


LEARNING OBJECTIVES
After reading this article, you will be able to:
• understand the putative mechanisms of action behind the antidepressant effects of vortioxetine and psilocybin, which target the serotonergic system
• recognise that there are several emerging target systems for treating depression, including the glutamatergic, GABAergic and inflammatory systems
• understand the effects of antidepressant drugs acting on these systems, at both the neurobiological and molecular level.
Major depressive disorder (MDD) is a common psychiatric disorder characterised by a variable mix of affective symptoms (such as sadness, feelings of guilt, low self-worth and suicidal ideation), reward-related symptoms (such as anticipatory, motivational and consummatory anhedonia), cognitive symptoms (such as an inability to concentrate) and vegetative disturbances (such as changes in weight, appetite, sleeping pattern, psychomotor activity and energy levels).
MDD has an estimated lifetime prevalence of 11% (Lim Reference Lim, Tam and Lu2018). It is a leading cause of disability worldwide, although there is a notable dearth of prevalence data from low-income countries. The prevalence of MDD is likely to increase over the coming years, owing to a combination of environmental, social and economic factors together with reduced stigma and a greater recognition of its symptoms by patients and healthcare professionals alike.
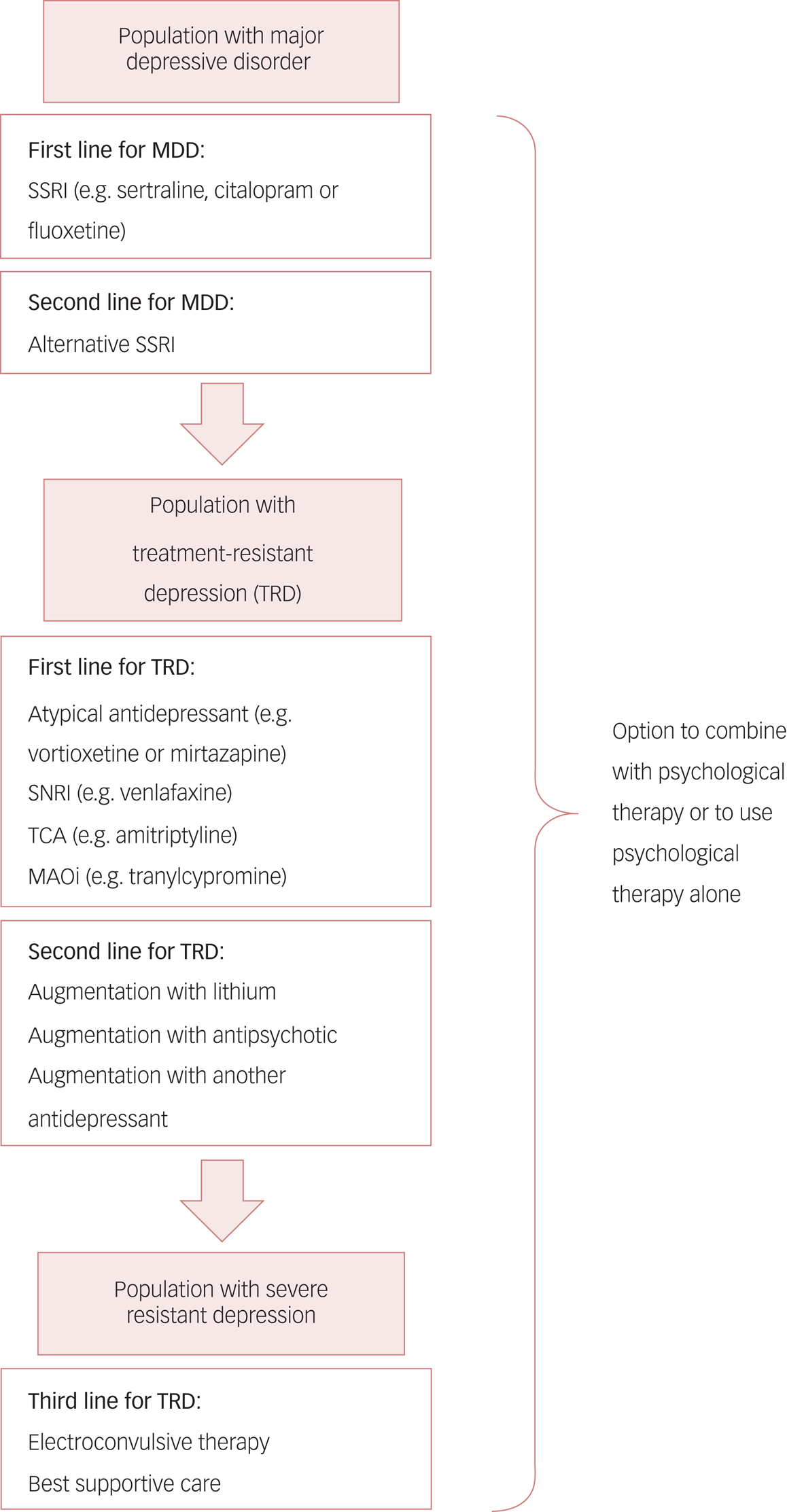
The current treatment pathway for MDD recommended by the National Institute for Health and Care Excellence (NICE) is outlined in Fig. 1. Although first-line psychological and pharmacological therapies are effective in many instances, approximately 20–30% of people fail to respond to two or more trials of first-line antidepressant medications (Rush Reference Rush, Trivedi and Wisniewski2006). These people are said to have treatment-resistant depression (TRD) (Al-Harbi Reference Al-Harbi2012). TRD has a particularly high associated morbidity and is highly recurrent, with as many as 80% of individuals relapsing within a year of achieving remission. For those with a protracted illness, the 10-year rate of recovery is only 40% (Fekadu Reference Fekadu, Wooderson and Markopoulo2009). The related concept of ‘difficult to treat depression' refers to depression that continues to cause significant burden despite usual treatment, and is sometimes used as an alternative term to TRD.
