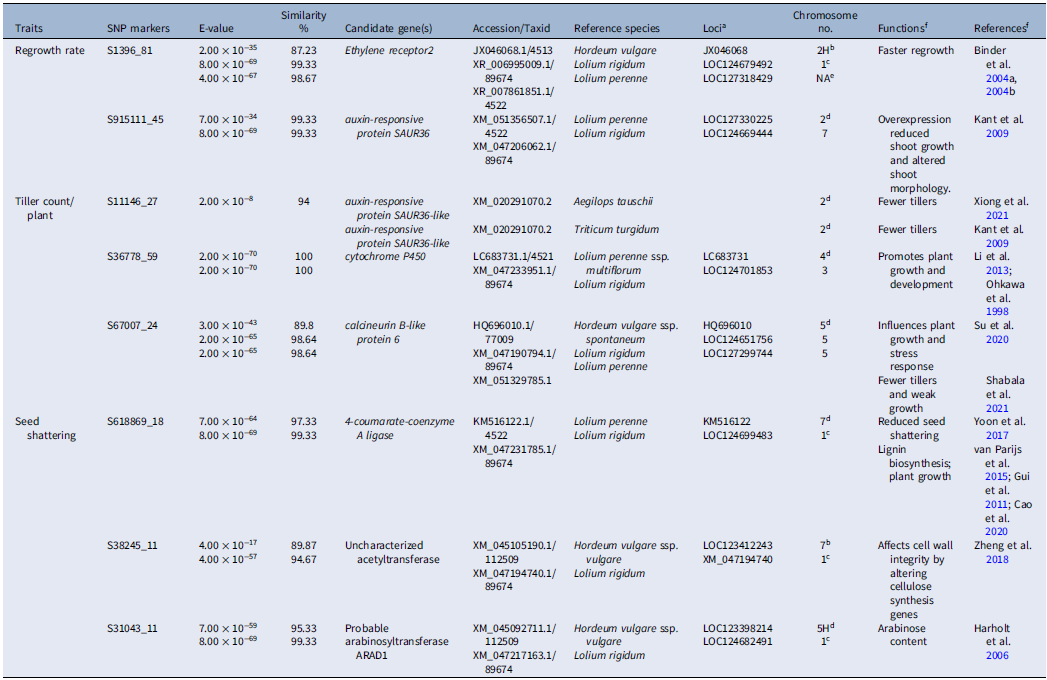
A total of 43 significant SNPs were identified in the GWAS for tiller production (P < 0.00001) based on Bonferroni’s correction of 3.27 × 10−6 (Figure 4). Among the significant SNPs, one QTL (SNP: S11146_27) with the E-value of 2.00 × 10−8 was found to match with the tiller production–controlling gene from the known sequence of A. tauschii and T. turgidum used as the reference species in BLAST analysis (Table 3). This QTL sequence matched with the important auxin-responsive protein small auxin-up RNAs (SAUR)36-like gene, which is located on chromosome 2 in L. perenne ssp. multiflorum (Kant et al. Reference Kant, Bi, Zhu and Rothstein2009; Xiong et al. Reference Xiong, Li, Zhang, Zhang, Liu, Peng, Chen and Zhao2021). Transgenic rice plants overexpressing the auxin-responsive gene SAUR39 produced significantly fewer tillers and leaves, with shorter plant height and lower yield compared with the wild-type rice (Kant et al. Reference Kant, Bi, Zhu and Rothstein2009). The SAUR gene family has been reported to be mainly expressed in elongation tissues of monocot species such as maize (Zea mays L.) and dicot species such as soybean [Glycine max (L.) Merr.], indicating their control on auxin-mediated cell elongation (Gee et al. Reference Gee, Hagen and Guilfoyle1991; Knauss et al. Reference Knauss, Rohrmeier and Lehle2003). SNP: S36778_59 matched with cytochrome P450 of L. perenne ssp. multiflorum that promotes plant growth and development as well as herbicide resistance in Lolium and related species (Li et al. Reference Li, Fang, Li, Zhang, Liu, Yang, Kang, Li and Wang2013; Ohkawa et al. Reference Ohkawa, Tsujii and Ohkawa1999). SNP: S36778_59 was found to be located on chromosome 4 in L. perenne ssp. multiflorum and chromosome 3 in L. rigidum. Another SNP, SNP: S67007_24, was found in a sequence matching the calcineurin B-like protein 6 gene in H. vulgare ssp. spontaneum, L. rigidum, and L. perenne that reduces tiller number and growth rate (Shabala et al. Reference Shabala, Alnayef, Bose, Chen, Venkataraman, Zhou, Shabala and Yu2021; Su et al. Reference Su, Huang, Ling, Mao, Huang, Su, Ren, Wang, Xu, Muhammad and Que2020). SNP: S67007_24 was found to be located on chromosome 5 of L. perenne ssp. multiflorum, L. perenne, and L. rigidum.
