


In the postwar period, cancer became a public enemy in American politics, a deadly threat that citizens expected their federal government to fight and conquer.Footnote 1 The US government invested massive sums in research on cancer, producing major advances in knowledge but relatively few clinical breakthroughs. The publication of Rachel Carson’s Silent Spring in 1962, alongside growing alarm about the health risks of ionizing radiation from nuclear power and weapons testing, focused attention on cancer’s environmental causes.Footnote 2 By the late 1960s, scientists attributed most cancer to genetic damage – i.e. mutations – caused by exposure to external agents, or carcinogens. This exposure might occur over decades at low doses, with cancer resulting from the cumulative genetic damage. In response to public anxiety and scientific consensus, the US government targeted cancer from mutation-causing chemicals as a key public-health problem. In 1968, the National Cancer Institute (NCI) launched a ‘Plan for Chemical Carcinogenesis and the Prevention of Cancers’. During subsequent years the agency publicized an estimate that as much as 90 per cent of human cancer was due to environmental agents.Footnote 3 But laboratory tests used to identify carcinogens in rodents took two years and cost $150,000 per chemical. If cancer was a result of DNA damage, scientists reasoned, many or even most carcinogens caused mutation – they were mutagens. In other words, every human cancer started with a mutation, which could be prevented through controlling exposure to carcinogens. While this somatic-mutation theory was not the only explanation for cancer, it could accommodate several known causes (including chemicals, radiation, viruses and even inherited susceptibility) and offered a clear avenue for prevention.Footnote 4
Geneticists had developed methods for detecting mutagenicity of chemicals in mice, but the most sensitive one, developed by William and Lianne Russell at Oak Ridge National Laboratory, required thousands of animals.Footnote 5 Consequently, a mouse mutagenicity test was not an affordable or feasible replacement for a rodent carcinogenicity test. Scientists including Marvin Legator at the Food and Drug Administration (FDA) and Heinrich Malling at Oak Ridge National Laboratory began developing simpler in vitro tests with microbes to identify mutagenic chemicals, advocating that such tests become part of routine toxicity testing for pesticides, drugs, food additives and commercial chemicals. In the early 1970s, biochemist Bruce Ames at the University of California, Berkeley introduced a particularly easy and quick test for mutagenicity, involving customized strains of bacteria on a petri dish. The assay could be done in two days and was cheap. He provided the test strains freely to anyone who requested them, and thousands of scientists at universities, companies and federal agencies began using the so-called Ames test.Footnote 6
In the 1970s, both companies and regulators were using Ames test data in evaluations of chemical toxicity. US government agencies, beginning with the FDA and the Environmental Protection Agency (EPA), revised their testing requirements for chemicals to include mutagenicity data.Footnote 7 But did this new generation of mutagenicity assays – such as the Ames test – provide a valid method to identify carcinogens? This was a lively topic of discussion among specialists.Footnote 8 The Ames test instantiated a theory of cancer causation (that DNA mutations cause tumours); in turn the assay was becoming part of the broader sociotechnical apparatus that regulators were developing to manage toxic chemicals. A critical step in its establishment for regulatory toxicology was validation of the test, particularly for carcinogenicity in animals. Several scientific organizations and regulatory bodies launched programmes to evaluate short-term mutagenicity tests, among which the Ames test was the most widely used.
Validation is a determination of whether a technical test actually measures what it purports to measure.Footnote 9 Drawing on EPA documents, Colleen Lanier-Christensen has distinguished helpfully between ‘method validation’ and ‘model validation’.Footnote 10 For method validation of mutagenicity testing, scientific and government entities sought to establish the reliability and reproducibility of genetic toxicological tests, codifying protocols and practices. The ‘ring tests’ set up by the Organisation for Economic Cooperation and Development (OECD) for their Special Chemicals Program are a good example of such interlaboratory validation.Footnote 11 In this case, identical samples of a chemical (usually unidentified) were sent to toxicologists in several institutions for testing, to see whether results were reproducible when practitioners in different laboratories (and different countries) used the same protocols. Such efforts were principally aimed at standardization and data quality.
‘Model validation’ addresses how well a test predicts its end point. Mutagenicity testing was employed to identify chemicals of concern for two different end points: (1) mutagenicity, i.e. that a chemical’s ability to cause mutations in bacteria or another test organism predicts its ability to damage DNA in humans, and (2) carcinogenicity, i.e. that mutagenicity of a chemical in bacteria (or another test organism) predicts that the substance induces cancer in animals. In both cases, validation involved establishing that a bacterial (in vitro) test was predictive for an animal (in vivo) test. Extrapolating from the animal tests to human health risk was a separate step, involving other assumptions (see Figure 1).
Diagram from David Brusick, ‘Evolution of testing strategies for genetic toxicity’, Mutation Research/Genetic Toxicology (1988) 205, pp. 69–78, 70. Copyright 1988, reproduced with permission of Elsevier. Note: the Ames test would fall under ‘Submammalian short-term tests’ in this diagram.

Figure 1 Long description
The diagram shows two approaches for selecting short-term tests to predict animal mutagens and carcinogens. On the left, the approach for evaluating mutation risk includes animal test results from the 'in vivo standards', namely the 'specific locus test', the 'heritable translocation test' and the 'rodent dominant lethal test'. Simpler short-term tests, including submammalian, mammalian in vitro and mammalian in vivo somatic cell tests, are predictive for animal test results. On the right, the approach for evaluating cancer risk includes animal test results from the 'in vivo standard,' the 'animal life-time tumorigenicity assay'. Simpler short-term tests, including submammalian, mammalian in vitro and mammalian in vivo somatic cell tests, are predictive for animal test results. Both approaches must be extrapolated to human health risk.
As other scholars have noted, regulatory science is different from academic research in its focus on standardization, marginalizing new lines of inquiry in the name of routinization.Footnote 12 For a few years after introducing his test, Ames was still improving his Salmonella strains to increase their sensitivity in detecting mutagenicity, but as the assay began to be adopted for regulatory purposes, the test strains and procedure were standardized.Footnote 13 Beyond procedural consistency, new scientific knowledge can change how a test is interpreted. For the Ames test, the growing attention paid to non-mutagenic carcinogens during the 1980s meant that mutagenicity was no longer seen as an adequate proxy for carcinogenicity. Even so, the Ames test remained highly successful in regulatory toxicology. In effect, processes of validation generated a trustworthy tool while qualifying its scientific meaning.