The current treatment pathway for major depressive disorder (MDD) as suggested by the National Institute for Health and Care Excellence (Reference National Institute2009).
First-line pharmacological treatment for MDD involves using a selective serotonin reuptake inhibitor (SSRI) such as sertraline, citalopram or fluoxetine; deciding between these agents is currently based on clinician and patient preference. If, despite dose adjustment, the first SSRI does not improve symptoms or is not tolerated, an alternative SSRI can be prescribed. Patients who do not respond to at least two first-line antidepressants have treatment-resistant depression (TRD). Treatment then may involve an atypical antidepressant such as vortioxetine; a serotonin and noradrenaline reuptake inhibitor (SNRI) such as venlafaxine; a tricyclic antidepressant (TCA); or a monoamine oxidase inhibitor (MAOi). If response is inadequate, clinicians can then consider augmentation with, for example, lithium or atypical antipsychotics. Finally, in patients with severe TRD, electroconvulsive therapy may be needed. ‘Best supportive care’ is a non-specific mix of treatment strategies in complex depression, including ongoing therapy with antidepressants, psychological support and social interventions. At all levels, there is the option to utilise psychological therapy (or to use psychological therapy as the only treatment modality).
The prevalence and associated disease burden of TRD, coupled with the disease burden associated with MDD more generally, means there is a pressing need to develop novel antidepressant therapies.
The multi-faceted psychopharmacology of MDD
The psychopharmacological account of MDD is one framework used to understand its aetiology and pathogenesis. It is not exclusive of psychological, cognitive, behavioural or sociological accounts and has been a fruitful avenue for the development of new medicines. A comprehensive account of the psychopharmacological aspects of MDD is beyond the scope of this article, so we summarise the main actors.
Monoamines and their receptors
The monoamine theory of depression originally asserted that the underlying pathophysiological basis of MDD is depletion in the levels of the neurotransmitters serotonin (5-HT, an indolamine), noradrenaline and/or dopamine (catecholamines) in the central nervous system (Everett Reference Everett and Toman1959). The roots of this hypothesis lay in simple observations: first, that depletion of monoamines seemed to induce feelings of depression and lassitude; and second, that antidepressants facilitated monoaminergic transmission.
In 1967, Alec Coppen proposed the serotonergic instantiation of the monoamine hypothesis: that depression is caused by low levels of serotonin in the central nervous system (Coppen Reference Coppen1967). The precise function of serotonin in the brain has remained unclear for several reasons: the extensive projections of dorsal raphe nucleus (DRN) neurons; the tendency for paracrine serotonergic transmission; and the diverse array of receptor subtypes. The relative contribution of indolamines and catecholamines to the aetiology and pathogenesis of depression is debated, and it is possible that they are linked to different aspects of MDD. It has been suggested that drugs increasing serotonergic transmission may be better at alleviating affective symptoms, whereas drugs increasing noradrenergic transmission may be better at alleviating motivational impairments (Nutt Reference Nutt, Demyttenaere and Janka2007).
The monoamine theory has led to the development of selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs). Nevertheless, the theory leaves some questions unanswered: if depleted levels of monoamines cause depression, why do both SSRIs and SNRIs – which acutely increase concentrations of monoamines in the synaptic cleft – take at least 1 week to improve mood? Why are they only effective for some patients? These questions have meant that there has been a shift away from focusing on levels of transmitters themselves towards understanding their action at monoamine receptors, which may be upregulated in response to depleted levels of monoamines. The monoamine receptor hypothesis posits that imbalanced monoamine receptor activation underlies MDD and it has received particular attention in the context of renewed interest in the antidepressant effects of psychedelic drugs which act on serotonin receptors.
The two most widely expressed serotonin receptors in the brain are the 5-HT1A and 5-HT2A receptors: 5-HT1A receptors are Gi-protein coupled and are inhibitory, whereas 5-HT2A receptors are Gq-protein coupled and are excitatory. In their bipartite model of brain serotonin function, Carhart-Harris & Nutt propose that serotonin mediates ‘adaptive responses to adversity’ via two distinct mechanisms (Carhart-Harris Reference Carhart-Harris and Nutt2017a). Post-synaptic 5-HT1A receptors in limbic structures mitigate the deleterious effects of stress and promote stress tolerance. This pathway is potentiated by chronic SSRI use, through desensitisation of 5-HT1A autoreceptors, thereby reducing negative feedback and promoting serotonin release in limbic structures. A 5-HT2A receptor pathway is post-synaptic, and in the cortex promotes plasticity, environmental sensitivity and adaptability. This pathway is potentiated by psychedelics such as psilocybin, which act as 5-HT2A receptor agonists. This bipartite view of serotonergic transmission is summarised in Table 1.
The bipartite model of serotonin (5-HT) function

DRN, dorsal raphe nucleus; SSRI, serotonin reuptake inhibitor.
After Carhart-Harris & Nutt (Reference Carhart-Harris and Nutt2017a).
Glutamate and GABA
The effect of all monoamines and their action on G-protein coupled receptors is to modulate excitability of downstream neurons in the wide areas of cortex that their axons innervate, in addition to effects on synaptogenesis and neural plasticity. Most cortical cells are glutamatergic (excitatory), followed closely by γ-aminobutyric acid (GABA)ergic neurons (inhibitory). These transmitter systems may be important ‘final common pathways’ leading to depressive symptoms.
