


Introduction
Salmon have generation lengths of just a few years and, as a result of homing to natal streams to reproduce after marine migrations, are expected to be closely adapted to contemporary freshwater environments (Quinn, Reference Quinn2005; Waples et al., Reference Waples, Pess and Beechie2008). However, studies in Alaska (Olsen et al., Reference Olsen, Crane, Flannery, Dunmall, Templin and Wenburg2011) and the Puget Sound region of Washington (Ruckelshaus et al., Reference Ruckelshaus, Currens, Graber, Fuerstenberg, Rawson, Sands and Scott2006) report only weak correlation between habitat and salmon genetic population structure, results that challenge our basic understanding of Pacific salmon evolutionary history.
Alternate hypotheses commonly suggested to explain divergent salmon populations include barriers to migration (Allendorf and Seeb, Reference Allendorf and Seeb2000; Habicht et al., Reference C, Seeb and Seeb2007), founder effects (Ostberg and Thorgaard, Reference Ostberg and Thorgaard1999; Allendorf and Seeb, Reference Allendorf and Seeb2000), and population bottlenecks (Marshall et al., Reference Marshall, Knudsen and Allendorf2004; Ramstad et al., Reference Ramstad, Woody, Sage and Allendorf2004), all incorporating random genetic drift to explain population divergence. Each of these cases may be explained by paleolake isolation with our current understanding of the distribution of paleolakes (Mann and Peteet, Reference Mann and Peteet1994; Kaufman and Stilwell, Reference Kaufman and Stilwell1997; Hamilton, Reference Hamilton2001; Duk-Rodkin et al., Reference Duk-Rodkin, Weber and Barendregt2002; Kaufman et al., Reference Kaufman, Young, Briner, Manley, Ehlers, Gibbard and Hughes2011; this study). This hypothesis is compatible with the effects of barriers to anadromy, population bottlenecks threatening extirpation, and thermal stress. Random processes alone do not explain many aspects of diversity in salmon biology (Varnavskaya et al., Reference Varnavskaya, Wood and Everett1994). Our hypothesis is consistent with non-random selective pressures affecting run timing (freshwater maturation) (Healy, Reference Healy, Groot and Margolis1991; Hard et al., Reference Hard, Kope, Grant, Waknitz, Parker and Waples1996; Johnson et al., Reference Johnson, Grant, Kope, Neely, Waknitz and Waples1997; Faustini, Reference Faustini1999; Price, Reference Price2001), age at maturation (WDFW-PNPTT, 2001; Sands et al., Reference Sands, Rawson, Currens, Graeber, Ruckelshaus, Fuerstenberg and Scott2009), spawning phenotypic traits (Ramstad et al., Reference Ramstad, Woody and Allendorf2010), body size (Smoker et al., Reference Smoker, Jensen, Heg, Fitzgerald, Williams, Bayliff, Rees and Johnson1953; Elmendorf and Krober, Reference Elmendorf and Kroeber1992; Weitkamp et al., Reference Weitkamp, Wainwright, Bryant, Milner, Teel, Kope and Waples1995; Hard et al., Reference Hard, Kope, Grant, Waknitz, Parker and Waples1996), egg size (Weitkamp et al., Reference Weitkamp, Wainwright, Bryant, Milner, Teel, Kope and Waples1995; Hard et al., Reference Hard, Kope, Grant, Waknitz, Parker and Waples1996), low-temperature embryonic development (Beacham and Murray, Reference Beacham and Murray1988), and residence versus anadromy (Jensen, Reference Jensen1956a, Reference Jensen1956b; Haw et al., Reference Haw, Wendler and Deschamps1967; Hard et al., Reference Hard, Kope, Grant, Waknitz, Parker and Waples1996).
The Pacific coast of North America from the Alaska Peninsula through southeastern Alaska and British Columbia to the northwestern Washington coast was inundated by Cordilleran glacial ice during the last glacial maximum (Armstrong et al., Reference Armstrong, Crandell, Easterbrook and Noble1965; Booth, Reference Booth1986; Mann and Peteet, Reference Mann and Peteet1994). The Bering Land Bridge and the interior of Alaska remained ice free during past ice advances, as did coastal areas south of British Columbia (Hopkins, Reference Hopkins and Hopkins1967; McPhail and Lindsey, Reference McPhail, Lindsey, Hocutt and Wiley1986). Conventional thought holds that salmon populations that remained open to anadromy (migration between fresh waters for reproduction and marine waters for growth) during glaciations served as the source for salmon recolonization from coastal habitat following the retreat of continental ice sheets (McPhail and Lindsey, Reference McPhail, Lindsey, Hocutt and Wiley1986; Quinn, Reference Quinn2005; Waples et al., Reference Waples, Pess and Beechie2008; Pitman et al., Reference Pitman, Moore, Sloat, Beaudreau, Bidlack, Brenner and Hood2020). If so, these populations should retain genetic evidence of gene flow between straying anadromous populations. Stepwise colonization into previously ice-covered habitat would be expected to create signatures of decreasing genetic diversity with nautical distance from ice-free refugia (Comps et al., Reference Comps, Gomory, Letouzey, Thiebaut and Petit2001). In contrast to this expected model of recolonization from refugia open to anadromy following deglaciation, pink salmon (Oncorhynchus gorbuscha) in upper Cook Inlet, Alaska, have been found to have a unique mitochondrial DNA (mtDNA) haplotype (a set of closely linked genetic markers that have not been altered by recombination and tend to be inherited together) found nowhere else, the degree of divergence suggesting isolation since about 98,300–53,600 yr BP (Churikov and Gharrett, Reference Churikov and Gharrett2002), predating the most recent glacial period. This degree of divergence and subsequent lack of gene flow suggest that continuity of anadromy and gene flow offers an incomplete model for salmon evolution and population structure.
Where glacial ice meets rising terrain, water is impounded and drainage diverted (Mangerud et al., Reference Mangerud, Jakobsson, Alexanderson, Astakhov, Clarke and Henriksen2004). Although frequently omitted from representations of glacial ice extent, the resulting proglacial (ice margin) lakes span a range of sizes and duration and can provide refugia for a wide variety of aquatic species (Högbom, Reference Högbom1917; Segerstrale, Reference Segerstrale1976, Reference Segerstrale1982; Carrivick and Tweed, Reference Carrivick and Tweed2013).
Here we investigate whether contemporary genetic population structure for Pacific salmon may have resulted from mixing among populations that maintained anadromy through the ice ages and others that were isolated in proglacial lakes. Our expectation is that populations surviving glacial advances in refugia with open access to the ocean (i.e., refugia open to anadromy) continued to utilize anadromy and would tend to exhibit reduced genetic diversity with nautical distance from those refugia due to founder events and restricted gene flow during dispersal and stepwise colonization or to local extirpations during this process in a dynamic landscape. In contrast, our expectation for salmon populations that may have survived as freshwater residents living in proglacial lakes (i.e., refugia closed to anadromy) is that genetic diversity would tend to increase with distance from formerly closed refugia as these populations interbreed with more diverse populations from refugia open to anadromy colonizing newly available habitat.
Chum salmon (Oncorhynchus keta), which range widely across the northern Pacific Rim from Oregon to Japan and Korea, are of special interest in addressing this question, because they are the only salmon species not known to have at least some populations that survive, mature, and reproduce entirely in fresh water (Salo, Reference Salo, Groot and Margolis1991; Johnson et al., Reference Johnson, Grant, Kope, Neely, Waknitz and Waples1997). If chum salmon populations survived ice ages living in freshwater refugia, it is plausible that other species of Pacific salmon did so as well.