This article compares several formal programmes – organized variously by academic scientists, government agencies, and international bodies – to validate mutagenicity tests (later termed genotoxicity tests). Debates over animal testing for carcinogens provided an important backdrop to these validation efforts.Footnote 14 In the 1970s, there were allegations of widespread fraud in commercial toxicology, which prompted US federal agencies to develop standards for ‘good laboratory practice’ (GLP) to certify that test data were reliable.Footnote 15 Despite the greater regimentation of animal carcinogenicity tests, they came under renewed attack by some scientists and industry spokespersons in the 1980s.Footnote 16 Rodent bioassays were not displaced from their role in risk assessment, but in the 1990s and 2000s toxicologists began to consider animal tumour data within a ‘mode-of-action’ approach that vetted positive carcinogenicity data based on whether a biological mechanism for tumour formation could be identified from other toxicology tests or structural information. In this context, mutagenicity became a criterion for interpreting the relevance of animal tumours, because mutation was a known mode of action for cancer. Whereas in vitro tests had been validated against animal tests, this turn of events shows that the direction of comparison at the heart of validation could be reversed, as in vitro assays could be used to evaluate data from rodent tests. In this sense validation remained a process open to new scientific information and regulatory approaches.
From bacteria to animals and back again
As soon as Bruce Ames established his bacterial screen for mutagens, he started assessing its validity for identifying known carcinogens.Footnote 17 The benchmark for establishing validity was not correlation with established human carcinogens; observational and epidemiological data on people were too limited.Footnote 18 Less than two dozen substances, out of tens of thousands of commercial chemicals, could be identified as human carcinogens through direct observation of higher-than-normal incidence of cancer. Usually this occurred in certain industries or professions where exposure was uniform and high; in many such cases specific chemicals were linked to cancer in specific organs, for example bladder cancer in aniline dye plants or leukemia among radiologists.Footnote 19 Instead, relevant data for most chemicals of concern came from animal tests. By 1941, 169 chemicals had been identified as carcinogenic in studies with laboratory animals; this had grown to over three hundred by 1950.Footnote 20
Even as the number of known chemical carcinogens increased, there was significant variability in experimental set-ups and species in animal tests used to identify them. Species variability alone could cause inconsistent results; a substance that caused tumours in one animal might not in another. Under the direction of Arnold J. Lehman, toxicologists at the FDA Division of Pharmacology developed a series of standard toxicity protocols. In 1955, a publication of these methods from his group included an animal test for carcinogenicity, contributed by Anne R. Bourke.Footnote 21 While acknowledging that a comprehensive carcinogenicity study might involve ‘large numbers of several species’, Bourke recommended exposing one strain of rats as well as one or more strains of inbred mice to make a determination of carcinogenicity.Footnote 22 A significant number of control animals were to be used, and all animals fully autopsied, to see whether they were tumour-bearing or tumour-free, with at least some microscopic pathology done on all animals.
This FDA protocol was further refined by the US National Cancer Institute, which became a hub of animal testing. In 1961, the NCI established a Carcinogenesis Studies Branch, to which an extramural contract system was added the following year. In 1969, the so-called Mrak report warned the US government that existing toxicity data for pesticides were seriously inadequate, leading the NCI to contract hundreds of carcinogenicity tests on pesticides and other suspect chemicals.Footnote 23 By focusing on carcinogenicity, this unit prioritized effects from chronic, low-dose exposures, which had not been the emphasis of traditional toxicology. It also aimed to put more carcinogenicity data in the public domain, given that most testing was conducted by industry and never published. The 1971 National Cancer Act gave a dramatic boost to the NCI’s budget; by 1974 the number of chemicals the agency was testing, most with the standard rodent carcinogenicity assay, reached five hundred.Footnote 24
During the years when the Carcinogenesis Studies Branch grew, the NCI’s leadership decided that this kind of regimented toxicology testing was not compatible with its mission of advancing understanding of cancer through basic research. In 1978, Joseph Califano Jr, the Secretary of Health, Education and Welfare under President Carter, moved the NCI’s carcinogen testing programme into a separate entity, the National Toxicology Program (NTP), with headquarters at the National Institute for Environmental Health Sciences (NIEHS) in North Carolina.Footnote 25 NTP test data were supposed to represent a gold standard for toxicology of chemicals, particularly for carcinogenicity.