The glutamate theory of depression suggests that the pathophysiological basis of depression lies in malfunction of mechanisms regulating the central clearance and metabolism of glutamate (Lener Reference Lener, Niciu and Ballard2017). GABAergic dysfunction in depression is thought to result from the deleterious effects of chronic stress, leading to hypothalamic–pituitary–adrenal axis disinhibition and further release of stress-associated glucocorticoids, potentiating further dysfunction (Lüscher Reference Lüscher and Möhler2019). Histological, neuroimaging and preclinical animal data support the notion that there are region-specific alterations to glutamatergic and GABAergic transmission associated with depression or depression-like behaviours (Lener Reference Lener, Niciu and Ballard2017). Both the glutamate and GABA hypotheses have been productive in the development of new antidepressants: examples include ketamine and brexanolone respectively.
Inflammation
Beyond specific neurotransmitter systems, there are also general disruptors of neurotransmission, which may have non-specific effects to upset monoaminergic, glutamatergic or GABAergic transmission across the cortex and therefore induce or potentiate depressive symptoms. One such example is inflammation. Peripheral activation of the immune system and the consequences this has within the brain has been implicated in deleterious mood changes in both humans and animals.
The brain monitors peripheral inflammation by two key pathways (Dantzer Reference Dantzer, O'Connor and Freund2008). In a neural pathway, locally produced cytokines activate the vagus nerve (e.g. during gastroenteritis) or the trigeminal nerve (e.g. during sinusitis) which relay afferent signals to their brainstem nuclei, and onwards to cortical regions. In a humoral pathway, systemically circulating cytokines can directly cross the blood–brain barrier (using saturable transporter mechanisms) and circulating pathogen-associated molecular patterns can activate toll-like receptors on macrophages in circumventricular organs. Both of these pathways lead to microglial activation within the brain and the production of pro-inflammatory cytokines, leading to a brain ‘image’ of the peripheral inflammatory response without the associated invasion of immune cells and tissue damage. Nevertheless, this may result in alterations to monoaminergic, glutaminergic or GABAergic neurotransmission.
There is substantial symptom overlap between sickness behaviour and depression, with the former recapitulating core symptoms of the latter. However, there are important qualitative differences: sickness behaviour is short lived and an adaptable response to illness, whereas depression is maladaptive and persists. The inflammatory theory of depression therefore suggests that some instances of depression are caused by a maladaptive version of cytokine-induced sickness behaviour. A correlative link between depression and inflammation is now well established, with higher levels of cytokines, acute phase reactants (such as C-reactive protein, CRP) and leukocyte subtypes in some individuals with MDD compared with controls (Lynall Reference Lynall, Turner and Bhatti2020).
There are many exciting and emerging treatment avenues for MDD and TRD; by necessity, this is a selective review of several advances in the field within each of these domains.
Modulation by monoamines
Vortioxetine
Vortioxetine is a novel multimodal modulator of the serotonergic system developed by Lundbeck (marketed under the name Brintellix® in the UK). Its description as a ‘multimodal’ derives from its action on more than one class of molecular target: both reuptake transporters and G-protein coupled receptors. Vortioxetine inhibits the serotonin reuptake transporter (SERT) – like conventional SSRIs – and has additional properties on serotonin receptors: it is an agonist at 5-HT1A receptors; a partial agonist at 5-HT1B receptors; and an antagonist at 5-HT1D, 5-HT3 and 5-HT7 receptors.
Vortioxetine's inhibition of SERT leads to elevation of serotonin in the synaptic clefts of DRN neuron terminals, resulting in downstream effects. However, the efficacy of SERT inhibition alone to increase synaptic serotonin is limited by three negative feedback mechanisms:
• recurrent collateral branches of DRN neurons synapse on the DRN cell body and local inhibitory interneurons: SERT inhibition results in excess serotonin at these terminals, accentuating negative feedback directly through 5-HT1A receptors (Gi-coupled) on DRN cell bodies, and indirectly through 5-HT7 receptors (Gs-coupled) on inhibitory interneurons;
• glutamatergic pyramidal neurons at cortical targets of DRN projections project back to local inhibitory interneurons within the DRN, further inhibiting serotonergic transmission;
• presynaptic 5-HT1B/1D receptors (Gi-coupled) are stimulated by the excess serotonin in the synaptic cleft, and act as negative feedback autoreceptors to reduce the presynaptic release of serotonin.
The pharmacological properties of vortioxetine – and its difference from SSRIs – enable it to ‘shut off’ these negative feedback loops, as described in Fig. 2.
The multimodal modulation of the serotonergic system by vortioxetine.
Representation of a serotonergic neuron of the dorsal raphe nucleus (DRN) synapsing onto the dendrites of a distant cortical pyramidal neuron, with serotonin depicted as semicircles and the effects of vortioxetine on cell activity indicated by arrows. Vortioxetine inhibits the serotonin reuptake transporter (SERT) to increase serotonin in the synaptic cleft, shown centrally in the figure at the synapse between the sertonergic neuron and the cortical pyramidal cell. This action is potentiated by vortioxetine's added ability to shut down three important negative feedback loops otherwise limiting serotonin release, indicated by boxes (a) to (c). (a) Recurrent collateral branches from serotonin neurons usually inhibit further serotonin release directly via Gi-coupled 5-HT1A receptors and indirectly via Gs-coupled 5-HT7 receptors on inhibitory interneurons. Vortioxetine is an agonist at 5-HT1A receptors on DRN cell bodies, which desensitise, and an antagonist at 5-HT7 receptors on inhibitory interneurons, therefore switching off both limbs of this negative feedback loop. (b) As depicted on its dendrites, most cortical cells co-express 5-HT1A and Gq-coupled 5-HT2A receptors. Serotonin release can stimulate cortical pyramidal cells, and these provide retrograde negative feedback to the DRN by stimulating inhibitory interneurons. Vortioxetine is an agonist on post-synaptic cortico–limbic 5-HT1A receptors, which do not desensitise, and it therefore inhibits pyramidal cells to switch off this feedback loop. (c) Gi-coupled 5-HT1B/1D receptors act as presynaptic autoreceptors to inhibit further serotonin release in response to the presence of the neurotransmitter in the synaptic cleft. Vortioxetine is an antagonist at 5-HT1B/1D receptors, and therefore switches off this negative feedback loop.

