


Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare and highly disabling neuroinflammatory condition with a prevalence cited as between 0.7/100,000 and 10/100,000 persons, depending on the region. Reference Papp, Magyari and Aktas1–5 In Canada, the prevalence of NMOSD remains uncertain. While there has been extensive research aimed at understanding regional differences in multiple sclerosis demographics and risk factors, Reference Huang, Kockum and Stridh6 a relative paucity of similar research exists for NMOSD.
Highlights
-
• This is the largest Canadian multicentre study describing demographic, clinical and treatment characteristics of AQP4+ NMOSD (N = 133).
-
• Participants were predominantly female (84.9%) with ethnic and racial variability; 63.3% were born outside Canada.
-
• Use of lower efficacy therapies, including azathioprine, remains common.
Most people with NMOSD test positive for serum aquaporin-4 IgG (AQP4) antibodies (70%–80%), Reference Wingerchuk, Banwell and Bennett7 a pathogenic antibody targeting AQP4 water channels located on astrocytes, which are enriched within ependymal tissues of the central nervous system. This results in an astrocytopathy with characteristic demyelinating lesions involving the optic nerve/chiasm, spinal cord and brainstem. Individuals presenting with NMOSD who are AQP4 antibody-positive (AQP4+) have a pathologically distinct disease compared to those with other demyelinating conditions, including those with AQP4 antibody-negative NMOSD. Furthermore, among the broader NMOSD population, those with AQP4+ disease tend to have a more predictable response to immunosuppressive/immunomodulatory therapies than their seronegative counterparts.
Given the distinct features of AQP4+ NMOSD, the current study was performed to examine the unique sociodemographic profile, clinical and imaging features and treatment approach for individuals with AQP4+ NMOSD in a multicenter Canadian study with representation from western, central and eastern provinces. A second objective was to describe patterns of disability in a contemporary AQP4+ NMOSD cohort, considering treatment developments over the past decade. Data for this study were collected as part of the CAnadian Neuromyelitis OPTIca Spectrum Disorder and other atypical demyelinating diseases Cohort Study (CANOPTICS), a prospective longitudinal study involving seven centers across Canada.
Methods
This study was a retrospective analysis of data collected at the baseline visit in CANOPTICS for individuals with aquaporin-4 IgG-positive (AQP4+) NMOSD. CANOPTICS is a multicenter, prospective longitudinal study of adults living with NMOSD and other atypical demyelinating diseases in Canada. Reference Rotstein, Freedman and Lee8 Healthcare in Canada is universal and predominantly publicly funded, with service delivery managed at the provincial and territorial level. There are interprovincial differences in healthcare infrastructure, including the approval and accessibility of certain medications. NMOSD care can vary accordingly – patients may be managed in general neurology settings in some provinces, whereas in others, care is centralized in tertiary care centers with specialized neuroinflammatory clinics. Referral patterns and access to specialized neuroinflammatory clinics also differ by region, influencing diagnosis and management pathways. Prior to initiation, study approval was obtained from each local Research Ethics Board. Consent for participation and retrospective data abstraction commenced in January 2021.
Participants
We identified participants aged 18 years and older who were enrolled in CANOPTICS and who met the 2015 diagnostic criteria for NMOSD Reference Wingerchuk, Banwell and Bennett7 with at least one serum antibody test (cell-based assay or ELISA) positive for AQP4 IgG. All cases with positive AQP4 IgG status were reviewed by the treating neurologist and CANOPTICS research team to ensure they were clinically consistent with NMOSD. There were eight participating centers in CANOPTICS at the time of this study, of whom seven (University of Alberta, University of Manitoba, University of Western Ontario [Western], University of Toronto – St. Michael’s Hospital, University of Toronto – Sunnybrook Health Sciences Centre, University of Ottawa, Dalhousie University) had enrolled AQP4+ NMOSD participants contributing data to this study. The distribution of centers involved in this study allowed for broad representation, including western, central and eastern provinces across the country.