We propose and evaluate the hypothesis of postglacial salmon colonization from ice-margin lakes by focusing on patterns of population genetic variation in chum salmon and their relationship to the glacial history of two North American regions: the southern Alaska Peninsula/upper Cook Inlet and the Salish Sea of northwestern Washington and southern British Columbia. We analyze available genetic data to assess whether extant chum salmon populations in these two regions are derived solely from ancestral anadromous populations restricted to open refugia or could also include colonization from ancestral populations surviving in cryptic, freshwater glacial refugia.
Background
Salmonids in proglacial lakes
Glacier-fed and glacier-dammed lakes have cold temperatures and high turbidity, with corresponding reductions in euphotic volume and overall biological primary productivity (Lloyd et al., Reference Lloyd, Koenings and LaPerriere1987; Pitman et al., Reference Pitman, Moore, Sloat, Beaudreau, Bidlack, Brenner and Hood2020). Nevertheless, studies in Alaska have shown that heavily glacier-fed water can host viable salmonid populations (Bird and Roberson, Reference Bird and Roberson1979; Young and Woody, Reference Young and Woody2007).
Chromosome studies of Atlantic salmon (Salmo salar) have shown that southeastern Baltic Sea populations have a unique pattern derived from isolated freshwater populations to the east and that extant Baltic Sea Atlantic salmon populations may be the result of both marine colonization from the west and freshwater colonization from the east (Svärdson, Reference Svärdson1945). Population genetic studies of European Atlantic salmon using allozymes (enzymes reflecting different genetic variants, or alleles, at a single site, or locus, in the genome) (Kazakof and Titov, Reference Kazakov and Titov1991; Koljonen et al., Reference Koljonen, Jansson, Paaver, Vasin and Koskiniemi1999) and mtDNA (Nilsson et al., Reference Nilsson, Gross, Asplund, Dove, Jansson, Kelloniemi and Kohlmann2001) identify genetically divergent southeastern Baltic Sea Atlantic salmon populations and propose that this reflects isolation in proglacial lakes during glacial advances. Finnegan et al. (Reference Finnegan, Griffiths, King, Machado-Schiaffino, Porcher, Garcia-Vazquez, Bright and Stevens2013) used DNA microsatellites (short tandem repeats of nuclear DNA sequences) and mtDNA to conclude that postglacial colonization of northern Europe by Atlantic salmon occurred from a cryptic refugium in northwestern France as well as the well-documented ice-free refugium farther south in the Iberian Peninsula. A range-wide study using DNA single nucleotide polymorphisms (SNPs, or genomic variants at a single nucleotide position in the DNA) suggested that an isolated glacial lake refugium is one possibility for the observed genetic pattern, but held that other environmental gradients may also play a part (Rougemont and Bernatchez, Reference Rougemont and Bernatchez2018). Based on allozyme genetic studies in the Russian Far East, Kamchatka sockeye salmon (Oncorhynchus nerka) population structure is thought to result from competition between two colonizing lineages, one of which survived the last ice age isolated by glacial ice in mountain lakes, the other lineage having maintained anadromy in areas open to the sea (Varnavskaya et al., Reference Varnavskaya, Wood and Everett1994).
Late Pleistocene glaciation in the southern Alaska Peninsula/upper Cook Inlet
The most recent ice advance along the southern Alaska Peninsula, which supports numerous chum salmon populations, occurred between 23,000 and 14,700 cal yr BP (Karlstrom, Reference Karlstrom, Karlstrom and Ball1969; Mann and Peteet, Reference Mann and Peteet1994). Ice accumulated initially along the precipitation maxima of seaward-facing coastal mountain slopes, with corresponding precipitation minima in the lee away from the sea. At Kodiak Island, coastal mountains created a precipitation minimum adjacent to the northwest corner of the island bordered by Shelikof Strait. Ice thickness of about 300 m impounded a lake with water depths approaching that thickness and areal extent of about 1000 km2 for up to 8000 years (Fig. 1). To the east, ice accumulated across the entrance to Cook Inlet, creating lakes of a range of sizes in areas to the north, including upper Cook Inlet (Karlstrom, Reference Karlstrom1964; Kaufman et al., Reference Kaufman, Young, Briner, Manley, Ehlers, Gibbard and Hughes2011). Accumulation of coastal and mountain ice also impounded a lake with an area of 8900–24,000 km2 and depths as much as 300 m in the interior of the Copper River (Wiedmer et al., Reference Wiedmer, Montgomery, Gillespie and Greenberg2010). This lake has been identified as an important refugium for fish that decanted into upper Cook Inlet both in normal proglacial lake outlets and in large outburst floods due to failure of ice dams between the watersheds.
The approximately 1000 km2 area on western Kodiak Island was surrounded by glacial ice from about 23,000 cal yr BP until about 15,000 cal yr BP. Most of this area was a lake cut off from the sea by glacial ice. Adapted from Mann and Peteet (Reference Mann and Peteet1994). A larger area near upper Cook Inlet was also impounded by glacial ice for a similar time (not shown) (Karlstrom, Reference Karlstrom1964; Wiedmer et al., Reference Wiedmer, Montgomery, Gillespie and Greenberg2010).

Late Pleistocene glaciation in the Salish Sea
About 18,000 cal yr BP, the Cordilleran Ice Sheet advanced south and west across northwestern Washington (Booth et al., Reference Booth, Troost, Clague, Waitt, Gillespie, Porter and Atwater2004), a region that supports chum salmon with exceptional genetic and life-history diversity. Alpine glaciation in the Cascade and Olympic Mountains was in decline and alpine ice extent in retreat before the surge of Cordilleran ice into the Puget lowland from the north (Armstrong et al., Reference Armstrong, Crandell, Easterbrook and Noble1965; Booth, Reference Booth, Ruddiman and Wright1987). This timing allowed for the continuous existence of ice-margin aquatic features in the Puget Lowland (Fig. 2). At the maximum extent, ice occupied the Puget Lowland and dammed tributary mountain valleys, forming smaller proglacial lakes on the north and east sides of the Olympic Mountains (Bretz, Reference Bretz1913; Tabor, Reference Tabor1975; Long, Reference Long and Hellwig2010) and in major valleys on the west slope of the Cascade Range (Bretz, Reference Bretz1913; Cary and Carlston, Reference Cary and Carlston1937; Mackin, Reference Mackin1941; Crandell, Reference Crandell1963; Rosengreen, Reference Rosengreen1965; Booth, Reference Booth1986). Large proglacial lakes developed again during the retreat of the ice sheet (2B) until deglaciation of the Puget Lowland was largely complete (Haugerud, Reference Haugerud, Waitt, Thackray and Gillespie2020) about 15,000 years ago (Bretz, Reference Bretz1913; Thorson, Reference Thorson1989).
The Salish Refugium: proglacial lakes in the Olympic and Cascade Mountains. (A) Reconstructed Cordilleran ice sheet in northwestern Washington at maximum ice extent about 16,000 cal yr BP. Contours are ice-surface elevation relative to present-day sea level (m). Modern oceanic shoreline is shown as a violet line. Geological evidence for proglacial lakes is indicated by numbers around the ice margin: 1, 2 (Tabor, Reference Tabor1975); 3, 4, 6 (Long, Reference Long and Hellwig2010); 5, 7 (Bretz, Reference Bretz1913); 8 (Crandell, Reference Crandell1963; Rosengreen, Reference Rosengreen1965); 9, 10 (Mackin, Reference Mackin1941; Booth, Reference Booth1986); 11, 12, 13, 14 (Booth, Reference Booth1986); 13 (Cary and Carlston, Reference Cary and Carlston1937). (B–D) Proglacial lakes during successive phases of ice-sheet decay. (B) South drainage of glacial Lake Russell through the Chehalis River valley to the Pacific Ocean. (C) North drainage of glacial Lake Bretz to the Strait of Juan de Fuca. (D) Marine incursion ca. 15,000 cal yr BP.