Clinical guidelines
NICE recommends vortioxetine (initially 10 mg orally once daily, adjusted according to response to 5–20 mg) as a possible treatment for adults having a first or recurrent episode of MDD if this has not responded to two antidepressants (i.e. meeting the most commonly used definition for TRD) (National Institute for Health and Care Excellence 2015). The recommendation was made on the basis of two clinical trials: one comparing vortioxetine with agomelatine for depression (Montgomery Reference Montgomery, Nielsen and Poulsen2014) and another comparing vortioxetine with escitalopram for treatment-emergent sexual dysfunction (Jacobsen Reference Jacobsen, Mahableshwarkar and Chen2015). An indirect treatment comparison study was also used to inform the recommendation, which compared vortioxetine with several other antidepressants and suggested that vortioxetine may be better tolerated than other antidepressants, such as sertraline and venlafaxine (Brignone Reference Brignone, Diamand and Painchault2016).
Efficacy and tolerability
Subsequently, a 2017 Cochrane meta-analysis assessed the efficacy and tolerability of vortioxetine compared with placebo and other oral antidepressant treatment for acute depression, analysing 17 trials (Koesters Reference Koesters, Ostuzzi and Guaiana2017). The meta-analysis found low-quality evidence to support a beneficial effect of vortioxetine over placebo for response rates (relative risk RR = 1.35, 95% CI 1.22–1.49), remission rates (RR = 1.33, 95% CI 1.15–1.53) and depressive symptoms (mean difference in change in Montgomery–Åsberg Depression Rating Scale (MADRS) score of 2.94 favouring vortioxetine, 95% CI 1.80–4.07), but in all cases the effect sizes were small. Compared with SNRIs, there were no differences in response rates (RR = 0.91, 95% CI 0.82–1.00) or remission rates (RR = 0.89, 95% CI 0.77–1.03) and a small advantage of SNRIs compared with vortioxetine on changes in depressive symptoms (mean difference in change in MADRS score of 1.52 favouring SNRIs, 95% CI 0.50–2.53). Important caveats related to the trials incorporated in the meta-analysis included:
• all the trials were of short duration, between 6 and 8 weeks
• the trials compared vortioxetine with venlafaxine (two), duloxetine (six) or placebo (seven) and no studies directly compared vortioxetine with SSRIs or other antidepressant classes.
Common adverse events associated with vortioxetine included nausea, headache, nasopharyngitis, diarrhoea and dizziness; however, adverse event reporting varied significantly across individual studies. Compared with SNRIs, there was evidence that fewer participants experienced adverse events when on vortioxetine (RR = 0.90, 95% CI 0.86–0.94). Its reduced propensity to induce sexual dysfunction may mean it is one appropriate ‘switch’ option for patients experiencing antidepressant-induced sexual dysfunction (Jacobsen Reference Jacobsen, Nomikos and Zhong2020).
Vortioxetine may additionally have beneficial effects beyond its action to improve mood: it seems to be effective in improving psychomotor speed, executive function, memory, attention and behavioural flexibility in depression (Bennabi Reference Bennabi, Haffen and Van Waes2019). Interestingly, Harmer and her team have demonstrated that in remitted individuals with prior MDD, vortioxetine modulates activity in regions of the brain involved in working memory (such as the dorsolateral prefrontal cortex) during an N-back task, demonstrating that the cognitive effects of vortioxetine may be dissociated from its antidepressant effects (Smith Reference Smith, Browning and Conen2018).
Current clinical use and further research
Vortioxetine is a promising antidepressant with multimodal actions on the serotonergic system to increase serotonin in the synaptic cleft and shut off negative feedback mechanisms limiting serotonin release. It has been approved for use in people with TRD, and may be better tolerated than alternative agents (Koesters Reference Koesters, Ostuzzi and Guaiana2017) and have beneficial effects on cognition (Smith Reference Smith, Browning and Conen2018). An important caveat is that vortioxetine's antidepressant efficacy has not been directly compared with several classes of commonly used antidepressants, such as SSRIs, monoamine oxidase inhibitors (MAOIs) and tricyclic antidepressants (TCAs). Further trials are required to determine whether vortioxetine is as effective, or as tolerated, as SSRIs when used as a first-line agent during MDD episodes.
Psychedelic drugs
Psychedelic drugs have been described as ‘compounds with appreciable serotonin 2A receptor agonist properties that can alter consciousness in a marked and novel way' (Carhart-Harris Reference Carhart-Harris and Goodwin2017b).