Data collection
Data for this study were collected from the baseline visit of CANOPTICS and comprised adults with AQP4 antibody-positive NMOSD with onset between 1990 and 2022. Data were derived from a review of individual medical records and were collected from January 5, 2021, to January 1, 2024. Data abstraction was carried out by trained research personnel using standardized forms. In cases where data were missing, participants were contacted directly when possible.
Study variables
Demographic variables examined included age at enrollment, date of birth, date of enrollment and at index attack, date of first positive serum AQP4 antibody test, sex at birth, ethnicity and race, country of birth, immigration status, age of immigration to Canada, smoking history, employment status at study entry, highest level of education attained at study entry, marital status at study entry, first attack localization and symptoms, hospitalization and recovery (none, partial, complete) with index attack. Dates were collected as month/year. Information regarding hospitalization at the time of the first attack was obtained from medical records.
Ethnicity and race data were extracted from the medical record. Participant ethnicity and race data were categorized according to North American or European White, African/Caribbean Black, East or Southeast Asian, First Nations/Inuit/Metis, Other and Multiracial and Multiethnic, the latter of which referred to participants belonging to multiple ethnic and racial groups. Employment data were categorized as employed full-time, employed part-time, unemployed due to disability and unemployed not related to disability, the latter of which captured students, homemakers and retired participants. The highest level of education was categorized as high school degree/GED or less, undergraduate or technical degree or postgraduate degree.
History of comorbid autoimmune disease was collected, including thyroid autoimmune disease, lupus, myasthenia gravis, celiac disease, rheumatic arthritis, Sjogren syndrome, inflammatory bowel disease, immune thrombocytopenia purpura and Other (antiphospholipid antibody-related disease, mixed connective tissue disease NYD, Raynaud’s syndrome, inflammatory polyarthritis NYD) within the study population. Personal history of other common non-autoimmune comorbidities, including type 2 diabetes mellitus, hypertension, malignancy and epilepsy, was also collected, as was family history of autoimmune disease in general and demyelinating disease in particular.
Index attack data included symptoms experienced by participants with their initial clinical attack, index attack type/syndrome and imaging features. Index attack type was categorized as transverse myelitis (TM) due to short segment lesions, long segment lesions and unknown lesion length; unilateral optic neuritis (ON) and bilateral ON; and area postrema syndrome, other brainstem/cerebellar syndrome and cerebral syndromes with and without encephalopathy. A given participant may have presented with multiple initial symptoms or syndromes, which were represented as the frequency of each presenting symptom within the total study population. Index attack severity was categorized as resulting in minimal functional impact with retained ability to perform activities of daily living (ADLs), significant functional disability not requiring hospitalization and significant disability requiring hospitalization. Data were collected for recovery from index attack and defined as either complete, partial or no recovery based on medical record descriptors of post-index attack recovery. Disease onset with a cluster of attacks was defined as having greater than one attack within 12 months of the index attack. Index attack imaging variables included regions imaged, as well as T2 lesion localization and presence of enhancing lesions.
Disability data were collected, including mean Expanded Disability Status Scale (EDSS), visual acuity and legally blind status defined as visual acuity of less than 20/200 in both eyes. EDSS was categorized in disability ranges: 0–1.5, 2–3.5, 4–4.5 and >6.
Treatment data
Treatment modalities, including first-line treatment (i.e., initial maintenance therapy) and second-line and third-line treatments, were categorized into the following treatment groups: azathioprine, MMF (mycophenolic acid or mycophenolate mofetil), rituximab, novel monoclonal antibodies (MAbs) (satralizumab and eculizumab) and Other (long-term/constant steroid, plasma exchange, tocilizumab, methotrexate, cyclophosphamide, intravenous immunoglobulin (IVIG), mitoxantrone, beta-interferons, glatiramer acetate). Incidence of adverse events/side effects to first-line treatments and indications for switching from a first-line to a second-line treatment were collected. Indications for changing from a first-line to a second-line treatment categorized as “Other” included family planning/pregnancy, lack of adherence, concern regarding long-term risk, patient choice and completed course of therapy.