To conduct an NTP standard carcinogen bioassay required hundreds of animals and more than two years of study. The first step involved pre-chronic toxicology studies, including determining the maximal tolerated dose (MTD, also known as the minimally toxic dose) for the test substance. To establish the MTD for a species or inbred strain, experimental animals were exposed to varying doses of the chemical to find the maximum amount (on average) that individuals could be exposed to without shortening the life span, causing the animal to lose more than 10 per cent of its weight, or producing overt signs of toxicity. To determine carcinogenicity, long-term (chronic) exposure tests would then be performed on a significant number of animals (usually dozens) from one or more species at dose levels at or below the MTD. The NTP’s standard protocol involved groups of fifty to a hundred males and females of two rodent species (from among standardized strains of mice, rats or Syrian hamsters) which were exposed, often through ingestion, over their lifetimes, from five to six weeks after birth to two years old. Usually the exposure level was set to the MTD and/or half the MTD. These were high doses, in many cases hundreds-fold higher than expected human exposure. However, given that chemical-induced carcinogenesis might well be a rare event at low dose, high doses were used in testing to maximize the chance of detecting the effect in a statistically significant number of animals.Footnote 26 Tests could be done at many doses if the investigators sought dose–effect curves. All tests, of course, included a control group of each species that were not exposed to the substance in question.Footnote 27
After two years of experimental exposure in such a carcinogenicity test, all surviving animals would be sacrificed and evaluated by histopathology for the presence of tumours, as compared with the usual incidence in that species.Footnote 28 As Harold C. Grice explained, a number of factors needed to be considered in evaluating this pathological data, including the number of animals tested, the number of tissues examined, the number of tumours and the presence of other pathological lesions.Footnote 29 It was often the case that the target organ for tumours in laboratory animals was not the same as the suspected site of cancer in humans. Laboratory mice are particularly susceptible to liver cancer, for example. Yet researchers demonstrated a correlation between the induction of liver cancer in mice and observations of human cancer in other organs.Footnote 30
It is worth stressing the regulatory centrality of these animal bioassays. For US government agencies such as the FDA, animal testing defined a cancer-causing chemical: ‘a carcinogen is a substance that when administered by an appropriate route, causes an increased incidence of malignant tumors in experimental animals as compared with a control series of untreated animals’.Footnote 31 International bodies also certified the long-term rodent test to determine carcinogenicity. At the International Agency for Research on Cancer (IARC), physician-turned-occupational-health-scientist Lorenzo Tomatis elevated long-term rodent tests as the preferred method to evaluate chemicals for carcinogenicity in humans.Footnote 32
The predictive power of a rodent carcinogenicity test rested on an assumption that one could extrapolate the results from animals to humans (as depicted in Figure 1). As Luther Carter (a science journalist) wrote in 1979,
there is general agreement among the agencies and their scientific advisers that a well-done bioassay or epidemiological investigation represents good science, even though (for obvious reasons) findings from tests with mice or rats cannot be verified by experiments with humans. The reason positive results from a bioassay with laboratory animals are generally accepted as strong evidence of potential human carcinogenicity is that virtually all known human carcinogens cause cancer in test animals.Footnote 33
As he observes, chemicals that tested positive in a rodent carcinogenicity test could not be tested on people to confirm the extrapolation of animal data to human health. In other words, the standard rodent carcinogenicity test itself could not be rigorously validated.Footnote 34
In sum, the standard animal carcinogenicity test was expensive, elaborate and lengthy.Footnote 35 By contrast, the Salmonella/microsome test developed by Bruce Ames to identify potential cancer-causing chemicals was quick and inexpensive. It involved exposing customized bacterial cells to the test chemical, in the presence of a rat liver (microsomal) extract that would generate the metabolic intermediates of the chemical found in the animal body. Each colony that grew on a test plate represented a mutation. If the number of colonies on a plate was greater than the control plate, which represented the rate of spontaneous mutations, the substance was scored as mutagenic (see Figure 2).
Schematic diagram of Ames test procedure. Histidine, CC BY-SA 3.0, at https://creativecommons.org/licenses/by-sa/3.0>, via Wikimedia Commons (accessed 9 March 2026).

Figure 2 Long description
The diagram illustrates the Ames test procedure. It starts with a Salmonella strain requiring histidine and a rat liver extract. These are combined with a possible mutagen and placed on a plate with media containing minimal histidine. The plate is incubated. A high number of revertants (his minus to his plus) on the test plate suggests the mutagen causes mutations. A control plate with natural revertants is also shown for comparison.
Beginning in the late 1960s, while Ames was still tinkering with his tester strains, he began sharing them with researchers in academe, industry and government. Errol Zeiger recalls being given early versions of the Ames test strains his first day of work in July 1969 at Marvin Legator’s unit at the FDA.Footnote 36 In 1972, the NCI set up a programme to compare how accurately existing mutagenicity tests could differentiate known carcinogens from non-carcinogens. The Ames test was among several systems being evaluated by the NCI; there were other microbial assays, as well as test systems based on insects (silkworms and flies), mice and mammalian cultured cells.Footnote 37 Initially, 103 compounds were selected for testing, to cover the major chemical groups of carcinogens, namely hydrocarbons, aromatic amines, nitrosamines, alkylating agents, and others. Testing was conducted through NCI contracts with Columbia University; the University of California, Berkeley; the Stanford Research Institute; and the Arthur D. Little Company. A similar Japanese national testing programme was going on in 1973 and 1974, involving workers in many laboratories.Footnote 38
In 1975, Ames and his co-workers published a paper based on a compilation of results from the three groups with NCI contracts: Ames’s at Berkeley, Herbert Rosenkranz’s at Columbia and the group at Stanford Research Institute. The paper also included some data from Japanese validation studies, and results from an EPA testing contract with Barry Commoner of Washington University. The only authors on this publication were members of the Ames laboratory, which angered some of the scientists at other institutions who had performed these assays.Footnote 39 The overall numbers were very encouraging: 90 per cent of Salmonella mutagens were also rodent carcinogens, and 89 per cent of known animal carcinogens were also bacterial mutagens.Footnote 40 The Ames test appeared to be a valid predictor of cancer in rodents.