Classic psychedelic drugs include lysergic acid diethylamide (LSD), mescaline and psilocybin. Psychedelics were used relatively frequently as adjuncts to psychotherapy for mood disorders in the mid-20th century, until their use was curbed by being placed on schedule I of the UN Convention on Narcotic Drugs in 1967. Nevertheless, their beneficial effect in mood disorders had been investigated at the time. A recent meta-analysis of 19 studies published between 1949 and 1973 investigating the efficacy of psychedelics in mood disorder treatment found that 79.2% of patients showed clinician-judged improvement after treatment with psychedelics (Rucker Reference Rucker, Jelen and Flynn2016). These early studies were often of poor quality: many were non-randomised trials, did not use standardised measures of symptom severity and did not use standardised diagnostic techniques.
In the late 1990s and early 2000s, there was a renewed interest in psychedelic drug research, with a growing appreciation that their effects could be investigated in accordance with the ethical and scientific standards required of medical research. In 2006, the first randomised controlled trial (RCT) investigating the safety and feasibility of psilocybin to treat obsessive–compulsive disorder was published (Moreno Reference Moreno, Wiegand and Taitano2006). Subsequently, pilot studies investigating the use of psychedelics such as psilocybin in TRD have been published, with results supporting their feasibility and efficacy in this population (Rucker Reference Rucker, Iliff and Nutt2018).
Psilocybin
Psilocybin is almost entirely converted to its metabolite psilocin during first-pass metabolism. Effects following oral administration of 12–25 mg psilocybin are noticeable within 40 min and last approximately 3–6 h after ingestion; its half-life is approximately 2.5 h. Psilocin has a multifunctional action on several classes of serotonin receptor: it is a potent agonist at post-synaptic 5-HT2A receptors and to a lesser extent at pre- and post-synaptic 5-HT1A receptors (Fig. 3). In the context of their bipartite model referenced earlier, Carhart-Harris & Nutt argue that psilocybin promotes active coping and adaptability through 5-HT2A-predominated means, enhancing cortical plasticity (Carhart-Harris Reference Carhart-Harris and Nutt2017a).
The mechanism of action of psilocybin.
Psilocybin (metabolised to psilocin) is a potent 5-HT2A receptor agonist: this receptor is widely expressed throughout the cortex and is the principal serotonergic excitatory receptor. Psilocin's hallucinatory effects are due to agonism at this receptor. Psilocin also turns off two of the negative feedback pathways described in Fig. 2: (a) recurrent collateral inhibition via 5-HT1A receptor desensitisation and (b) inhibition mediated by cortical pyramidal cell synapses onto inhibitory interneurons, through 5-HT1A receptor agonism. SERT, serotonin reuptake transporter

Efficacy and tolerability
Recently, a phase II RCT found that two 25 mg doses of psilocybin separated by 3 weeks were non-inferior to a 6-week course of escitalopram (coupled with a 1 mg ‘placebo-like’ daily dose of psilocybin) as measured by scores at week 6 on the Quick Inventory of Depressive Symptomatology (QIDS-16) (Carhart-Harris Reference Carhart-Harris, Giribaldi and Watts2021). The overall incidence of adverse events was similar in both groups, although psilocybin had a lower incidence of emotional blunting and sexual side-effects. Promising clinical trial results must be tempered by an acknowledgement of the challenges faced when designing trials investigating the antidepressant effects of psychedelics. Trials suffer from the problem of unmasking (‘unblinding’) because the acute psychoactive effects of treatment doses of psilocybin are substantial and profound. Additional problems include expectancy bias (Barnby Reference Barnby and Mehta2018) and that psilocybin is often administered with psychological support (psychedelic-assisted psychotherapy), and so it can also be difficult to disentangle the drug's effect from general care effects. Possible routes to mitigate some of these confounds include:
• active placebos, such as niacin, 3,4-methylenedioxymethamphetamine (MDMA) or ketamine; although the subjective experience following these drugs differs from psilocybin, these could reduce unmasking
• comparing different doses of psilocybin, such as 10 v. 25 mg, which are reported to have qualitatively different effects
• having placebo arms that include psychotherapeutic support, to dissociate effects of psilocybin from general care effects.
Early-phase clinical trials are promising, but larger multicentre trials are still needed so the results can be generalised. The potential benefit associated with psilocybin use in MDD is still, effectively, hypothetical and the hypothesis should be tested using phase III RCTs.
Accumulating data support psilocybin's safety when used in a medically controlled setting in combination with psychological support. In the most recent 2021 RCT comparing psilocybin with escitalopram, no serious adverse events were observed in individuals taking treatment doses of psilocybin, and the total proportion of patients reporting adverse events was similar across both the psilocybin (87%) and escitalopram (83%) arms (Carhart-Harris Reference Carhart-Harris, Giribaldi and Watts2021). Adverse events associated with psilocybin typically occur within 24 h of dosing, the most common being headache (67%), nausea (27%) and fatigue (7%) (see supplemental Table S5 of Carhart-Harris et al (Reference Carhart-Harris, Giribaldi and Watts2021) for a detailed overview of the spectrum of side-effects).
During acute administration of psilocybin, there is the possibility of having a distinctly negative psychological experience or ‘bad trip.’ The factors determining whether a trip will be good or bad are not fully understood: although positive experiences seem to be dose dependent, negative ones are not (Hirschfeld Reference Hirschfeld and Schmidt2021). It is widely acknowledged that users’ experience of psilocybin is dependent on the context, and so it would make intuitive sense that giving psilocybin in a controlled, well-supported environment would reduce the likelihood of negative experiences. Studies that have explored the acute subjective effects of psilocybin show that it induces a psychosis-like state (van Amsterdam Reference van Amsterdam, Opperhuizen and van den Brink2011), and so current trials have excluded individuals with a family or personal history of a psychotic disorder, because of the risk that psilocybin may precipitate or exacerbate psychosis (although a causal relationship is yet to be established).