As a similar (inverse) drainage history likely characterized the ice advance phase, we interpret the earlier observations to indicate the persistence of proglacial lakes with labyrinthine connections to each other and intermittent connections to the ocean throughout the most recent glacial cycle. At maximum ice extent, gradients and flow rates of ice-margin drainage were high and episodically variable (Booth, Reference Booth1986) and unsuitable for anadromy. We further infer that a similar history played out during each of the at least six times northwestern Washington was glaciated during the last 2 million years. We hypothesize that salmon inhabited these proglacial lakes during maximum ice extent and then radiated to other drainages as the ice retreated.
Methods
Pacific Rim
A genetic dataset common to both southern Alaska Peninsula/upper Cook Inlet and Salish Sea chum salmon comprises SNP samples from 310 populations across the known range of chum salmon around the Pacific Rim (Dann, Reference Dann2023; Fig. 3C). Methods of sample collection, laboratory analyses, and population statistical analyses are detailed in DeCovich et al. (Reference DeCovich, Dann, Rogers Olive, Liller, Fox, Jasper, Chenowith, Habicht and Templin2012). Target sample size was 95 individuals for each population (range: 41–200; total 32,817 individuals from 310 populations). After extensive development and testing, a final set of 91 SNP loci was used in the analysis. As this extensive dataset was collected for management purposes in Alaska, sample density is highest there. Tissue samples from an earlier allozyme dataset (Seeb and Crane, Reference Seeb and Crane1999) were available for SNP resampling in Alaska, but not in the Salish Sea.
Chum salmon range-wide between-population genetic distance and within-population diversity based on single nucleotide polymorphisms (SNPs). (A) Unrooted dendrogram of pairwise genetic distance showing continental-scale genetic structure. “Limbs” of the tree reflect a spectrum of divergence (F ST; Weir and Cockerham, Reference Weir and Cockerham1984) ranging from single populations such as Sturgeon-Kitoi (diamond symbols) (Kodiak Island, in black) to the regional divergence of the Salish Sea (Puget Sound and Vancouver Island, in red and blue, respectively). (B) Within-population diversity paired with mean between-population divergence. Kodiak Island and the Puget Sound areas have high levels of contrast in genetic variability parameters within relatively small geographic areas. (C) Color-coded key for SNP range-wide genetic variability.

Portions of the SNP dataset have previously been analyzed and published as part of regional population structure analyses, including populations from Washington, USA, and British Columbia, Canada (Small et al., Reference Small, Rogers Olive, Seeb, Seeb, Pascal, Warheit and Templin2015), southwestern Alaska (Petrou et al., Reference Petrou, Seeb, Hauser, Witteveen, Templin and Seeb2014), western Alaska (Garvin et al., Reference Garvin, Kondzela, Martin, Finney, Guyon, Templin, DeCovich, Gilk‐Baumer and Gharrett2013), and Japan (Sato et al., Reference Sato, Templin, Seeb, Seeb and Urawa2014). Here we have visualized range-wide structure for all 310 populations using conventional phylogenetic trees (branching diagrams that depict evolutionary relationships between populations based on their common ancestry and subsequent genetic differentiation; Rambaut, Reference Rambaut2010) and a scatter plot of within-population diversity (expected heterozygosity, H e, a measure from 0 to 1 of genetic variation within a population that reflects the probability that an individual has multiple alleles at a given locus) and mean pairwise population structure (fixation index, F ST, a measure from 0 to 1 of the degree of genetic isolation between two putative populations, with 1 being complete isolation and 0 being completely random mating between the two, in this case at the level of the SNP). We calculated mean pairwise population F ST and within-population H e (Weir and Cockerham, Reference Weir and Cockerham1984) using the R package hierfstat v. 0.5-7 (Goudet, Reference Goudet2005). These parameters are expected to be inversely related for the case of sequential founder effects (Ramachandran et al., Reference Ramachandran, Deshpande, Roseman, Rosenberg, Feldman and Luca Cavalli-Sforza2005). We sought to identify geographic regions with high contrast (i.e., a broad range of values) in diversity and divergence to elucidate the underlying mechanisms for variation in salmon population structure.
Southern Alaska Peninsula/upper Cook Inlet
The Alaska Peninsula and Kodiak Island region is an area of high contrast in genetic variation in chum salmon for both allozymes (Seeb and Crane, Reference Seeb and Crane1999) and SNPs (Petrou et al., Reference Petrou, Seeb, Hauser, Witteveen, Templin and Seeb2014). We extend previous analyses to include upper Cook Inlet, in light of documented ice age refugia at Kodiak Island (Fig. 1) (Karlstrom, Reference Karlstrom, Karlstrom and Ball1969; Mann and Peteet, Reference Mann and Peteet1994), upper Cook Inlet (Karlstrom, Reference Karlstrom1964; Kaufman et al., Reference Kaufman, Young, Briner, Manley, Ehlers, Gibbard and Hughes2011), and the adjacent Copper River basin (Wiedmer et al., Reference Wiedmer, Montgomery, Gillespie and Greenberg2010; Kaufman et al., Reference Kaufman, Young, Briner, Manley, Ehlers, Gibbard and Hughes2011).
For 59 chum salmon sample populations in the southern Alaska Peninsula/upper Cook Inlet, we used Bayesian lineage clustering to evaluate the likely number of founding populations represented by the SNP dataset (STRUCTURE; Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Falush et al., Reference Falush, Stephens and Pritchard2003). An earlier genetic study of chum salmon in western Alaska, including the Alaska Peninsula, identified two major lineages contributing to population structure there: a Beringian lineage and another from the Gulf of Alaska (Seeb and Crane, Reference Seeb and Crane1999). We evaluated a range from 1 to 12 lineages (K = 1–12) to describe the colonization of the southern Alaska Peninsula. Because of the relatively low rate of homing, we believed 12 to be a reasonable maximum number of lineages for this analysis (Tallman and Healey, Reference Tallman and Healey1994; Brenner et al., Reference Brenner, Moffitt and Grant2012; Keefer and Caudill, Reference Keefer and Caudill2014). We evaluated the most likely number of lineages (K) according to the change in variance of likelihood at each level of K (Evanno et al., Reference Evanno, Regnaut and Goudet2005). See the Supplementary Material for details of the analysis. We also conducted a principal component analysis (PCA) to visualize patterns of genetic differentiation using the R (R Core Team, 2019) package adegenet v. 2.1.3 (Jombart, Reference Jombart2008).
We visually evaluated the geographic distribution of the rare allozyme allele mAAT-2r* (Seeb and Crane, Reference Seeb and Crane1999), because its occurrence in chum salmon is widespread in the Alaska Peninsula area but undetected elsewhere except for a single individual sampled in northwestern Alaska.
Salish Sea
We examined chum salmon population structure in the Salish Sea because of high contrast in genetic variation (Fig. 3) as well as diverse run timing variation (Phelps et al., Reference Phelps, LeClair, Young and Blankenship1994) in a limited geographic area that was subject to extensive glaciation.
Some summer-run chum salmon populations in the Salish Sea region experienced severe declines in the early 1990s (Johnson et al., Reference Johnson, Grant, Kope, Neely, Waknitz and Waples1997). An allozyme dataset (Phelps et al., Reference Phelps, LeClair, Young and Blankenship1994) is available for which sampling predates many of those population minima, whereas most of the SNP samples were taken after those minima (Small et al., Reference Small, Rogers Olive, Seeb, Seeb, Pascal, Warheit and Templin2015). Analysis of the impacts of hatchery supplementation on within-population genetic diversity (H e) of summer-run chum salmon from the region using DNA microsatellites shows evidence of loss of diversity, especially associated with population minima (Small et al., Reference Small, Currens, Johnson, Frye and Von Bergen2009). The allozyme dataset has about twice the sample density as the SNP dataset, and those samples are concentrated in the area of interest, the range of summer-run Salish Sea chum salmon. For example, in southwestern and west-central Puget Sound (SW and WC PS) the allozyme dataset has 16 chum salmon populations, while the SNP dataset has only 4; in the Discovery-Sequim Bay area, the allozyme dataset has 3 populations and the SNP dataset only 1 population. Because of the strong geographic component to chum salmon population structure in the Salish Sea and the history of recent summer-run chum salmon population bottlenecks, our Salish Sea analyses rely primarily on the allozyme dataset. Genotypic data are not available for the Salish Sea allozyme dataset, but the geographic concentration of samples and sampling before potential population bottlenecks provide compelling reasons for relying on the allozyme dataset for Salish Sea biogeographic analysis.