Analysis
Data were described using simple summary statistics and analyzed at two different time points: (a) date of the index attack and (b) date of study enrollment in order to characterize the Canadian AQP4+ NMOSD population at their initial disease presentation and at present. For categorical variables, we used frequency counts and percentages, and for continuous variables, median and range. Missing data were not imputed except for where the month of a participant’s index attack was unknown; it was imputed as January 1 of the year listed.
Results
Baseline demographic data and disability measures
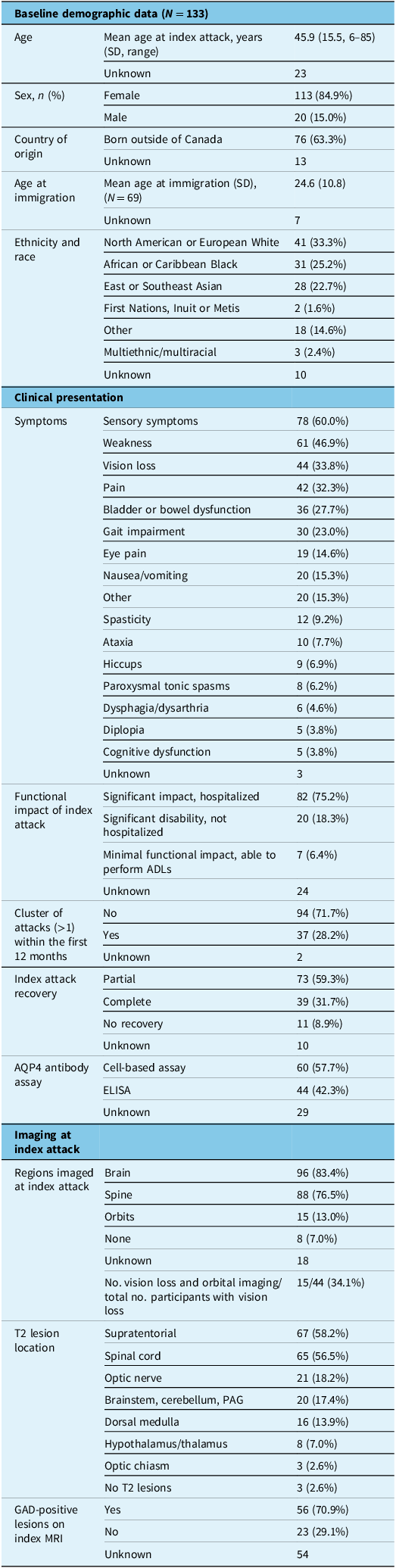
Overall, 133 participants with AQP4+ NMOSD were included in this study, with a mean age (SD, range) of 52.0 (15.0, 18–87) years at enrollment. Disease onset date ranged from 1990 to 2022. Participants were mostly female (84.9%), and the largest affected ethnic and racial groups were North American or European White (33.3%), African or Caribbean Black (25.2%) and East or Southeast Asian (22.7%). Most participants were born outside of Canada (63.3%), with a mean age (SD) at immigration of 24.6 (10.8) years. First attack was after arrival in Canada in 63/69 (91.3%) immigrants for whom the date of arrival in Canada was available, and all but one had AQP4+ status confirmed in Canada. Comorbid autoimmune disease was observed in 24.8% of participants; the most common comorbid autoimmune diseases were systemic lupus erythematosus and thyroid autoimmune disease. Eight percent of participants had two or more comorbid autoimmune diseases/conditions. Of note, only 58% of participants had AQP4 antibody positivity identified through cell-based assay testing, while 42% had been identified using only the less reliable ELISA assay.