The neurobiological correlates of psilocybin's antidepressant effects have been explored using functional magnetic resonance imaging (fMRI). In 19 people with TRD, imaging before and 1 day after 25 mg psilocybin showed a reduction in blood flow to the amygdala, which was correlated with a substantial antidepressant response (Carhart-Harris Reference Carhart-Harris, Roseman and Bolstridge2017c). Five weeks post-dose, differences in connectivity between the ventromedial prefrontal cortex and the lateral parietal cortex (part of the default mode network) could differentiate responders from non-responders. This study did not have a control arm, so the differences in neuroimaging correlates associated with its therapeutic response cannot be unequivocally attributed to psilocybin.
Current clinical use and further research
There remain several barriers to psilocybin's routine clinical use: it remains unclear at which stage in the MDD treatment pathway psilocybin would best sit; an appropriate dosing strategy has yet to be determined; clear guidance is needed on the appropriate psychotherapeutic modality to use in conjunction with psilocybin; and there remains an ongoing need for long-term data reviewing its efficacy and safety.
Excitation and inhibition
Targeting glutamate receptors with intranasal S-ketamine
The N-methyl-d-aspartate (NMDA) receptor antagonist ketamine has shown significant promise as a rapidly acting, potent glutamate-based antidepressant effective in treatment-resistant populations. The impetus to trial ketamine as an antidepressant resulted from several drivers: (a) an implication of glutamate in the pathophysiology of depression; (b) the efficacy of NMDA receptor antagonists in animal models of depression (Trullas Reference Trullas and Skolnick1990); (c) the effect of monoaminergic antidepressants on NMDA receptor function (Paul Reference Paul, Nowak and Layer1994); and (d) data suggesting efficacy of weak NMDA receptor antagonists (such as amantadine) in people with depression (Vale Reference Vale, Angeles Espejel and Cesar Dominguez1971). The first RCT exploring the use of intravenous racemic ketamine in TRD (an intravenous infusion of 0.5 mg/kg over 40 min) was in 2000 (Berman Reference Berman, Cappiello and Anand2000), with more since.
The mechanism of ketamine's antidepressant effect remains unclear. At the molecular level, ketamine's classic action is as an NMDA receptor antagonist, blocking the channel pore and preventing the influx of sodium and calcium ions. At the level of brain regions, both preclinical and clinical evidence implicates the anterior cingulate cortex, lateral habenula, striatum and hippocampus as being important in ketamine's antidepressant, or (in the case of animal models) antidepressant-like, effects (Alexander Reference Alexander, Jelen and Mehta2021).
Ketamine's region-specific effects on brain activity may reflect the relative expression of NMDA receptors on pyramidal cells and on inhibitory interneurons. In circuits predominated by interneurons, ketamine-induced NMDA receptor blockade is thought to trigger an acute glutamate surge, including in subregions of the prefrontal cortex (Lener Reference Lener, Niciu and Ballard2017). In circuits predominated by pyramidal cells, such as the hippocampal formation and different subregions of the prefrontal cortex, there seems to be a reduction in glutamate release (Stan Reference Stan, Alvarsson and Branzell2014). Ultimately, there are downstream changes in protein kinase activity, leading to phosphorylation of transcription factors, changes in the level of gene expression, synaptogenesis and neural plasticity. An overview of ketamine's key hypothesised mechanisms of action at the synaptic level is shown in Fig. 4.
Ketamine's action at a cortico–cortical glutamatergic synapse.
Hypothesised cellular effects of ketamine are grouped in the diagram. +, agonism or stimulatory effects; −, antagonism or inhibitory effects. Figure based on discussion in Jelen et al (Reference Jelen, Young and Stone2021). The glutamate surge hypothesis (left) supposes that ketamine causes an acute glutamate surge in circuits predominated by interneurons due to antagonism on presynaptic NMDA receptors, leading to a reduction in inhibition and increased glutamate release. Ketamine and ketamine metabolites (such as (2R,6R)-hydroxynorketamine) also have effects on AMPA receptors (centre), which increase intracellular calcium through activation of voltage-gated calcium channels and cause exocytosis of vesicles containing brain-derived neurotrophic factor (BDNF) owing to post-synaptic depolarisation. Neurotrophic effects (lower right) are mediated through BDNF signalling. BDNF activates tropomyosin receptor kinase B (TrkB) autoreceptors, which activate downstream protein kinases. Calcium-dependent (phospholipase C [PLC]/inositol-3-phosphate [IP3]/calmodulin [CaM]/calmodulin-dependent kinase [CaMKII]) and calcium-independent (rat sarcoma virus [Ras]/rapidly accelerated fibrosarcoma [Raf]/mitogen-activated protein kinase [MEK]/extracellular signal-related kinase [ERK] and Ras/phosphoinositide-3-kinase [PI3K]/protein kinase B [Akt]/mammalian target of rapamycin complex-1 [mTORC1]) signalling pathways result in transcription factor phosphorylation (such as cyclic AMP response element binding protein [CREB] and eukaryotic elongation factor-2 [eEF2]) and alterations in gene expression.

Understanding ketamine's mechanism of action is further complicated by (a) different actions of its two constitutive enantiomers, R-ketamine (arketamine) and S-ketamine (esketamine), (b) actions of its metabolites and (c) its actions on other neurotransmitter systems.
(a) S-ketamine is a more potent NMDA receptor antagonist than R-ketamine. Based on this, S-ketamine would presumably have a more potent antidepressant effect. However, a recent open-label pilot study of R-ketamine in seven individuals with TRD suggests that it may rapidly improve depressive symptoms with minimal dissociative and haemodynamic side-effects (Leal Reference Leal, Bandeira and Correia-Melo2021).