The Phelps et al. (Reference Phelps, LeClair, Young and Blankenship1994) dataset of chum salmon in the Pacific Northwest consists of collections of more than 13,000 individuals representing 105 populations from the Oregon coast northward to the upper Strait of Georgia east of Vancouver Island. An issue for biogeographic analyses in the Salish Sea is the history of transplants of hatchery fish throughout the area. For chum salmon this was commonly the export of fry from hatcheries in the Hood Canal area of Puget Sound to other areas, often to streams draining the western slope of the Cascade Mountain range. We relied on published data and firsthand knowledge to remove populations likely to have been influenced by this activity from the allozyme dataset (Phelps et al., Reference Phelps, LeClair, Young and Blankenship1994; Johnson et al., Reference Johnson, Grant, Kope, Neely, Waknitz and Waples1997; Supplementary Table S2). Similar quality control measures limited the number of allozyme alleles used in the analysis (Supplementary Table S3); details are provided in the Supplementary Material.
We created a tree based on continuous maximum likelihood (CONTML program; Felsenstein, Reference Felsenstein1973) and plotted it on a geographic map using the program GenGIS 2 (Parks et al., Reference Parks, Mankowski, Zangooei, Porter, Armanini, Baird, Langille and Beiko2013). We obtained similar results with a neighbor-joining tree using Nei’s (Reference Nei1972) distance (not shown).
Using the filtered allozyme dataset from Phelps et al. (Reference Phelps, LeClair, Young and Blankenship1994), we conducted analyses of within-population genetic diversity (H e) and allelic richness standardized at population sizes of 40 individuals (Ar40) (El Mousadik and Petit, Reference El Mousadik and Petit1996) for 90 populations of chum salmon from the Pacific Northwest and southern British Columbia, including the Salish Sea. The 90 chum salmon populations represent as many as 12 distinct subregional groups for fishery management purposes based on geography and adult run timing (summer, fall, winter), as identified in Phelps et al. (Reference Phelps, LeClair, Young and Blankenship1994). We conducted both analyses of variance (ANOVAs) of within-population diversity and an analysis of covariance (ANCOVA) of H e and Ar40, testing for the influence of metapopulation (a group of spatially separated populations that interact) structure on Salish Sea chum salmon genetics to better understand its evolutionary history. We conducted all these analyses in base R v. 4.3.2 (R Core Team, 2023; see the Supplementary Material for analytical details).
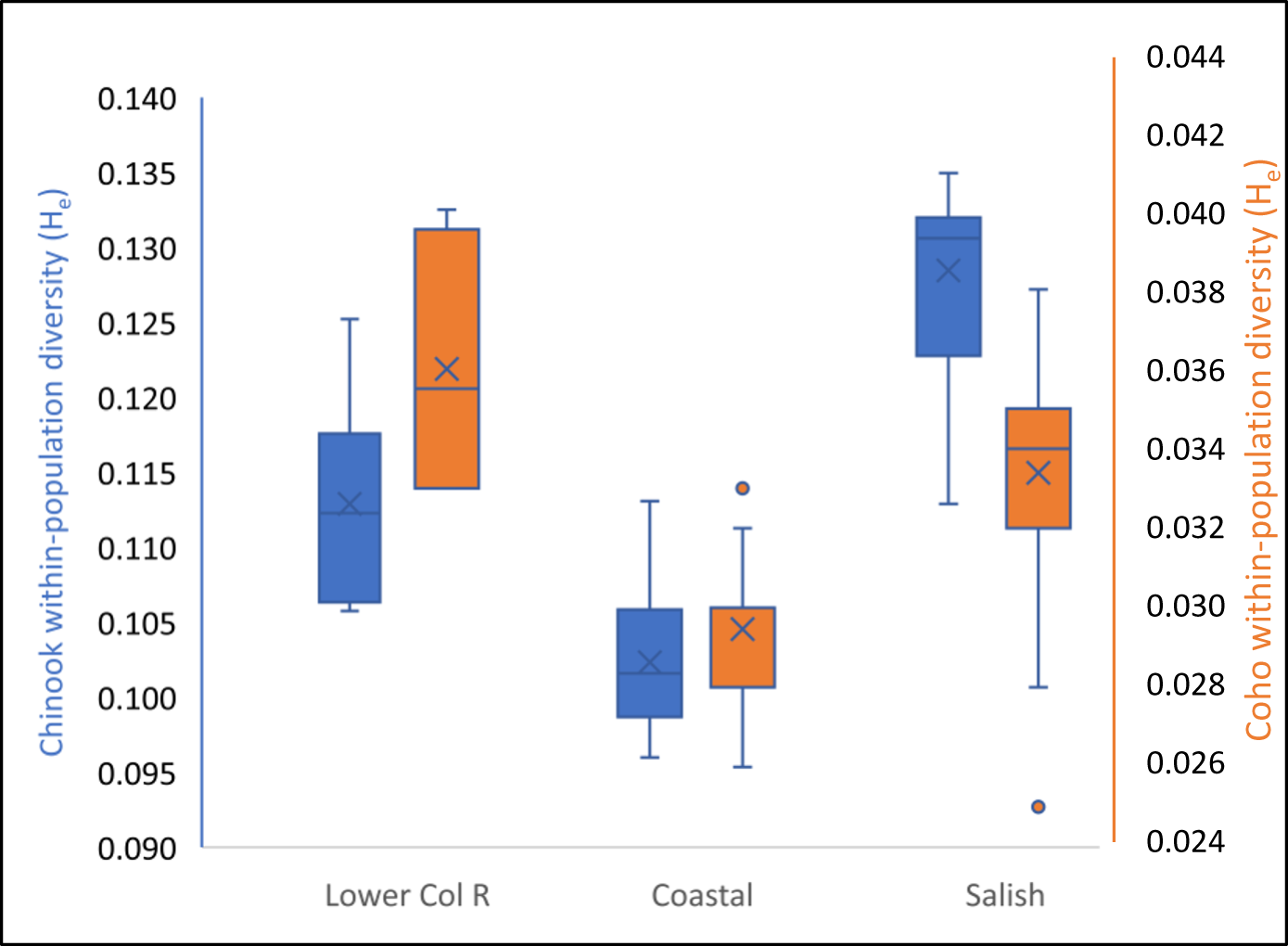
To provide biological and geographic context, we also examine allozyme genetic data for both Chinook (Oncorhynchus tshawytscha) and coho (Oncorhynchus kisutch) salmon in the region. The coho salmon data are as published in Teel et al. (Reference Teel, Van Doornik, Kuligowski and Grant2003). The Chinook salmon data are the dataset analyzed in Waples et al. (Reference Waples, Teel, Myers and Marshall2004), for which we calculated within-population diversity using the methods given earlier for chum salmon.