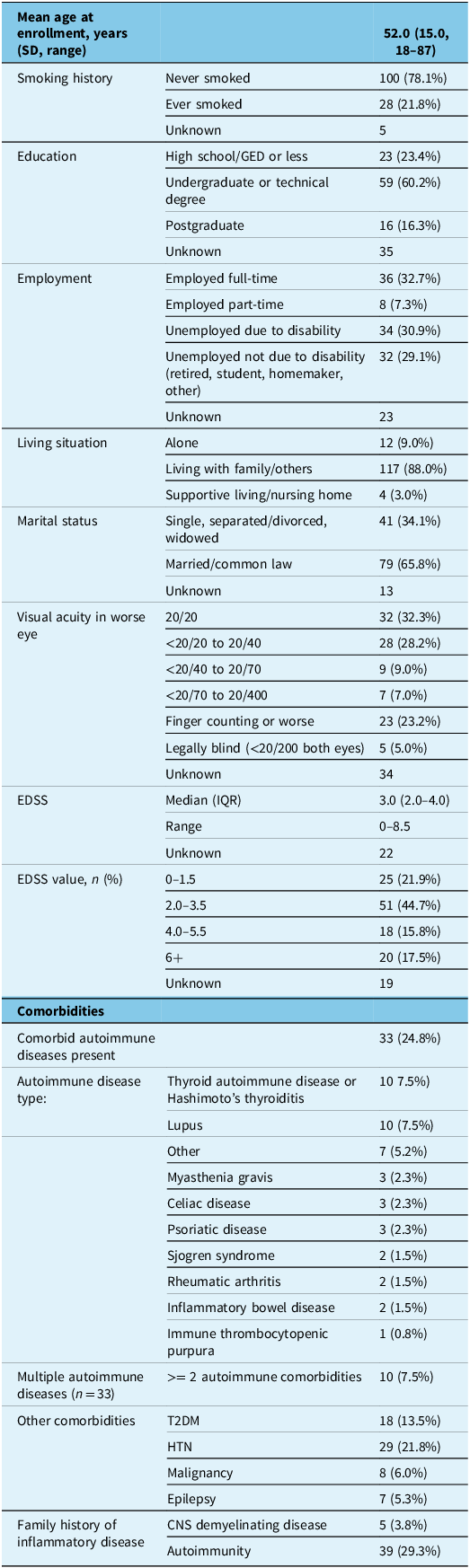
The median EDSS (IQR) at study enrollment was 3.0 (2.0–4.0). Over two-thirds of participants had an EDSS of 3.5 or less (i.e., moderate disability in a single functional domain or minimal disability in several domains) with retained ability to ambulate independently. With respect to disability due to vision loss, 32.3% of participants had normal visual acuity at enrollment, while 23.2% had profound vision loss of finger counting or less in the worse eye; 5.0% were legally blind, that is, vision of less than 20/200 in both eyes (Table 1). Forty percent of participants were employed full-time (32.7%) or part-time (7.3%), while 29.1% of participants were unemployed due to disability (Table 2).
Baseline demographic, clinical and imaging characteristics at index attack in Canadian adults with AQP4+ NMOSD

Note: “Other” ethnicity and race includes Latin/Central/South American, Middle Eastern/West Central Asian and Oceanian. “Other” index attack symptoms includes Other = seizure, decreased LOC= level of consciousness, hyponatremia, trigeminal neuralgia, fever, fatigue, vertigo, autonomic dysfunction and other/unknown symptoms. Note that index attack symptoms and T2 lesion localization are represented as a percentage of the total number of participants with data for each (i.e., frequency of each symptom presentation) as participants may have presented with multiple symptoms and lesion localizations at onset. Disease onset date ranged from 1990 to 2022. ADLs = activities of daily living; AQP4+ NMOS = aquaporin-4 IgG-positive neuromyelitis optica spectrum disorder; GAD = gadolinium, PAG = periaqueductal gray.
Baseline demographic and clinical characteristics at enrollment for Canadian adults with AQP4+ NMOSD, including sociodemographic, disability and comorbid disease measures

Note: AQP4+ NMOSD = aquaporin-4 IgG-positive neuromyelitis optica spectrum disorder; EDSS = Expanded Disability Status Scale; T2DM = type 2 diabetes mellitus; HTN = hypertension; CNS = central nervous system.
No participants had a personal history of type 1 diabetes, sarcoidosis or vitiligo, and as a result, these conditions are not represented in the tables.