Manhattan plots showing significant single-nucleotide polymorphisms (SNPs) associated with (A) regrowth rate, (B) seed shattering, and (C) tiller production in wild Lolium multiflorum populations. The horizontal green and red dotted lines indicate the Bonferroni-adjusted significant and suggestive threshold levels, respectively.

List of significant sequence matches from BLAST analysis of single-nucleotide polymorphism (SNP) markers associated with seed shattering, tiller production, and regrowth rate of Lolium perenne ssp. multiflorum wild populations in the study.

a Protein coding.
b In the absence of chromosome information for these SNPs in L. perenne ssp. multiflorum genome, chromosome number was assigned according to H. vulgare.
c Chromosome number in L. rigidum.
d Chromosome number according to L. perenne ssp. multiflorum.
e NA, not available.
f Functions of the gene (s) based on published studies
Regrowth Rate
GWAS identified a total of 19 significant SNP markers (P ≤ 0.00001) for regrowth rate using Bonferroni’s correction and false positive discovery rate fixed, respectively, at 3.27 × 10−6 and P < 0.01 (Figure 4). For regrowth rate, one QTL (SNP: S1396_81) with the E-value of 2.00 × 10−35, and two more hits with L. perenne and L. rigidum were identified to match with a gene sequence of these reference species in BLAST analysis (Table 3). The flanking sequence of this SNP matched with the Ethylene receptor2, that is, the ETR2 gene, which promotes faster regrowth in foxtail millet [Setaria italica (L.) P. Beauv.] and other species (Binder et al. Reference Binder, Mortimore, Stepanova, Ecker and Bleecker2004a, Reference Binder, O’Malley, Wang, Moore, Parks, Spalding and Bleecker2004b). Because ethylene regulates the developmental processes, including the growth of etiolated seedlings, mutations in ETR genes significantly delayed recovery from reduced growth rate in the species (Binder et al. Reference Binder, Mortimore, Stepanova, Ecker and Bleecker2004a, Reference Binder, O’Malley, Wang, Moore, Parks, Spalding and Bleecker2004b). It is reported that plants with an etr1-6; etr2-3; ein4-4 triple loss-of-function mutation took more time to recover than single mutants (Binder et al. Reference Binder, Mortimore, Stepanova, Ecker and Bleecker2004a). The other marker–trait association was detected for SNP: S915111_45, which matched auxin-responsive protein SAUR36 (SAUR, Small auxin-up RNAs) in L. perenne and L. rigidum found to control shoot growth and morphology in several species, such as Tausch’s goatgrass [Aegilops tauschii ssp. strangulata (Eig) Tzvelev] (Kant et al. Reference Kant, Bi, Zhu and Rothstein2009). Both these QTLs are located on chromosome 2 of L. perenne ssp. multiflorum, as per the sequence annotation suggested by Chen et al. (Reference Chen, Kölliker, Mascher, Copetti, Himmelbach, Stein and Studer2024) and Frei et al. (Reference Frei, Veekman, Grogg, Stoffel-Studer, Morishima, Shimizu-Inatsugi, Yates, Shimizu, Frey, Studer and Copetti2021). But in L. rigidum, the ETR2 gene was annotated to chromosome 1.
Seed Shattering
GWAS identified a total of 54 significant SNP markers (P ≤ 0.00001) for seed shattering using Bonferroni’s correction and false positive discovery rate fixed, respectively, at 3.27 × 10−6 and P < 0.01 (Figure 4). The details of the position and estimates and significant matches found from the BLAST analysis are presented in Table 3. One QTL (SNP: S618869_18) was detected with an E-value of 7.00 × 10−64 for seed shattering from the known sequence of a seed shattering–controlling gene reported in L. perenne (chromosome 7) and L. rigidum (chromosome 1) (Table 3). This QTL sequence matched with the important 4-coumarate-coenzyme A ligase gene, 4CL (Yoon et al. Reference Yoon, Cho, Antt, Koh and An2017), which is a major gene in the lignin biosynthesis pathway. Suppression of this gene interferes with the production of p-coumaroyl-CoA, a precursor for the biosynthesis of p-coumaryl alcohol that ultimately produces lignin (Yoon et al. Reference Yoon, Cho, Antt, Koh and An2017). Cao et al. (Reference Cao, Huang, Luo, Zheng, Zhong, Sun, Gui and Li2020) reported that cell-specific suppression of this gene had a differential effect on lignin: downregulation of the gene reduced lignin content in xylem tissue (Gui et al. Reference Gui, Shen and Li2011; Kajita et al. Reference Kajita, Katayama and Omori1996; Lee et al. Reference Lee, Meyer, Chapple and Douglas1997).