Results
Pacific Rim
The range-wide SNP dataset produced a highly structured field of population diversity and divergence for chum salmon (Fig. 3). Prominent ranges of correlated low diversity and high divergence extend down the North American and Asian coasts. The most diverse and least divergent chum salmon populations are found in Bering Sea drainages (Fig. 3B). Ecoregions also have a range of diversity–divergence contrasts within them. The most extreme contrast in diversity and divergence is associated with chum salmon populations from Kodiak Island, south of the Alaska Peninsula. Sturgeon-Kitoi populations from Kodiak Island maintain genetic variability parameters (diversity and divergence) similar to those from Japan (Fig. 3B, black ellipse, H e = 0.274, F ST = 0.185), although surrounded by relatively diverse local populations (H e = 0.350, F ST = 0.068; Fig. 3B). Chum salmon populations from nearby upper Cook Inlet also form a group characterized overall by low diversity and high divergence, while other Alaska populations feature higher diversity and lower divergence. Chum salmon populations from a very restricted geographic range in Puget Sound, on the order of 100 km of linear distance, also exhibit a considerable contrast of diversity–divergence values, a range of values greater than that of the entire Yukon River, comparable to the range of values for Korea and Japan or for the Okhotsk-Kamchatka eco-region of eastern Russia (Fig. 3C). The similarly high contrast in genetic variability parameters within these localized geographic areas suggests that there may be an underlying biogeographic mechanism related to genetic variability and common to all areas.
Southern Alaska Peninsula/upper Cook Inlet
An unrooted phylogenetic tree for the range-wide SNP dataset shows high mean pairwise divergence for upper Cook Inlet and Sturgeon-Kitoi chum salmon populations and high contrast in divergence within Kodiak Island (Fig. 4A). The result of our STRUCTURE analysis for 5133 individuals from 59 populations from the southern Alaska Peninsula supports K = 4 distinct groups with two single-lineage groups isolated (Sturgeon-Kitoi and Upper Cook Inlet) and the remaining 49 populations consisting of an admixture of the other two lineages (Fig. 4B; see details in the Supplementary Material). Most individuals from Kodiak Island show a similar level of mixing, but those from Sturgeon-Kitoi exhibit 98% single-source genetic ancestry. Chum salmon from eight populations from upper Cook Inlet totaling 568 individuals also have 98% single source composition, but from a different genetic ancestry than Sturgeon-Kitoi and the rest of the southern Alaska Peninsula.
Chum salmon population structure south of the Alaska Peninsula. (A) An unrooted phylogenetic tree (based on single nucleotide polymorphisms [SNPs]) of 310 chum salmon populations from Korea to the Columbia River in North America showing mean between-population divergence (F ST; Weir and Cockerham, Reference Weir and Cockerham1984) of Sturgeon-Kitoi (black) in Kodiak and Upper Cook Inlet chum salmon (bright green). (B) SNP STRUCTURE analysis (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000; Falush et al., Reference Falush, Stephens and Pritchard2003) for 5133 individuals from 59 chum salmon populations south of the Alaska Peninsula showing mixed genetic composition of 49 of those populations. There has been limited gene flow out of Sturgeon-Kitoi (black) and upper Cook Inlet (green) to other nearby populations around Kodiak Island and along and across the Alaska Peninsula and little gene flow into Sturgeon-Kitoi and upper Cook Inlet. (C) Distribution of the rare mAAT-2r* allozyme allele for many of the same populations. The 35% concentration at Sturgeon-Kitoi (red box) declines rapidly in nearby populations (Seeb and Crane, Reference Seeb and Crane1999). The location of upper Cook Inlet SNP samples are also shown (green). (D) Principal component analysis (PCA) for 5133 individuals of 59 chum salmon populations: Sturgeon-Kitoi (black triangles) and eight upper Cook Inlet chum salmon populations (green dots) are divergent from other populations along the southern Alaska Peninsula (yellow dots) and from other Kodiak Island populations (black dots).

The rare allozyme allele mAAT-2r* has a restricted distribution around the southern Alaska Peninsula and diminishes rapidly with distance from the Sturgeon River, where it occurs in 35% of the sampled fish. It occurs at 27% and 20% within 180 km of Sturgeon River and at less than half the Sturgeon rate at 21 other locations (Fig. 4C).
A PCA of SNP allele frequencies for 5133 individual chum salmon resolves three metapopulation clusters consistent with the STRUCTURE analysis: upper Cook Inlet, Sturgeon-Kitoi, and all other individuals from Kodiak Island and the southern Alaska Peninsula (Fig. 4D). The majority of Kodiak Island individuals group with the rest of chum salmon from the southern Alaska Peninsula and separate from the Sturgeon-Kitoi group on Kodiak Island.
These results for the southern Alaska Peninsula and upper Cook Inlet indicate that these two distinct areas have some chum salmon populations that have experienced very little gene flow from other populations in the region. Taken together, the STRUCTURE and PCA results support postglacial colonization by the two major lineages found in the earlier allozyme study, Beringian and Gulf of Alaska, and by two additional lineages, Sturgeon-Kitoi and upper Cook Inlet. The latter two lineages appear to be relatively isolated from the former two and from each other. The rare allozyme allele mAAT-2r* for the Sturgeon-Kitoi population does appear at lower rates in 23 other populations, however, demonstrating that there has been some gene flow from Sturgeon-Kitoi. Most chum salmon populations on Kodiak Island are not reproductively isolated from other southern Alaska Peninsula chum salmon.
Salish Sea
Phylogenetic tree construction for the Pacific Northwest chum salmon allozyme dataset shows that coastal fall-run populations are genetically unlike fall-run populations from the eastern Olympic area and least like Hood Canal and Discovery-Sequim Bay summer-run chum salmon (Fig. 5A). Despite close geographic proximity (about 50 km) between chum salmon of the coastal Chehalis River and southern Puget Sound across the drainage divide between them, the marine migratory distance around the Olympic Peninsula is about an order of magnitude greater. Chum salmon populations from the Chehalis River are genetically distinct from those of southern Puget Sound, and populations from the inland marine waterways of the Salish Sea are genetically intermediate between them (Fig. 5B).
Pacific Northwest chum salmon allozyme genetic and geographic population structure. (A) An unrooted phylogenetic tree (based on F ST) showing affinity of coastal and Columbia River populations to populations in the Strait of Juan de Fuca, Georgia Strait and the Fraser River and the difference between Eastern Olympic (southwestern and west-central Puget Sound, Hood Canal-Discovery-Sequim Bay) and both Coastal and Cascade metapopulations. (B) Geographically referenced version of the tree shown in A.

An ANCOVA of within-population allozyme gene diversity (H e) and allelic richness (Ar40) showed support for two metapopulations for chum salmon in the Salish Sea region: a 50 population Coastal group and a separate 40 population group from the eastern Olympic area (P < 0.0001; Fig. 6A and B), but not for a Cascades group. Details are in the Supplementary Material.
Within-population genetic diversity distribution at different scales for chum salmon. (A) Map of metapopulation within-population gene diversity variation above and below average, showing higher allozyme gene diversity concentrated east of the Olympic Mountains. (B) Within-population gene diversity versus allelic richness. Within-population gene diversity of the Eastern Olympic metapopulation exceeds that for Coastal populations from a much larger area, based on an analysis of variance (ANOVA) and pairwise Kramer-Tukey comparisons of the adjusted means of H e (see text). (C) Map showing the locations for mid-scale grouping of chum salmon populations in D. (D) Within-population gene diversity first decreases along the colonization route from the open coastal refugia, and then increases progressively toward the eastern Olympic area. The x-axis positions do not represent quantitative distances. See A for locations. If the tapered notches of boxes do not overlap, the medians of the respective groups differ significantly (P < 0.05). Hood Canal fall-run and SW & WC Puget Sound chum salmon are the only groups with within-population diversity greater than the South Coast and Columbia River group (Supplementary Table S4).