Index attack characteristics and imaging
The mean age (SD, range) at index attack was 45.9 (15.5, 6–85) years. TM was the most frequent type of index attack and was observed in 53.8% of participants, with 43.8% of all participants presenting with longitudinally extensive TM, 4.6% with short segment TM and another 4.6% with TM of unspecified length. ON was the second most frequent index attack and was observed in 34.6% of all participants (27.7% unilateral ON and 6.9% bilateral ON). Brainstem syndromes were observed in 14.6% of participants at index attack and included area postrema syndrome (10.8%) and other brainstem/cerebellar (3.8%) syndromes (Figure 1).
Index attack type for Canadians with aquaporin-4 IgG-positive neuromyelitis optica spectrum disorder represented as a percentage of all participants (n = 133). Participants may have had multiple attack types at index attack.

Index attacks were highly disabling, with 75.2% of participants requiring hospital admission due to severe disability and another 18.3% not requiring hospitalization but documented to have significant disability impairing their ability to participate in activities of daily living (ADLs). Only 6.4% of participants experienced minimal functional impact with preserved ability to perform ADLs at index attack. Some degree of functional recovery from index attack was observed in nearly all (91.1%) participants, with 31.7% experiencing full recovery and 59.3% partial recovery. Less than 10% had no recovery following the initial attack (Table 1).
Imaging at index attack
Imaging with MRI was performed in 93.9% of participants at index attack. Most participants underwent MRI of the brain (83.4%) and/or spinal cord (76.5%). Orbital imaging was less frequently performed, with 13.0% of all participants and 34.1% of subjects with documented vision loss undergoing orbital MRI at index. T2 hyperintense lesions were observed most often in the supratentorial white matter (58.2%) and in the spinal cord (56.5%) on the index scan, with 70.9% demonstrating at least one gadolinium-enhancing (GAD+) lesion on index MRI. A minority of participants (2.6%) did not have any T2 lesions identified on MRI performed at index attack (Table 1).
Disease activity
A minority (28.2%) of participants experienced two or more attacks within the first 12 months of their disease. At study enrollment, the median (IQR) number of attacks experienced by participants was 2 (1–4) with a range of 1–7 prior attacks; the annualized relapse rate was 0.27 (95% CI: 0.25–0.29). The median (IQR) disease duration was 8 (3–14) years at enrollment. The proportion of participants with any previous history of TM or ON consistently increased with disease duration, but most brain attacks occurred early in the disease in this cohort.
Treatment utilization
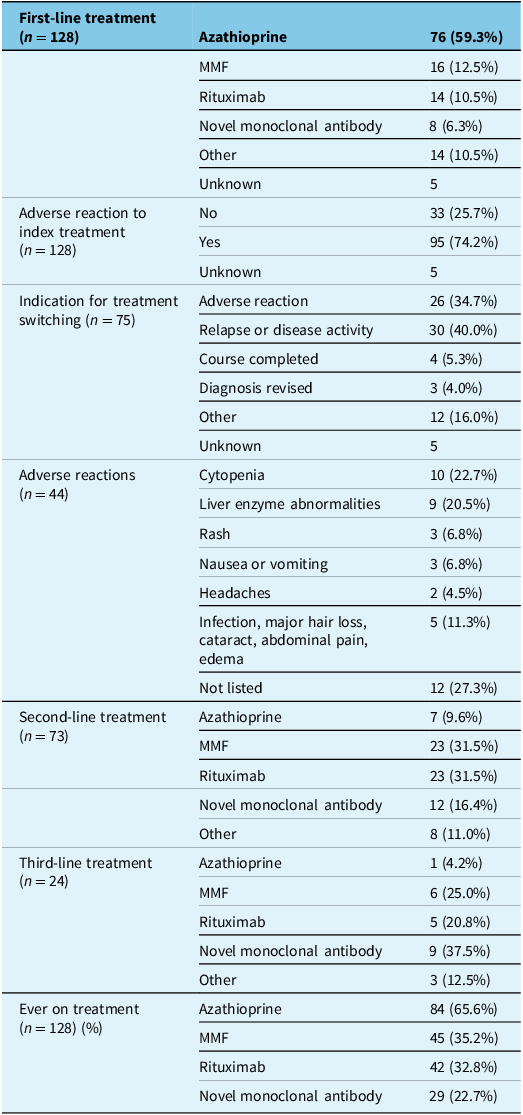
The majority of participants were treated with azathioprine as their initial NMOSD maintenance therapy (59.3%), followed by MMF (12.5%), rituximab (10.5%) and other treatment modalities. Novel MAbs were utilized least frequently as first-line treatment, in only 6.3% of participants. When treatments were analyzed by the year/epoch of treatment initiation, azathioprine remained the most commonly utilized first-line therapy across epochs; however, in more recent years, the use of MMF, rituximab and MAbs as first-line therapy has increased (Table 3 and Figure 2A and B). None of the participants with index attack before 2008 were initiated on rituximab as a first-line therapy. Treatments categorized under the “Other” category included long-term steroids, methotrexate or agents started for an initial diagnosis of MS that was later confirmed to be NMOSD (i.e., interferon beta, glatiramer acetate, mitoxantrone). MMF and rituximab were the most commonly used treatments as a second- or third-line treatment; rituximab was the most utilized second- or third-line treatment in participants with index attack within the last 5 years (2018–2023), followed by novel MAbs (Figure 2B). For the participants who were immigrants and for whom data were available, 63/65 (96.5%) started their first NMOSD maintenance therapy after arrival in Canada.