(b) Metabolism of racemic ketamine to hydroxynorketamine (HNK) is necessary for ketamine's antidepressant-like effects in rodents (Zanos Reference Zanos, Moaddel and Morris2016). (2R,6R)-HNK was associated with more substantial antidepressant effects than (2S,6S)-HNK, and the former's antidepressant effects depended on α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors rather than NMDA receptors. There have been no clinical trials exploring the antidepressant efficacy of (2R,6R)-HNK in humans.
(c) Ketamine has a broad action on several other neurotransmitter systems, including the monoaminergic, cholinergic and opioid systems, likely downstream of its effects on glutamatergic transmission.
Efficacy, tolerability and licensing
Although intravenous ketamine has been most extensively studied, challenges associated with the intravenous route mean that alternative routes of administration are of interest, and so an S-ketamine intranasal spray (Spravato®) has been developed by Janssen Pharmaceuticals. In 2019, Spravato® was approved by the US Food and Drug Administration (FDA) for use as an augmentation agent with another oral antidepressant for TRD under a risk evaluation and mitigation strategy (REMS). The REMS stipulates that:
• Spravato® comes with a boxed warning for: sedation; impairments with attention, judgement and thinking (dissociation); suicidal thoughts and behaviours; and misuse
• the spray is self-administered in a certified doctor's office or clinic – it cannot be taken at home
• the patient must be monitored for at least 2 h after receiving their dose.
These stipulations relate to the acute side-effects of intranasal S-ketamine, which can induce dissociation, dizziness and nausea. It may also lead to transient hypertension, meaning it may not be suitable for individuals with poorly controlled hypertension or pre-existing aneurysmal disease.
Several clinical trials have evaluated the use of intranasal S-ketamine as an adjunct to an oral antidepressant – all relatively short-term trials. A 2020 meta-analysis (Zheng Reference Zheng, Cai and Xiang2020) of four RCTs found that adjunctive intranasal S-ketamine resulted in a greater rate of study-defined response (RR = 1.39, 95% CI 1.18–1.64) and study-defined remission (RR = 1.42, 95% CI 1.17–1.72) at trial end-points. Results were significant for a greater response with S-ketamine at 2 hours (RR = 2.77, 95% CI 1.62–4.76), peaking at 24 h (RR = 5.42, 95% CI 1.38–21.20) and lasting for at least 28 days (RR = 1.36, 95% CI 1.16–1.58). The authors of this meta-analysis concluded that intranasal S-ketamine has ultra-rapid antidepressant effects, lasting for at least 28 days. In 2021, a Cochrane meta-analysis of nine studies using S-ketamine (six of which were intranasal) found that S-ketamine was more efficacious than placebo in terms of response at 24 h (odds ratio OR = 2.11, 95% CI 1.20–3.68), at 1 week (OR = 1.60, 95% CI 1.09–2.34), at 2 weeks (OR = 1.57, 95% CI 1.09–2.28) and at 4 weeks (OR = 1.84, 95% CI 1.44–2.37; Dean Reference Dean, Hurducas and Hawton2021). There was no difference found at 72 h (OR = 1.34, 95% CI 0.92–1.96), which may reflect that some of the studies were underpowered to detect a difference.
The FDA has approved Spravato® for use, but NICE technology appraisal consultations have not approved intranasal S-ketamine as an adjunctive therapy for TRD (National Institute for Health and Care Excellence Reference National Institute2018). The reasons given for this decision included: the short duration of several of the trials, which limits generalisability to the long-term treatment of depression; the exclusion criteria for trials (such as the presence of psychiatric comorbidities and the presence of suicidal ideation in the previous 6 months), which could exclude patients with TRD and limits generalisability to ‘naturalistic’ samples; and safety concerns, including the risk of misuse and the risk of withdrawal syndromes after ketamine's cessation.
In 2017, the UK's Royal College of Psychiatrists released a position statement on the use of ketamine to treat depression (Royal College of Psychiatrists 2017). It recommended that mental health practitioners ‘proceed with caution when treating patients with ketamine’ (p. 3), citing concerns regarding the long-term efficacy and safety of ketamine use. Additionally, the College highlighted a lack of clarity regarding the optimal mode of drug administration to achieve sustained antidepressant effects; indeed, the appropriate frequency of administration for long-term maintenance treatment is not clear, although ketamine is typically administered once per week or once every other week.
R-ketamine
Based on data supporting the antidepressant effect of ketamine's other enantiomer, R-ketamine (Leal Reference Leal, Bandeira and Correia-Melo2021), it may yet prove valuable as an antidepressant, although it is a significantly earlier stage of development. One biopharmaceutical company is currently investigate safety and tolerability of differing doses of R-ketamine in healthy volunteers before exploring its potential in depression (Perception Neuroscience 2020), and a trial comparing the efficacy and safety of R-ketamine, S-ketamine and racemic ketamine in TRD is underway in China (Zhang Reference Zhang2018).
GABAergic modulation and brexanolone in post-partum depression
GABAergic transmission undergoes particularly substantial changes during pregnancy, owing to pregnancy-associated increases in neurosteroids derived from progesterone. Neurosteroids are exogenous and endogenous steroids that alter neural activity through rapid, non-genomic actions, including via the GABA receptor. Allopregnanolone is an endogenous neurosteroid that acts as a positive allosteric modulator of the GABAA receptor: it potentiates GABAergic transmission by slowing down the ‘off’ time of the receptor, leading to a prolonged inhibitory current (Belelli Reference Belelli and Lambert2005).