There were limitations to how these laboratory results could be extrapolated to humans. As the paper by Ames and his colleagues indicates, classifying carcinogens is not straightforward, because some carcinogens are strong and others are weak in their effect – this is referred to as ‘potency’. Even if a high percentage of carcinogens in laboratory rodents were also carcinogens in humans, there are species-specific differences in potency to consider. The Salmonella mutagenicity test could not predict carcinogenic potency, as John Ashby and J.A. Styles at Imperial Chemical Industries observed in a 1978 letter to Nature.Footnote 41 They also pointed out that the liver microsomal extract that was a key ingredient of the Ames test was not sufficiently standardized with respect to its enzyme constituents for results to be considered quantitative. Consequently, they viewed the Ames test as more suitable as an ‘early warning system rather than the final arbiter of animal and possibly human carcinogenicity’.Footnote 42 It might be noted that Ashby and Styles were developing their own in vitro mutagenicity test, one that used mammalian cell cultures rather than bacterial cells.Footnote 43
By 1976 Ames’s test strains were being used by seventy-five industrial and drug companies.Footnote 44 Commercial toxicology companies began offering to perform the Ames test for two hundred dollars per chemical, a pittance compared to cost of a rodent carcinogenicity test.Footnote 45 Academic researchers were also avid adopters of the Ames test. By 1981, Ames test data were published on more than a thousand chemicals.Footnote 46 The Ames test went on to become a popular tool in the emerging field of genotoxicology. Between 1970 and 2010, more than ten thousand papers using the Ames test had appeared in the scientific literature.Footnote 47
How predictive of carcinogenicity did Ames test data need to be for regulators to use it to take action on potential carcinogens? As Frederick de Serres observed at a 1974 meeting on new mutagenicity methods, there was not a metric for sufficient validity ‘before being recommended as a basis for legislation. Would 85% positive on carcinogens and 10% positive on non-carcinogens be acceptable?’Footnote 48 In 1976 the Environmental Defense Fund asked the US Consumer Product Safety Commission to label children’s garments containing the flame retardant Tris, given that Arleen Blum and Ames had demonstrated the compound to be mutagenic, which was a ‘highly reliable predictor’ for carcinogenicity.Footnote 49 After receiving preliminary results from animal carcinogenicity tests on Tris, in April 1977 the agency banned the sale of garments containing it. The compound had only been introduced a few years prior, to meet 1973 federal regulations for decreased flammability in children’s sleepwear. As Ames noted,
It seems clear that many more chemicals will be added to the list of human carcinogens, as we are being exposed to an increasing flood of chemicals that have not been tested before use for carcinogenicity or mutagenicity, from flame retardants in our children’s pajamas to pesticides accumulating in our body fat. … The solution is prevention: identifying environmental mutagens and minimizing human exposures. Rapid, accurate in vitro tests, such as the Salmonella/microsome test, should play a crucial role in realizing this goal.Footnote 50
Questions about the validity of mutagenicity testing were no longer academic.
Scientists band together to validate short-term tests
The broad concern with exposures to mutagens had already prompted scientists to start meeting and publishing on the topic.Footnote 51 The Environmental Mutagen Society (EMS), founded in the US in 1969, focused on scientific communication (notably through Mutation Research) and compiling existing data in an Environmental Mutagen Information Center (EMIC) at Oak Ridge National Laboratory. In contrast to the animal toxicologists who populated the established Society of Toxicology, many of the members of EMS were trained as biochemists, microbiologists, geneticists or molecular biologists. Organizations similar to the EMS were founded in other countries, and an International Association of Environmental Mutagen Societies arose in 1973. Its leaders established an International Commission for the Protection against Environmental Mutagens and Carcinogens (ICPEMC), which first met in 1977 in Research Triangle, North Carolina, hosted by Frederick J. de Serres of the NIEHS. The Institut de la vie, a private non-profit organization in France dedicated to issues of science and society, agreed the fund the organization.Footnote 52
The ICPEMC was modeled on the International Commission of Radiological Protection (ICRP), an organization of scientific experts that promulgated safeguards for exposure to ionizing radiation. The ICRP’s standards provided the basis for radiation safety regulations enacted by many national governments and were also adopted by bodies such as the International Atomic Energy Agency. The ICPEMC had similar aspirations for informing safety regulation for chemicals, but the half-century difference in timing mattered. The ICRP was established by radiologists at an international congress in 1928, before most national governments had set up regulatory bodies to oversee safety around ionizing radiation.Footnote 53 By contrast, at the time of the ICPEMC’s founding, most national governments, and some international bodies, already regulated toxic chemicals and set safety-relevant standards.
To produce recommendations that could be taken up by regulatory bodies, the ICPEMC set up set up committees to address the following topics:
(1) the development, validation, application and comparison of short-term screening systems for the identification and characterization of chemical mutagens and carcinogens;
(2) the relation between carcinogenesis and mutagenesis;
(3) the establishment of a registry of national regulatory principles and actions;
(4) the formulation of general principles for evaluating risk estimate procedures and developing dose exposure limits;
(5) a survey of epidemiological studies on those sections of the human population that are being exposed to mutagenic and carcinogenic agents;
(6) a continued review of the state of our knowledge with regard to the levels of chronic and acute exposure to specific chemicals.Footnote 54
These topics reflect a mixture of scientific (e.g. 2) and policy (1 and 3) objectives. Committee 3 involved specialists in international law and policy, and its efforts overlapped with concurrent activities at the OECD.Footnote 55 Boundaries among the committees were blurry. Committees 1 and 2 were both addressing the validity of short-term mutagenicity tests; their members struggled to differentiate their work, with Committee 1 deciding to deal with comparisons between different tests and issues of reproducibility and Committee 2 taking up the state of the science on whether mutagenicity tests were (or would be expected to be) predictive of carcinogenicity.Footnote 56 A committee for topic 6 was apparently not constituted for lack of funds.Footnote 57
Committee 2 tackled whether mutagenicity tests were predictive of carcinogenicity.Footnote 58 They drew on performance indices set out by John Cooper, Rudolpho Saracci and Philip Cole, namely sensitivity, specificity and predictive value.Footnote 59 Sensitivity is a measure of whether a result is a true positive, calculated as the number of carcinogens scoring positive in a mutagenicity test divided by the total number of carcinogens tested. Specificity measures true negatives, calculated as the number of non-carcinogens that are negative divided by the total number of non-carcinogens tested. The predictive value is the number of positive results from true carcinogens divided by the total number of positive results. The committee selected ten mutagenicity tests to evaluate from the more than a hundred reported in the literature. The next step was to ascertain whether ‘adequate validation studies have been completed to enable the selection of a test for its particular performance criteria’.Footnote 60 Only the Ames test was considered fully established by this criterion, in part due to the wealth of published data using it.Footnote 61 Yet there was considerable variability in the literature on the Ames test when it came to validation. Reports of its sensitivity ranged from approximately 50 to 90 per cent, its specificity from 50 to 93 per cent, and its predictive value from 65 to 95 per cent. The committee did not try to adjudicate these differences, which it attributed to many factors, including the selection of chemicals used in each validation study. The final report closed by urging greater scientific research into the processes of mutagenicity and carcinogenicity, although accumulating knowledge did not necessarily account for the shortcomings of existing tests.Footnote 62
The ICPEMC’s activities were oriented around not only these six committees but also four further task groups, composed of members of the commission and aimed at producing specific policy statements, such as whether smoking carries genetic risks or how populations accidentally exposed to a suspected mutagen should be monitored.Footnote 63 Task Force 2 was to recommend a ‘strategy for using the best short-term screening systems available at present’.Footnote 64 Its report, ‘Advice on screening of chemicals for mutagenicity’, was published in Mutation Research.Footnote 65 It pointed the reader to a number of handbooks for protocols and provided contact information for representatives of the European, American, Indian and Japanese environmental mutagen societies, but avoided ranking different tests or methods.