Treatment modality use in Canadians with aquaporin-4 IgG-positive neuromyelitis optica spectrum disorder represented based on (A) overall treatment modality use and first-line treatment modality as a percentage of all participants and (B) first- and combined second/third-line treatment modality use over time relative to index attack date range (prior to 2008, 2008–2017 and 2018–2023). Treatment utilization is represented as a percentage of the number of participants in each time period cohort. MMF = mycophenolate mofetil.

Treatment history in Canadian adults with AQP4+ NMOSD (n = 133)

Note: “Other” treatment category included plasma exchange, mitoxantrone, cyclophosphamide, interferon beta, glatiramer acetate, IVIG and prednisone. AQP4+ NMOSD = aquaporin-4 IgG-positive neuromyelitis optica spectrum disorder; MMF = mycophenolate mofetil.
Most participants (74.2%) experienced an adverse reaction to their first-line treatment, the majority of which were mild. The most common adverse reactions were laboratory abnormalities (cytopenia and abnormal liver enzymes: 22.7% and 20.5%, respectively). At enrollment, 43.0% of participants remained on their first-line treatment, while 57.0% had required a change in treatment. The most common indications for a treatment switch from first-line therapy were relapse or disease activity (40.0%) and adverse reactions (34.7%).
Discussion
This study serves as the first descriptive analysis of AQP4+ NMOSD in Canadian adults, outlining demographic characteristics, disability and treatment utilization in a contemporary cohort. Our patient population, similar to other North American cohorts, was predominantly female with disease onset in middle age and included a higher proportion of Black and Asian participants compared to the general population 9 , consistent with previous studies showing greater representation of racial and ethnic minority groups. Reference Lee, Smith, Li, Iordanova and Langer-Gould10–Reference Rato, Chen, Francis, Leite, Palace and Geraldes11 Interestingly, there was a high number of immigrants in our study population, in keeping with previous findings that the risk of NMOSD is increased in immigrants compared to those born in Canada and in Asian and Black individuals in Canada. Reference Dababneh, Lee and Stratos12–Reference Rotstein, Wolfson and Carruthers13 The age of disease onset for our study was similar to, but slightly older than, other reported cohorts, including a recent meta-analysis of sex ratio and age distribution in AQP4+ NMOSD Reference Arnett, Chew and Leitner14 (45.9 years in our study vs. 41.7 years). This may be partly accounted for by ethnic and racial differences in disease onset, given that prior studies have found that individuals identified as “White” tend to have an older age of disease onset than other populations. Bukhari et al. Reference Bukhari, Lechner and Saqqur15 found that White populations had an age of disease onset from 40 to 45 years versus 20–30 in East Asian participants. Similar findings were noted in a national case-control study of NMO demographics in a Canadian population, with Black and Asian participants having an age of disease onset in their 30s versus White participants tending to experience disease onset in their mid 40s. Reference Rotstein, Wolfson and Carruthers13 Another consideration for our reported older age of onset is that many of our participants were born abroad (63.3%), and the observation period in immigrants is limited by their age of arrival. Most had disease onset after arrival in Canada, which may reflect barriers to entry in those with preexisting disability.