Two other SNPs, namely S38245_11 and S31043_11, matched with acetyltransferase and arabinosyltransferase ARAD1, respectively, in H. vulgare ssp. vulgare (chromosomes 7 and 1, respectively) and L. rigidum (chromosomes 5 and 1, respectively). In plants, these two genes are involved in the cellulose and arabinose synthesis pathways, respectively, which are integral to cell wall formation and degeneration. These genes represent the monolignol biosynthesis gene family, which can decrease lignin in L. perenne (van Parijs et al. Reference van Parijs, Ruttink, Boerjan, Haesaert, Byrne, Asp, Roldán-Ruiz and Muylle2015) and also can regulate pectic arabinan, which controls cell wall integrity (Hussey et al. Reference Hussey, Mizrachi, Spokevicius, Bossinger, Berger and Myburg2011). Because these compounds play an important role in seed shattering in almost all plant species, manipulation of their biosynthetic pathway significantly influences the level of seed shattering across plant species, including Lolium ssp. (Elgersma et al. Reference Elgersma, Leeuwangh and Wilms1988; Fu et al. Reference Fu, Song, Zhao and Jameson2018; Harholt et al. Reference Harholt, Jensen, Sørensen, Orfila, Pauly and Scheller2006; Lee et al. Reference Lee, Yoon, Lee, Jeon, Lee, Lee, Chen, Yun, Oh and Wen2018; Seymour et al. Reference Seymour, Ostergaard, Chapman, Knapp and Martin2013; Zheng et al. Reference Zheng, Liu, Lin, Liu, Wang, Xin, Yao, Peng, Zhou, Ni and Sun2019).
Considerable genetic diversity was observed among the wild populations of L. perenne ssp. multiflorum from the Texas Blackland Prairies (Figures 1 and 2), suggesting the occurrence of gene flow in the region. Significant SNP markers (marker–trait associations) associated with adaptive traits, such as tiller production, regrowth rate potential, and seed shattering, were identified in this study using GWAS. These results could be further improved by aligning the ddRADseq reads to a reference genome. Ongoing genome sequencing efforts in L. perenne ssp. multiflorum will facilitate such alignment in the future, enabling the identification of a large number of SNP markers and their chromosomal locations. Given that L. perenne ssp. multiflorum. exhibit high outcrossing, with high heterozygosity, a haplotype-based genome assembly and GWAS approach would provide deeper insights into allelic variation across both haplotypes and their associations with key traits. This information on marker–trait association can be used for multiple applications: breeding high biomass–yielding (high-tillering) forage cultivars through marker-assisted selection or targeting reduced tillering to decrease their competitive ability and overall biomass for improved weed management.
Increasing seed retention (low-seed shattering) would be helpful for higher commercial seed yields in breeding programs. On the other hand, promoting seed retention at harvest would enable harvest weed seed control strategies (weed management). Robust regrowth potential after grazing or cutting would improve forage productivity, whereas slower regrowth potential after cutting or herbicide application would be used in weed management. Regrowth rate and tillering are complex, polygenic traits influenced by both genetic and environmental factors. Further studies are required to identify additional associated markers, explore their interactions, and functionally characterize the genes underlying these traits. In the next step, GWAS targeting other adaptive traits in L. perenne ssp. multiflorum, such as seed dormancy, leaf morphology (color and blade width), awn length, and plant biomass, will provide broader insight into the gene(s) controlling these adaptive traits in this species.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/wsc.2026.10101
Funding
AM acknowledges funding support from the Netaji Subhash ICAR- International Scholarship awarded by the Indian Council of Agricultural Research, and the Tom Slick Graduate Research Fellowship from Texas A&M AgriLife Research. The sequencing effort was supported by a seed grant awarded to NKS by Texas A&M AgriLife Genomics & Bioinformatics Services.
Competing interests
The authors declare that no conflict of interest exists.




 Open access
Open access