One cannot help but be struck by the duplication of effort (two committees, one task force) within this one organization, which also laboured in an increasingly crowded field of expert groups. The ICPEMC, which sent representatives to meetings of the ICRP, was also in contact with the IARC.Footnote 66 It sought to coordinate its efforts with the United Nations Environmental Programme and the World Health Organization, but those two bodies were co-sponsoring with the International Labour Organization a separate validation programme through the International Programme on Chemical Safety (IPCS), which published several major reports in the 1980s.Footnote 67 Needless to say, the same experts in environmental toxicology tended to be involved in all of these organizations, as well as in relevant national bodies. It was not clear who could adjudicate different determinations by these various groups.Footnote 68
In the mid-1990s, the ICPEMC folded due to lack of funding.Footnote 69 Leaders of the ICPEMC recognized that certifying testing regimes was not sufficient for effective regulation. ‘The fear exists that an enormous amount of expensive data may be generated, filed, and forgotten.’Footnote 70 This self-organized commission of scientists lacked ultimate authority to classify carcinogens or validate tests.
Validation at US federal agencies
In the 1970s and 1980s, several US agencies launched validation efforts for mutagenicity tests. As mentioned above, the first example of this was at the NCI, which contracted with scientists to do validation studies of mutagenicity assays in the early 1970s.Footnote 71 Concurrently, the Department of Health, Education, and Welfare (DHEW, the US agency that included the NCI) appointed a group of scientists to validate the new genetic toxicology tests. In 1974, the DHEW Subcommittee of Environmental Mutagenesis of the Committee to Coordinate Toxicology and Related Programs set up an ad hoc committee to study how effective in vitro genetic tests such as Ames’s were at identifying mutagens – but not carcinogens (covered by another subcommittee). It was staffed by leading scientific specialists, several at government agencies. Frederick de Serres (chair), Heinrich Malling, Gary Flamm, Virginia Dunkel and Errol Zeiger began surveying data on the Ames test, the E. coli WP2 test (methodologically similar to the Ames test but using mutant strains of a different microbe), and a mouse cell culture assay (the L5178Y mouse lymphoma assay). There were initially two other cultured-mammalian-cell tests under consideration, one using Chinese hamster ovary cells, and the other using human lymphoblasts. Regarding the latter, the group decided it would be ‘inappropriate for the U.S. Government to support the validation of a patented test when there were equally promising tests in the public domain’.Footnote 72 The other test, derived from hamster cells, was set aside in part because a member of the committee (Flamm) had developed it. The NCI’s unpublished studies supported the decision taken by the EPA and FDA to use the Ames (Salmonella/microsome) and mouse lymphoma tests. Although the mouse lymphoma test was not yet widely employed, the committee’s selection of it gave it a major boost among rivals.Footnote 73
The EPA established its own validation programme in the late 1970s. Shortly after the agency’s creation in 1970, the EPA had inherited the registration of pesticides from the USDA, a process which included submission of extensive toxicity data. The passage of the Toxic Substances Control Act (TSCA) in 1976 extended the reach of EPA oversight to all commercial chemicals not already covered under statutes for drugs, food additives, pesticides and radiochemicals. A high priority for the agency was determining which new toxicology methods, such as mutagenicity testing, would be used in its decision making. The availability of short-term mutagenicity assays (like the Ames test) had been cited in the Congressional debates over the TSCA as evidence that the tens of thousands of commercial chemicals on the market could be subjected to at least preliminary screening for carcinogenicity. In turn, the EPA expected that its implementation of the TSCA would involve specifying which tests industry would need to perform for the premanufacturing notification forms for new commercial chemicals (and, in some cases, for existing chemicals). As it turned out, chemical trade associations were able to block the EPA’s requirement of specific tests for most commercial chemicals, though the agency still provided guidance on test systems for new chemicals – and requirements for pesticides.Footnote 74
In 1979, the EPA established a programme called Gene-Tox to evaluate the new in vitro genotoxicity test methods, including mutagenicity assays like the Ames test.Footnote 75 It was administered through the EPA Office of Pesticides and Toxic Substances by an Office of Testing and Evaluation created to specify new regulatory requirements for chemicals. The first phase of the Gene-Tox programme involved setting up so-called work groups of experts assigned to evaluate methods based on the published literature. As Errol Zeiger put it, Gene-Tox was ‘an extensive validation-by-literature-search effort that examined the performance of dozens of tests and endpoints’.Footnote 76 Initially, twenty-three assays were selected for evaluation; the Ames test was the most widely used among them. These work groups set out to evaluate each ‘assay’s ability to discriminate between mutagens and nonmutagens and/or carcinogens and noncarcinogens’ and ‘the system’s performance with chemicals of various classes and identification of chemicals whose effects are not adequately described’.Footnote 77 The starting point was a list of all citations for a given bioassay in the EMIC database.
Each Gene-Tox work group report was published in the open literature. The findings of these work groups were also reviewed by an assessment panel, which was charged with evaluating each method for hazard identification and risk estimation, both of which are steps of formal risk assessment, which guided EPA decision making.Footnote 78 Assays like the Ames test were aimed at hazard identification, the initial step of finding the chemicals that needed regulatory oversight; subsequent steps of risk assessment involved complex extrapolation models for estimating (human) exposure and dose.