Analysis of variance of mid-scale within-population genetic diversity shows a geographic pattern of intermediate diversity on the open coast with diversity initially decreasing in intracoastal areas but then reaching a maximum for Hood Canal fall-run chum salmon in the eastern Olympic area (F 6,83 = 49.939, P < 0.0001; Fig. 6C and D, Table 1). Summer-run chum salmon in the eastern Olympic area have lower diversity than nearby fall-run chum salmon populations, based on pairwise post hoc Tukey’s Honestly Significant Difference (HSD) comparisons (P < 0.0001; Fig. 6D). Only two groups have significantly greater diversity than the South Coast and Columbia River group: Hood Canal fall-run chum salmon and the SW and WC PS group (P < 0.0001, Table 1).
Analysis of allozyme variance for 90 chum salmon populations in the Salish Sea and Olympic Peninsula. showing results of post hoc Kramer-Tukey pairwise comparisons with adjustments for multiple comparisons for chum salmon within-population allozyme genetic diversity.a

a Significance at P < 0.05 in bold. Groups in the location column compared to subsequent columns: higher diversity italicized and lower diversity not.
Allozyme genetic diversity for chum salmon in SW and WC PS is greater there than in the Cascade Mountains (P = 0.0006), an area closer in migratory distance to Coastal populations (Fig. 6D, Table 1). The source of this additional genetic variation in SW and WC PS was previously unexplained.
The Salish Sea analyses show a complex pattern of geographic distribution of genetic diversity for chum salmon. There is strong support (Fig. 6B; P < 0.0001) for two large metapopulations: a Coastal metapopulation that extends well into the Strait of Georgia and Puget Sound and an Eastern Olympic metapopulation that extends across several marine areas at and near Hood Canal that were connected early in deglaciation to the eastern margin of the Olympic Mountains (Bretz, Reference Bretz1913; Thorson, Reference Thorson1989). The highest genetic diversity is associated with the eastern Olympic area rather than the Coastal area. Higher gene diversity for Hood Canal fall-run chum salmon reflects gene flow from both coastal fall- and summer-run chum salmon populations. The eastern Olympic area coincides with the limited range of summer-run chum salmon in the Salish Sea.
These analyses also showed that SW and WC PS summer- and fall-run chum salmon are alike in terms of diversity, supporting the decision to combine them for the diversity analysis, but allelic heterogeneity tests do support separation between summer- and fall-run SW and WC PS chum salmon (Phelps et al., Reference Phelps, LeClair, Young and Blankenship1994, pg. 68, p. 73). Summer-run chum salmon there are also distinct in terms of geographic distribution: in SW and WC PS they are found exclusively in watersheds that very early in deglaciation produced outwash features around the southwest margin of the ice occupying Hood Canal. Our examination of these features using digital elevation models and LIDAR imaging confirmed the north-south drainage pattern early in deglaciation (Fig. 7) and the localized distribution of these outwash features. These landforms are remnants of drainage around and beneath the ice early in deglaciation (Fig. 8).
(A) Map of southern Puget Sound showing summer-run chum salmon range in the region. (B) Digital elevation model of southern Puget Sound highlighting the paleo-outlet to the Chehalis River valley. Through early stages of deglaciation, water flowed from north to south, including across local drainage divides via outwash channels such as the Kent Lake and Mason Lake spillways, and the entire Puget Lowland drained into the Chehalis River. The present distribution of summer-run chum salmon outside Hood Canal is restricted to these early deglaciation features. Lake Cushman is located immediately below the legend for the Mason Lake spillway, and the fossil sockeye salmon location is just south of Lake Cushman.

(A) LIDAR-derived image of the Kent Lake spillway in southern Puget Sound. Striations in purple are glacial landforms incised by drainage features at successively lower levels around the margin of decaying ice during deglaciation. Summer-run chum salmon outside Hood Canal are found only in remnants of these features. Notice the perched meander, a semi-circle on the upland immediately east of the start of the spillway and between two incised stream valleys. This elevated fluvial feature suggests that ice was still present in the Skokomish River valley and Hood Canal at the time the Kent Lake spillway was formed (Polenz et al., Reference Polenz, Contreras, Czajkowski, Legorreta Paulin, Miller, Martin and Walsh2010), that is, early in deglaciation. (B) LIDAR-derived image of the Mason outwash channel (Mason Lake spillway) and Mason Lake with drainage via Sherwood Creek to southern Puget Sound. Sherwood Creek summer- and fall-run chum salmon spawn in the headwaters of the Sherwood Creek–Mason Lake drainage in Schumacher Creek, which enters Mason Lake from the west (not shown). The incision of the Mason outwash channel across glacial flutes suggests that it was initiated under decaying ice, that is, early in deglaciation.

Within-population diversity values for Pacific Northwest Chinook and coho salmon are given in Supplementary Table S4 and shown in Figure 9. Diversity for Salish Sea Chinook and coho salmon is greater than that of Coastal populations and comparable to that of populations from the lower Columbia River.
Pacific Northwest Chinook and Coho salmon allozyme within-population gene diversity. Gene diversity within Pacific Northwest Chinook and coho salmon populations is greater within the Salish Sea and lower Columbia River than along the open coast, similar to results for Salish Sea East Olympic chum salmon, which also have greater diversity than on the open coast (Figure 6). One possibility is that these elevated diversities are the result of secondary contact between colonizing coastal and resident lake-isolate populations in the Salish Sea and Columbia River.

x
Discussion
Salmon population structure and glacial history
Most explanations for contemporary population structure in Pacific salmon are based on environmental and life-history factors that are observable today. Anadromy and the larger-than-trout body size conferred by growth and maturation in the marine environment are widely considered the defining characteristics for salmon and are directly related to their biological, economic, and cultural significance. The limited record of naturally occurring freshwater-resident life histories for Pacific salmon other than sockeye has been considered little more than anecdotal where it has been considered at all (but see Arostegui and Quinn, Reference Arostegui and Quinn2019). Sockeye salmon have a well-known freshwater morph, kokanee. Atlantic salmon have a similar freshwater form (Guiry et al., Reference Guiry, Royle, Orchard, Needs-Howarth, Yang and Szpak2020). Steelhead (Oncorhynchus mykiss) are the anadromous form of rainbow trout. Both sockeye and coho salmon have freshwater-resident residual forms (Ricker, Reference Ricker1938; Forrester and Ricker, Reference Forrester and Ricker1953); there are reports of freshwater-resident coho salmon (Kurenkov et al., Reference Kurenkov, Gorshkov and Tolstyak1982; Kirillova et al., Reference Kirillova, Kirillov, Malyutina, Kuzishchin, Gruzdeva and Pavlov2021), and a report of a likely freshwater-resident pink salmon (Hennick et al., Reference Hennick, Edfelt and Eaton1968). Aside from these naturally occurring freshwater-resident forms, Chinook, coho, and pink salmon and steelhead have been introduced to the Great Lakes, where they remain resident, demonstrating the capacity to adapt to freshwater-only environments. These Great Lakes freshwater pink salmon have also been found to display additional life-history polymorphism, developing a multiyear age structure never seen in truly anadromous populations (Wen-Hwa and Lawrie, Reference Wen-hwa and Lawrie1981).
Representations of ice age maximum glacial extent in the Salish Sea usually omit smaller proglacial lakes (McPhail and Lindsey, Reference McPhail, Lindsey, Hocutt and Wiley1986; Smith et al., Reference Smith, Montgomery, Peterson and Crowley2007; Waples et al., Reference Waples, Pess and Beechie2008). These omissions may have contributed to fish biologists frequently equating the presence of ice to the absence of fish (Waples et al., Reference Waples, Pess and Beechie2008)—not just marine and anadromous species, but freshwater species as well (McPhail and Lindsey, Reference McPhail, Lindsey, Hocutt and Wiley1986). The implications of the ubiquitous occurrence of proglacial lakes around ice sheets at maximum ice extent have not been considered for Pacific salmon life history, as most studies have simply focused on the consequences of glacial retreat on downstream salmon habitat (e.g., Pitman et al., Reference Pitman, Moore, Sloat, Beaudreau, Bidlack, Brenner and Hood2020). Thus, the possibility for survival of salmon living with a trout life history in proglacial lakes through maximum ice extent remains at the margins of our understanding of both salmon biology and ice age glacial environments. The process of colonization accompanying ice retreat has been presumed to be immigration from large refugia, and in the case of salmon, from unglaciated areas open to the sea (Quinn, Reference Quinn2005; Waples et al., Reference Waples, Pess and Beechie2008).