Historically, NMOSD has been demonstrated to be a highly disabling condition with worse functional outcomes compared to other demyelinating conditions, including MS. Our study demonstrated a lower median EDSS and a smaller proportion of participants with severe vision loss than previous studies, Reference Pandit, Asgari and Apiwattanakul16–Reference Paolilo, Hacohen and Yazbeck17 which could partly reflect improved diagnostic and treatment approaches in recent years. The relatively favorable disability outcomes observed in our study may also reflect shorter overall disease duration in this cohort, with two-thirds having had disease onset within the last 10 years. Nevertheless, the impact of disability within the cohort was still substantial, with nearly one-third of participants reporting unemployment due to disease-related disability, thus highlighting the need for improved management strategies supporting improved clinical outcomes.
Despite evolving evidence that rituximab and novel MABs have higher efficacy and lower adverse events compared to other evidence-based treatments, Reference Magdalena, Clarissa and Sutandi18–Reference Yin, Qiu and Duan20 we observed that Canadians with AQP4+ NMOSD are more likely to be treated with less effective immunomodulatory therapies, such as azathioprine and MMF, both as first-line treatment options and with respect to overall lifetime treatment utilization. However, we observed a shift in treatment patterns over time, with increased utilization of higher-efficacy first-line treatments, including rituximab and novel MAbs, in more recent years. Use of lower-efficacy off-label treatments for AQP4+ NMOSD has decreased in recent years, likely representing more accurate and earlier diagnosis of the disease as well as improvements in access to evidence-based treatments. Only two of the four currently approved MAbs for AQP4+ NMOSD were utilized in our study population (satralizumab and eculizumab) likely due to timing; ravulizumab and inebilizumab were only approved in late 2023, and data collection was completed by January 1, 2024.
This study enhances the understanding of the Canadian AQP4+ NMOSD patient population and the evolution of diagnostic and management strategies over the past decades. Several key findings underscore the need for ongoing improvements in NMOSD care. First, we must aim to diagnose AQP4+ NMOSD as early and accurately as possible. Achieving early diagnosis hinges on recognizing typical clinical symptoms and signs, performing appropriate neuroimaging and sending the serum AQP4 antibody test. The cell-based assay for the AQP4 antibody has both a higher sensitivity (76% vs. 65%) 21–Reference Ruiz-Gaviria, Baracaldo, Castañeda, Ruiz-Patiño, Acosta-Hernandez and Rosselli22 and specificity (>99% vs. 90–95%) Reference Prain, Woodhall and Vincent23 compared to the ELISA assay and should be sent as early as possible in any suspected case of NMOSD. Our study shows that many Canadian patients with seropositive NMOSD diagnoses were only tested with the older ELISA assay (42%) versus cell-based assay testing (58%) likely due to the availability of only the ELISA assay when they were first diagnosed. We would recommend re-checking serology with the cell-based assay in cases where a diagnosis was previously made with only the ELISA assay. Reliance on ELISA assays is a significant limitation due to the potential for misclassification bias and the potential for false positive and false negative results. Accurate identification of individuals with seropositive (AQP4+) NMOSD is crucial for ensuring access to appropriate therapies, as certain therapies for multiple sclerosis can exacerbate NMOSD. Reference Kim, Kim, Li, Jung and Kim24–Reference Pikor, Prat, Bar-Or and Gommerman25 Therefore, expanding access to precise diagnostic tools – particularly cell-based assays – should be widely prioritized across all centers and recognized as a national clinical priority. Our investigation also revealed suboptimal utilization of orbital MRI during the initial NMOSD attack, particularly for patients presenting with ON symptoms. This is relevant given that ON due to NMOSD is often distinguished from other conditions, such as MS, by features such as longer optic nerve lesions that are closer to and/or involving the optic chiasm, which can be a relevant clinical clue to prompt AQP4 and MOG antibody testing.