Gene-Tox established its own database of chemicals, including known (‘evaluated’) carcinogens.Footnote 79 The validity of any short-term system to predict human cancer rested on (1) the adequacy of the animal carcinogenicity test to establish that end point and (2) the reliability of the classification of mutagenic and carcinogenic chemicals against which a short-term test was validated. In general, data from the NTP and the IARC provided the basis for identified carcinogens, but agencies used this information in different ways. For example, the FDA focused on animal carcinogenicity; the EPA also counted oncogenicity, which included chemical substances that cause benign neoplasms.Footnote 80
The main outcome of the Gene-Tox programme was the winnowing down of many proposed in vitro mutagenicity tests to those that were supported by published data, the few ‘regulatory tests’.Footnote 81 (The organizers of Gene-Tox noted that Committee 1 of the ICPEMC was engaged in a similar endeavour, and made available to that committee information coming out of Gene-Tox.Footnote 82) As Errol Zeiger recalled, the enthusiasm among genetic toxicologists for developing new tests meant that the literature was flooded with poorly tested assay methods. A well-validated test correctly identified non-carcinogens as well as carcinogens. Published data for non-carcinogens are not easy to find, because negative results often go unpublished. Gene-Tox also demonstrated that not enough chemicals had been evaluated in most test systems to make real comparison possible.
While Gene-Tox was working on data in the literature, the NTP was producing new data. Between 1984 and 1986, the NTP released results of rodent carcinogenicity tests for ‘a large number of chemicals not previously available for in vitro test system validation’.Footnote 83 The NTP conducted mutagenicity tests alongside its animal bioassays, turning this into a blind test of whether carcinogenicity correlated with mutagenicity. A sizable group of the chemicals that the NTP tested scored positive for mutagenicity but could not be identified as carcinogens in rodents. Others proved carcinogenic but were not mutagenic. Taking into account the new NTP data decreased the estimated predictive value of several mutagenicity test systems for carcinogens, because these tests were not as specific as previously thought.Footnote 84
EPA scientists and regulators struggled with how to classify and regulate non-mutagenic carcinogens, referred to as non-genotoxic or epigenetic (here referring to effects not attributable to changes in DNA sequence via mutation or chromosomal damage).Footnote 85 In effect, as Michael Waters has observed, the very category of ‘non-genotoxic carcinogens’ came out of using the Ames test to identify and classify carcinogens.Footnote 86 Organochlorine pesticides (such as DDE, a metabolite of DDT) provided examples of carcinogens that were not mutagenic.Footnote 87 EPA officials under Reagan suggested that non-genotoxic carcinogens might be regulated using thresholds, which were never assumed for mutagenic carcinogens. Such a policy was not implemented at the time, but this classification of ‘non-genotoxic carcinogens’ remained in place and became a disputed aspect of the EPA’s risk assessment guidelines.
Challenging the animal bioassay
Concurrently, the validity of the standard rodent carcinogenicity test itself came under question during the 1980s – and, indeed, under attack. This was due in part to new research on the mechanisms for liver tumours in rodents, but the critique was also pushed by industry-funded groups as part of a broader fight against chemical regulation.Footnote 88 Bruce Ames, who had lost faith that reducing consumer exposure to chemical contaminants could change human cancer rates, became a prominent critic of rodent tests. With co-worker Lois Gold, he published a 1990 paper on ‘too many rodent carcinogens’ that were being identified by chronic rodent bioassays. They were persuaded by new research that exposure level alone (testing at or near the ‘maximum tolerated dose’) was responsible for the result that half of chemicals tested via the rodent carcinogenicity assay were positive, including natural as well as synthetic substances.Footnote 89 This made him doubt that chemicals which were not mutagenic in bacteria were human carcinogens at all. Species differences were also at issue. Scientists estimated that only 84 per cent human carcinogens proved to be carcinogenic in rodents.Footnote 90 Unfortunately, attempts to improve that number (the rodent test’s sensitivity) came at the expense of its specificity. Because the rodent cancer test was the bedrock against which mutagenicity tests were validated for the end point of carcinogenicity, critiques of this regulatory standard wobbled the entire short-term testing regime.
In 1986, Raymond Tennant and ten co-authors published a paper in Science that attracted a great deal of attention.Footnote 91 Called ‘Prediction of chemical carcinogenicity in rodents from in vitro genetic toxicity assays’, it was seen by many as an assault on the popular Ames test, because it showed the concordance between mutagenicity on the Ames test and carcinogenicity on the standard rodent test to be significantly lower (60–70 per cent) than Ames and co-authors had suggested in 1975 (∼ 90%). The paper looked at four major genotoxicity tests: (1) mutagenicity in the Ames test, (2) mutagenicity in mouse lymphoma cells, (3) chromosomal aberration in Chinese hamster ovary (CHO) cells, and (4) sister chromatid exchange in CHO cells. The paper confirmed the reliability of these four tests, which produced ‘reproducible results in interlaboratory trials with coded chemicals’.Footnote 92 In other words, method validation was strong. In addition, there was remarkable concordance of positive results between the four different tests. More disappointing, from a regulatory point of view, was that the tests were not complementary: they tended to give similar results, despite their slightly different end points (e.g. mutation via DNA base-pair change versus chromosomal damage). Most agencies were recommending or requiring multiple tests, one a bacterial test (the Ames test or its counterpart in E. coli) to look for induced base-pair mutations and one a mammalian test (usually with cells, not animals) for chromosomal aberration. The frequent agreement between bacterial and mammalian test system results called into question the value of requiring multiple tests.