We argue that the influence of impoundment of salmon in proglacial lakes on population persistence during ice ages is underappreciated but necessary to understand present-day population structure. Chum salmon are a model system to address this question, because we know of no wild chum salmon that are entirely freshwater resident, which means their sequestration in proglacial lakes for several thousand years would suggest that all salmon species could share a similar capacity. Because chum salmon spend less time in fresh water than most other species of salmon, they have reduced opportunities for local adaptation among populations; this has resulted in shallower differentiation in population structure than for other salmon species. However, ice age lake isolation of chum salmon is expected to lead to a higher degree of population divergence. We argue that long-term isolation in proglacial lakes could have directly promoted the patterns of genetic differentiation we observe in these populations today.
Southern Alaska Peninsula/upper Cook Inlet
In Alaska, at Kodiak Island and in upper Cook Inlet, chum salmon populations occur with little evident genetic connection to surrounding populations. One possibility is that these are outlier colonizers from distant areas; another is that they are descendants from populations that survived locally living like trout in proglacial lakes. These populations are genetically as distinct as are those from Japan in term of diversity and divergence (Fig. 3B) but not close to Japanese chum salmon in terms of pairwise distance (Fig. 3A, 4A); thus, it is difficult to identify appropriate ancestral donor populations. The rare allozyme allele (mAAT-2r*) for Sturgeon-Kitoi chum salmon at Kodiak Island further complicates the challenge of finding an ancestral donor population for Sturgeon River. If such genetic anomalies are to be expected from recolonization of newly emerging habitat by anadromous straying, then such rare alleles should be more common, suggesting parallel colonization from a distant source, rather than steeply declining with distance, suggesting a single nearby source at Sturgeon River (Fig. 4C). The fact that the Sturgeon River watershed coincides with the area of a proglacial lake of about 1000 km2 and as much as 8000 years duration provides a potential refugial source (Fig. 1).
Could chum salmon have survived there cut off from the sea? Although there are no known sustaining freshwater-resident populations of chum salmon, either natural or artificial, there are cases where chum salmon have been raised in freshwater confinement for more than a year (Chin and Kuroda, Reference Chin and Kuroda1935; Andrews, Reference Andrews1963), and a single small 4-year-old landlocked subadult has been reported (Peden and Edwards, Reference Peden and Edwards1976). Eight thousand years of isolation in an ice-dammed proglacial lake could have created the observed midsummer-run timing, minimal diversity, high mean pairwise divergence, and rare allozyme allele found for Sturgeon River chum salmon.
Only three populations of lake-spawning chum salmon have been documented in North America: Kelly Lake in the Kotzebue area (DeCovich et al., Reference DeCovich, Dann, Rogers Olive, Liller, Fox, Jasper, Chenowith, Habicht and Templin2012), Kluane Lake in the Canadian upper Yukon River (Wilson, Reference Wilson2011), and Carmen Lake in upper Cook Inlet (DeCovich et al., Reference DeCovich, Dann, Rogers Olive, Liller, Fox, Jasper, Chenowith, Habicht and Templin2012). Each of these locales is the site of an ice age lake (Karlstrom, Reference Karlstrom1964; Hamilton, Reference Hamilton2001; Duk-Rodkin et al., Reference Duk-Rodkin, Weber and Barendregt2002) with populations near the end of a limb on the chum salmon SNP genetic tree (Fig. 3A).
The traditional model of Pacific salmon range expansion as the Cordilleran ice sheet receded posits that anadromous populations colonized newly available ice-free habitats from large ice age refugia that had retained access to the ocean. But the persistence of some salmon populations living as trout in proglacial lakes is more consistent with the evidence of population structure for some present-day anadromous chum salmon populations in Alaska. These formerly isolated populations contribute significantly to the overall genetic diversity currently observed within the species.
Salish Sea
Summer-run chum salmon from the Puget Sound region of the Salish Sea are outliers in terms of the geography of run timing, an exception to the cline of earlier run timing with increasing latitude. There are no other North American summer-run chum salmon south of northern British Columbia. Based on allozyme genetic data, they have been identified as an ancient lineage but with no suggestion for the origin of this divergence (Phelps et al., Reference Phelps, LeClair, Young and Blankenship1994; Johnson et al., Reference Johnson, Grant, Kope, Neely, Waknitz and Waples1997). Direct genetic evidence for ancestral freshwater residence of Salish Sea chum salmon has been found in the contrast of pairwise genetic difference between summer- and fall-run chum salmon populations at chromosome loci associated with residence for steelhead (Waples et al., Reference Waples, Seeb and Seeb2017), again without explanation of the origin of this finding.
In contrast to the situation with unique chum salmon populations in Alaska, there appears to have been gene flow within Salish Sea chum salmon populations. The genetic SNP tree shows progressive population-pair transitions encompassing a wide range of mean pairwise divergence (F ST) (Fig. 3A). This is accompanied by a similar wide range in within-population diversity (Fig. 3B). The greater detail of the allozyme tree (Fig. 5A) indicates that coastal chum salmon populations did not colonize Puget Sound directly via the Chehalis River outlet during deglaciation. This conclusion agrees with that for Chinook salmon (Waples et al., Reference Waples, Pess and Beechie2008). Both the metapopulation analysis (Fig. 6A and B) and mid-scale geographic distribution of within-population gene diversity (Fig. 6C and D) highlight an area of high diversity east of the Olympic Mountains. Genetic diversity of chum salmon populations from the open coast is lower than in Puget Sound, which is inconsistent with the South Coast being the single source of colonization. Diversity is lower still along the Strait of Juan de Fuca, as would be expected for stepwise colonization of the first areas to emerge from beneath the Cordilleran ice cover. Rather than continuing to decrease with distance from the open coast, diversity increases progressively toward the eastern Olympic area. The additional genetic variation represented by the observed increase in within-population diversity might have several possible sources: colonizing chum salmon populations from distant areas, rapid postglacial evolution, or another local chum salmon refuge. Similar to the situation for the Alaskan examples, the available evidence does not support distant sources for the observed genetic signatures, nor is there any evidence for postglacial generation of new genetic variation within Eastern Olympic chum salmon.
High within-population gene diversity coincides with the localized range of summer-run but not fall-run chum salmon, evidence that summer-run chum salmon are the source of the additional genetic variation reflected in higher within-population diversity and that an ice age refugium for summer-run chum salmon is nearby (Fig. 7A). See the Supplementary Material for additional discussion of Salish Sea chum salmon biogeography. We suggest that this refugium was at proglacial Lake Cushman in the Olympic foothills just northwest of the Great Bend of Hood Canal (Bretz, Reference Bretz1913; Fig. 7B). Traditional environmental knowledge of the First Peoples Twana culture includes the specific diminutive term “little dog salmon” for the historical and distinct early run of chum salmon to the Skokomish River, the outlet for Lake Cushman (Elmendorf and Kroeber, Reference Elmendorf and Kroeber1992). They are not known to be present in modern times and are not included in any enumeration or genetic sampling. Runoff from proglacial Lake Cushman would have contributed to the outwash flow that created the Kent Lake spillway and the Mason outwash channel, potentially seeding freshwater-resident chum salmon into those watersheds. The fact that summer-run chum salmon in SW and WC PS are found today only in the streams that occupy the terrain defined by former spillway and outwash channels supports this interpretation.