Second, early use of high-efficacy therapy for NMOSD is imperative for improving outcomes. While there has been increased adoption of higher-efficacy first-line therapies, such as rituximab and novel MAbs, use of these treatments still trails behind older, less effective and less well-tolerated therapies. We observed that high-efficacy treatments for NMOSD are typically reserved for second- or third-line use following treatment failure or intolerance to previous therapies. We hypothesize that this practice likely stems from therapeutic inertia in caring for patients with NMOSD, a phenomenon that has been well described, Reference Cobo-Calvo, Gómez-Ballesteros and Orviz26–Reference Thon, Sharkus, Thakkar, Hunter, Siegler and Thon27 as well as barriers to accessing costly high-efficacy treatments, which are often not approved by provincial drug access programs. Reference Wingerchuk, Weinshenker, McCormick, Barron, Simone and Jarzylo28 Rituximab has been more widely funded and used to treat NMOSD in recent years; therefore, the high use of oral off-label therapies in this study also reflects the historic nature of the cohort. We suspect that expanded use of high-efficacy treatment accounts for the reduced incidence of disabling attacks and disability in our study sample compared to older studies and further supports efforts to increase clinician confidence in initiating high-efficacy treatment and advocacy efforts to achieve more widespread drug accessibility.
Strengths of this study include the multicenter, geographically representative national cohort and the uniform approach to data collection through a single protocol. Limitations of the current study include the retrospective study design, which is associated with potential biases in the data, including missing data for participants with more remote disease onset. We did not have data available regarding the grading of serious adverse events or cytopenias in the CANOPTICS database. Additionally, given that our sample comprised a substantial number of immigrants who immigrated during early adulthood, there may be additional gaps in completeness of data for these participants prior to immigration (i.e., index attacks occurring in their home country); this may have contributed to inaccuracies in our estimate of disease duration based on the date of the index attack and may have contributed to the slightly older mean age of onset observed in the current study relative to prior studies, although this difference is relatively minor. Recruitment rates into CANOPTICS were high, and most patients with NMOSD are followed at specialized demyelinating disease clinics, but we cannot rule out the possibility of selection bias.
Conclusions
In conclusion, this comprehensive analysis of AQP4+ NMOSD in Canadian adults provides valuable insights into the demographic profile, disease severity and treatment landscape of this complex disease over a span of nearly three decades. Our findings highlight the ethnically and racially varied and predominantly female population affected by this condition. Disability outcomes with respect to both EDSS and visual impairment were considerably more favorable than in previous reports.
Author contributions
AIM and DRL performed data analysis and interpretation. AIM drafted the original manuscript. DLR supervised the study. AK contributed to data collation and extraction. DLR, MSF, GF, LL, RAM, RAM, SAM, JAM, NEP, PS and CSC provided critical feedback and edits to the manuscript.
Funding statement
The authors have no competing interests to declare.
This study received funding from MS Canada (EGID 918124).



 Open access
Open access
Target article
Demographic and Clinical Characteristics of Aquaporin-4 Antibody-Positive Neuromyelitis Optica Spectrum Disorder in Canadian Adults
Related commentaries (3)
Reviewer Comment on Momen et al. “Demographic and Clinical Characteristics of Aquaporin-4 Antibody Positive Neuromyelitis Optica Spectrum Disorder in Canadian Adults”
Reviewer Comment on Momen et al. “Demographic and Clinical Characteristics of Aquaporin-4 Antibody Positive Neuromyelitis Optica Spectrum Disorder in Canadian Adults”
Reviewer Comment on Momen et al. “Demographic and Clinical Characteristics of Aquaporin-4 Antibody-Positive Neuromyelitis Optica Spectrum Disorder in Canadian Adults”