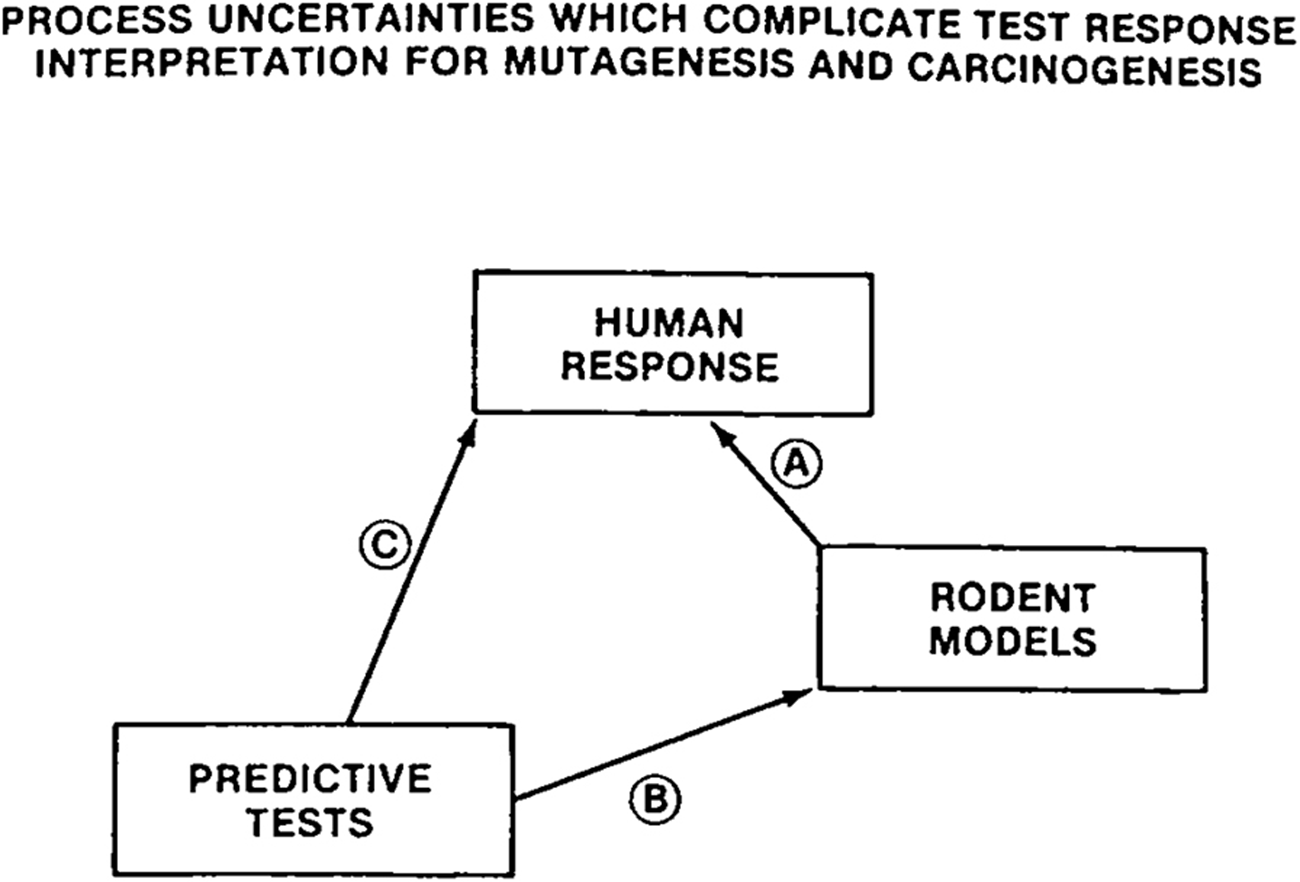
When interviewed about the paper a decade later, Tennant said that the pessimism about short-term tests in response to the paper was an overreaction. In his view, the Ames test did a good job of identifying carcinogens, and none of the other tests did any better. For a company toxicologist or regulator, the Ames test produced valuable information: if the chemical tested positive on the Ames test, it was likely going to be a carcinogen in animals.Footnote 93 The problem with short-term tests was that expectations for their predictive value were too high. Two years later, genetic toxicologist David Brusick echoed the sense that unrealistic hopes had guided the field. ‘Faced with the uncertainty concerning the generality of mutagenic mechanisms and the existing limitations for method validation, the prospects for selecting a universal genetic toxicity testing approach on the basis of concordance between predictive tests and human or other mammalian models appear poor’.Footnote 94 In this paper he included a schematic diagram showing the complexities of correlating in vitro tests, rodent models and human response (see Figure 3). In the end, the validation programmes of the 1970s and 1980s qualified the certainty with which mutagenicity tests could be used to identify cancer-causing substances. Regulators still utilized both in vitro mutagenicity tests and animal carcinogenicity tests but were aware of their limitations.
‘(A) Accuracy in predicting human response with rodent models cannot be defined although false responses can be documented. The exact frequency of false responses is unknown. (B) Accuracy in predicting rodent models with in vitro test results can be approximated. False responses for the animal can be documented but this information does not permit assessment of accuracy in predicting human responses due to the uncertainties in the reliability of the animal models. (C) Accuracy in predicting human response cannot be defined although false responses can be documented. Documented false responses for predictive tests and rodent models are not congruent. Some false responses against rodent standards will therefore be correct responses for humans’. Diagram and caption from David Brusick, ‘Evolution of testing strategies for genetic toxicity’, Mutation Research/Genetic Toxicology (1988) 205, pp. 69–78, 71. Copyright 1988, reproduced with permission of Elsevier.

Figure 3 Long description
The diagram illustrates the process uncertainties complicating test response interpretation for mutagenesis and carcinogenesis. It features three main components: human response, rodent models and predictive tests. Arrows labeled A, B and C indicate the relationships between these components. The arrow labeled A points from human response to rodent models, B points from rodent models to predictive tests and C points from predictive tests to human response. The title at the top reads 'Process Uncertainties Which Complicate Test Response Interpretation for Mutagenesis and Carcinogenesis.'.
However, there remained – and remains – significant dissatisfaction with rodent carcinogenicity assays. In the twenty-first century, government agencies, international bodies, animal welfare groups and industry all contend that improved in vitro assays and computational methods should replace experimental animals.Footnote 95 This is part of a broader commitment, associated with the terms ‘mode of action’ and ‘adverse outcome pathways’, to better utilize basic research on the molecular changes associated with chemical exposure in risk assessment and regulatory decision making.Footnote 96
At the current time, these newer regulatory strategies provide not so much a replacement for animal testing as new ways to interpret laboratory results in light of toxicological science and other test data, especially during risk assessment. That mutations can cause cancer remains an accepted scientific theory, hence when animal carcinogens are also genotoxic, researchers or regulators may infer that DNA damage is the mechanism by which a chemical induces cancer.Footnote 97 On this basis, in 2005 the EPA introduced an extra child-protective safety factor to the risk assessment of mutagenic carcinogens.Footnote 98 When an animal carcinogen is not genotoxic (as determined by in vitro tests or predicted by computer models), its risk level as a human carcinogen might be questioned, unless there is another identified mode of action for carcinogenicity.Footnote 99 This trend in regulatory toxicology has turned earlier mutagenicity-to-carcinogenicity validation efforts on their head, as genotoxicity data are now used to assess the relevance of animal tumour data.