The advance of Cordilleran ice from the north blocked Puget Sound and confined all species of Oncorhynchus in a cold freshwater environment. Chinook salmon and steelhead in the Salish Sea both exhibit genetic anomalies. Chinook salmon have unusual sex-linked allele frequencies at allozyme allele PEPB-1 (Marshall et al., Reference Marshall, Knudsen and Allendorf2004), and Salish Sea steelhead have 60 chromosomes, while coastal Washington populations have 59 or 58 (Ostberg and Thorgaard, Reference Ostberg and Thorgaard1999). There are no known outside potential source populations with either the Chinook salmon sex linkage or for that 60-chromosome steelhead karyotype.
Temperature shock during embryonic development has been shown to perturb sex (Craig et al., Reference Craig, Foote and Wood1996) and to induce change in chromosome number in salmonids (Svärdson, Reference Svärdson1945). Upwelling springs in proglacial lakes and glacier-fed streams would provide an environment conducive to thermal shock of embryos. The geographic extent of these two independent chromosome anomalies is remarkably similar, reaching between the Salish Sea and the open coast, and consistent with an early advantage in recolonization for lake-isolate founding populations of Chinook salmon and steelhead (Waters et al., Reference Waters, Fraser and Hewitt2013). The examples of genetic anomalies for Chinook salmon (Marshall et al., Reference Marshall, Knudsen and Allendorf2004) and steelhead (Ostberg and Thorgaard, Reference Ostberg and Thorgaard1999) in the Salish Sea together with a similar case for pink salmon in upper Cook Inlet (Churikov and Gharrett, Reference Churikov and Gharrett2002) are sufficiently abnormal that the respective authors did not attribute them to conditions that we observe today. Instead, Churikov and Gharrett (Reference Churikov and Gharrett2002) suggested isolation in ice age lakes as the mechanism for genetic divergence. Chum salmon are closely related to pink salmon and share minimal freshwater residence life history and the capacity for brackish-water spawning and rearing. We suggest the same lake isolation mechanism for chum salmon, as well as the other members of Oncorhynchus. The possibility that loci related to residence and run timing, mtDNA haplotype, and chromosome and sex-linkage anomalies, might all be indicators of ancestral freshwater isolation is an unexplored avenue of research that warrants further examination.
The regional pattern of within-population diversity for Chinook and coho salmon mirrors that for chum salmon (Fig. 9). Salish Sea populations have greater diversity than populations from the open coast and similar to that of populations from the Columbia River. The Columbia River is known to have been a refuge for fish throughout the ice ages (McPhail and Lindsey, Reference McPhail and Lindsey1970; Waples et al., Reference Waples, Pess and Beechie2008). The common level of diversity across species suggests that the Salish Sea area was also a refuge for fish through the ice ages. We suggest that elevated within-population diversity for chum, Chinook, and coho salmon in the Salish Sea and Columbia River could result from secondary contact between lake-isolate populations and colonizing populations from the open coast.
Finally, fossil evidence shows that sockeye salmon spawned in glacially turbid water of a proglacial lake in the lower Skokomish River during an ice advance about 1 million years ago (Smith et al., Reference Smith, Montgomery, Peterson and Crowley2007). These fossiliferous lake sediments are about 11 km from the location of proglacial Lake Cushman, along the course of the North Fork Skokomish River (Fig. 7). The geological and biological processes presented here have been active for long enough to substantially shape the life history and genetic population structure of salmon in the region.
Conclusion
Our results suggest that the well-documented diversity encompassing both lake type and anadromous metapopulations of Baltic Sea Atlantic salmon and Kamchatka sockeye salmon may be expressed in all Pacific salmon and that this duality is likely responsible for some of the bimodality observed in salmon population structure (Waples et al., Reference Waples, Pess and Beechie2008). In 1958, Rounsefell observed that “freshwater forms of anadromous salmonids normally use a lake as a miniature sea, ascending its tributaries to spawn” (Rounsefell, Reference Rounsefell1958, p. 173). Another way of saying this is that anadromous forms of freshwater salmonids use the ocean as a big lake, descending to the ocean to maximize growth and colonize new breeding habitat. In this way, we might think of salmon as trout with a sense of adventure. There is now sufficient correlative information to propose that the population structure of Pacific salmon is the product of long-standing competition between and contributions from ancestral ocean-going and lake-isolate lineages. The capacity to survive freshwater isolation in unique environments at small, but stable or growing, population sizes can favor establishment of beneficial mutations (Kimura, Reference Kimura1962; Otto and Whitlock, Reference Otto and Whitlock1997) and subsequently their expansion as first colonizers (Seeb and Crane, Reference Seeb and Crane1999; Marshall et al., Reference Marshall, Knudsen and Allendorf2004; Ostberg and Thorgaard, Reference Ostberg and Thorgaard1999; Waters et al., Reference Waters, Fraser and Hewitt2013), thereby enhancing the adaptability of Oncorhynchus and potentially contributing to the plasticity for which they are known. The Pacific salmon genome has evolved over 107 years, yet these fish exist in a landscape that is replumbed—by glaciation, landslides, and volcanism—over 104 to 105 year timescales. Populations with the capacity to switch between anadromy and whole-life freshwater residence would have an evolutionary advantage in the dynamic environment found along the coast of western North America (Montgomery, Reference Montgomery2000; Waples et al., Reference Waples, Pess and Beechie2008).
Salmon have adapted to survive in extreme ice age lake conditions and to colonize deglaciated habitat from ice-bound freshwater refugia as well as from open coastal refugia. Newly available habitat would have been colonized from two sources: one via freshwater-resident salmon populations from the headwaters, the other via anadromous populations that retained access to or matured in the ocean. As a result, pre-contact salmon population structure carried the imprint of Pleistocene habitats. Salmon that evolved to cope with repeated glacial advances throughout the Quaternary period may be imperfectly adapted to interglacial periods such as the present. If the fraction of salmon diversity that remains were lost, it likely would only be restored over geologic timescales. In the Salish Sea region, many salmon populations are threatened, endangered, or already lost, especially those with distinctive life-history phenotypes involving unusual migration timing or habitat use. The secret of their ice age past may have kept us from fully recognizing their uniqueness and vulnerability to modern stresses of industrialization, over-exploitation, and climatic warming.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/qua.2025.10070.
Acknowledgments
We acknowledge the efforts of teams who collected samples from about 78,000 chum salmon from 1985 to 2011 on two continents across six countries, including two provinces and four states in North America, without which this study would not be possible. Likewise, sample curation and laboratory analyses conducted primarily for fishery management purposes at the Alaska Department of Fish and Game and the Washington Department of Fish and Wildlife were essential to this study. We are grateful for all their efforts. We thank Penny Crane, Tony Gharrett, Chris Habicht, Jill Coyle, Andy Barclay, Eleni Petrou, Dan Prince, George Pess, and David Teel for their comments and assistance. We thank Tyler Dann for analysis of the SNP dataset, making the baseline available, and comments. We thank the authors’ affiliate organizations for their administrative support. PCM thanks Concerned Area M Fishermen for its support. Finally, we thank the associate editor Mary Edwards and anonymous reviewers for their helpful comments in improving this paper.
Data availability statement
SNP data as used in Fig. 1 are available from Dann (Reference Dann2023) under Dryad entry https://doi.org/10.5061/dryad.q2bvq83r1. Genotypic SNP data as used in the STRUCTURE and PCA analyses are available from Petrou et al. (Reference Petrou, Seeb, Hauser, Witteveen, Templin and Seeb2014) under DRYAD entry https://doi.org/10.5061/dryad.ds304.
Competing interests
The authors declare no conflicts of interest.












 Open access
Open access