The debate over the carcinogenicity of glyphosate, the active ingredient of the commercial herbicide Round-Up, illustrates how mutagenicity data matter to current regulatory decision making. The EPA released a comprehensive report on the carcinogenicity of glyphosate in 2016, which – using a weight-of-evidence approach and based on GLP test results – confirmed the agency’s long-standing conclusion that glyphosate is ‘“not likely to be carcinogenic to humans” at the doses relevant for human health risk assessment’.Footnote 100 By contrast, the 2015 IARC monograph on glyphosate had classified the substance as category A2, or probably carcinogenic to humans.Footnote 101 There are several differences between the data sets evaluated by the EPA and by the IARC in their reports. The EPA considered mostly data submitted by companies, which was certified as GLP, whereas the IARC considered data published in the scientific literature. In addition, the EPA considered studies of the active ingredient (glyphosate), whereas the IARC also considered toxicity studies of the product formulations, which often include other compounds that may enhance the toxicity of the active ingredient.Footnote 102
Strikingly, a key disagreement concerns whether glyphosate is mutagenic (or genotoxic). In all but one study considered by either the EPA or the IARC, glyphosate registers negative on the Ames test.Footnote 103 The IARC also considered many published data from other genotoxicity tests which use mammalian cells, some of which are susceptible to false positives due to cytoxicity.Footnote 104 By only considering GLP data and favouring the best-validated tests (especially the Ames test), the EPA disregarded positive mutagenicity data that were key to the IARC’s evaluation.Footnote 105 In addition, IARC considered a second mode of action, namely oxidative stress, that the EPA did not consider relevant in its risk assessment. For both bodies, mode-of-action reasoning made mutagenicity data critical to interpreting evidence of possible carcinogenicity (e.g. epidemiological data from agricultural workers and gardeners who used Round-Up). Decisions about which tests are valid guide these organizations as they include or exclude scientific studies from consideration of risk.
Conclusions
In the 1960s through the 1980s, public alarm that human cancer was caused by chemical exposure prompted scientists and US officials to develop technocratic ways of identifying hazardous chemicals and assessing their risks.Footnote 106 Given the high cost of animal carcinogenicity tests, researchers devised other methods to identify potential carcinogens in pesticides, commercial chemicals, food additives and drugs. The Ames test exemplified this trend in regulatory toxicology, and its advocates sought to justify the test’s predictiveness and validity. This article has recounted some representative programmes to validate mutagenicity testing as a way to identify and control cancer-causing chemicals. The prominence of international organizations in these validation programmes reflected the efforts by Americans and Europeans to harmonize regulatory requirements to expand global trade, but, in the realm of chemicals, ‘soft law’ was not initially as effective as national regulatory bodies (such as the EPA) in setting safety standards.Footnote 107
Validation opens up the uncomfortable issue of how tests relate to reality. This has been as true for the rodent carcinogenicity test as for the Ames test. Mice and rats are not humans, and the doses at which animals are exposed to test substances in the laboratory are high compared to expected human exposures. Could toxicological test systems with bacteria and cultured animal cells become well validated enough to spare the lives of millions of rats and mice consumed in standard carcinogenicity tests? In the summer of 2021, I put this question to the toxicologist Daniele Mandrioni and his answer struck me. He said that animal tests would remain essential to toxicology because ‘the mouse doesn’t care about your hypothesis’. What he meant was that every short-term carcinogenicity assay embodies a theory about what causes cancer, and if the theory is wrong or incomplete, so is the test. With the mouse, either it gets cancer from a test substance or it does not, but that outcome is not based on a biological theory of carcinogenesis. The Ames test definitely had a theory (that mutations cause cancer); this was both its strength and its vulnerability.
Yet once the Ames test was validated, shifts in cancer biology that qualified or even challenged the somatic-mutation theory of cancer did not necessarily displace mutagenicity testing. Indeed, the adoption of mode-of-action reasoning for risk assessment for carcinogens acknowledges that cancer may have several causes while giving mutagenicity data a role in certifying the human relevance of animal tumours. In addressing the question whether a simple bacterial test can identify human carcinogens, validation became a two-way street.
Acknowledgements
I thank the many colleagues who offered critical comments when I presented earlier versions of this paper in the Program in History of Science and the Department of History at Princeton University; SPHERE, CNRS & Université Paris Cité, Paris; the Validation and Regulation in the Sciences of Health Working Group, MPIWG, Berlin; and the International Congress of the History of Science and Technology, Dunedin, New Zealand. Karine Chemla, Mathilde Cohen, Jean-Baptiste Grodwohl, Ariane Hanemaayer, Ben Hegarty, Justine Holzman, Lara Keuck, Colleen Lanier-Christensen, Anne Le Goff, Anin Luo, Nicole Nelson, Sharad Pandian, Chris Phillips, Valentin Thomas, Sara Tridenti, Austen van Burns and two anonymous referees provided invaluable suggestions for improving the essay. Discussions with Errol Zeiger, Michael Waters and Dan Levy were immensely helpful, and I thank Scott Frickel for sharing tapes of his 1990s interviews with genetic toxicologists. This article benefited from a FIAS fellowship at the Institut d’études avancées, Paris (2024–5), funded from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 945408 and from the French state programme Investissements d’avenir, managed by the Agence nationale de la recherche (ANR-11-LABX-0027-01 Labex RFIEA+). I gratefully acknowledge other funding from Princeton University, the National Endowment for the Humanities and the Guggenheim Foundation.
Competing interests
The author declares none.



 Open access
Open access