The interaction between neurosteroids and the GABAergic system is thought to be relevant to the aetiology of post-partum depression. Increases in allopregnanolone during pregnancy are thought to cause homeostatic decreases in GABAergic transmission. After parturition, levels of neurosteroids return to normal rapidly and GABA receptor expression subsequently returns to normal. Post-partum depression may be related to defective GABAergic plasticity during this critical period; namely, an inability of the GABAergic system to return to the pre-pregnancy state.
Further renewed interest in the GABAergic hypothesis has stemmed from the discovery that brexanolone – allopregnanolone available as an intravenous preparation – showed benefit in individuals with post-partum depression. In a phase II RCT involving 21 female in-patients with post-partum depression, a 60 h intravenous infusion of brexanolone significantly reduced scores on the Hamilton Rating Scale for Depression (HRSD) at the 60 h time point (Kanes Reference Kanes, Colquhoun and Gunduz-Bruce2017). Significant and meaningful reductions in HRSD scores were subsequently replicated in a phase III study (Meltzer-Brody Reference Meltzer-Brody, Colquhoun and Riesenberg2018). Secondary end-points from this trial highlighted that brexanolone had antidepressant effects evident within 48 h of commencing the infusion and were sustained up to 30 days later for higher doses (similar to the rapid and sustained effects of ketamine).
Brexanolone (as Zulresso®) was approved by the FDA in 2019 for post-partum depression as the first such treatment licensed specifically for this condition (Food and Drug Administration 2019). It was approved with a REMS requiring patients to be monitored for the entirety of the 60 h infusion, with continuous pulse oximetry because of the risk of syncope; other side-effects include somnolence, flushing and dry mouth. It is not yet approved by NICE for use in post-partum depression, and barriers to its use include its complex method of administration (a 60 h continuous intravenous infusion) together with its high estimated costs (up to $34 000 for one infusion).
Disrupting the balance: inflammation as a novel target in depression treatment
There have been several meta-analyses exploring the antidepressant effects of common anti-inflammatory agents either as add-on therapies or as monotherapy, compared with placebo add-on or monotherapy (e.g. Bai Reference Bai, Guo and Feng2020). These meta-analyses include a wide array of compounds (including non-steroidal anti-inflammatory drugs, various cytokine inhibitors and antibiotics such as minocycline) and generally support a beneficial effect of anti-inflammatory agents. However, results from individual RCTs are typically variable and modest, with non-sustained antidepressant effects. The link between peripheral inflammation and depression is undoubtedly complex, and it is possible that conventional anti-inflammatory agents do not optimally target the mechanisms by which peripheral (or central, glial-mediated) inflammation affects mood.
Minocycline has received particular attention as a promising treatment of MDD associated with inflammation. It is a tetracycline antibiotic with good blood–brain barrier penetrance and is already in use as an antibiotic. A 2018 meta-analysis concluded that there may be an antidepressant effect associated with minocycline as either an add-on therapy or monotherapy, but conclusions that can be drawn are limited by the heterogeneity of the individual studies (Rosenblat Reference Rosenblat and McIntyre2018). Most recently, a 2021 RCT investigated whether baseline inflammatory status (as measured by high-sensitivity CRP, hsCRP) could influence the response to minocycline augmentation in individuals with TRD (Nettis Reference Nettis, Lombardo and Hastings2021). Stratified using a cut-off of hsCRP <3 mg/L (hsCRP−) or ≥3 mg/L (hsCRP+), hsCRP+/minocycline patients showed a significantly greater improvement in HRSD scores following augmentation compared with hsCRP+/placebo, hsCRP−/minocycline and hsCRP−/placebo patients.
Interestingly, the beneficial effect of minocycline did not correlate with reductions in peripheral hsCRP or interleukin 6 (IL-6), suggesting an antidepressant mechanism beyond improving peripheral inflammation. The study's authors hypothesise that this could be linked to minocycline's ability to suppress microglial activation, thereby reducing central inflammation. There are also data to suggest that minocycline may act centrally via an NMDA receptor-dependent mechanism: cultures of murine cortical neurons treated with minocycline are protected from excitotoxicity induced by extracellular glutamate (Lu Reference Lu, Yang and Chen2021), thereby linking this drug with the glutamate hypothesis of depression.
It is important to note that not all people with depression will have inflammation as a cause or contributor to their symptoms; rather, there are likely subgroups of individuals with ‘inflamed depression’, who have elevated levels of inflammatory cytokines and/or leukocyte subsets and may therefore be potentially responsive to anti-inflammatory therapy. Even within this group, individuals will differ depending on the aetiology of their inflammatory response and the resulting profile of humoral and cellular immune responses. This has implications for the treatment of ‘inflamed depression’, suggesting that a one-size-fits-all approach may not be adequate.
Conclusions
In this article we have highlighted several novel psychopharmacological developments in the treatment of MDD and post-partum depression, focusing on (a) agents targeting the modulatory monoamines such as vortioxetine and psilocybin; (b) agents targeting the balance between excitation and inhibition, such as S-ketamine and brexanolone; and (c) agents targeting inflammation, such as the antibiotic minocycline. The landscape of novel pharmacological agents for the treatment of depression is vast and varied. Further work is needed to understand which groups of patients will best respond to which antidepressants and how best to integrate novel antidepressants into current treatment pathways while ensuring equitable access to treatment.
Data availability
Data availability is not applicable to this article as no new data were created or analysed in this study.
Author contributions
Both authors contributed equally to this manuscript, including all elements of conceptualisation, writing, editing and responding to reviewer comments.
Funding
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Declaration of interest
A.H.Y. has delivered paid lectures or sat on the advisory boards for the following companies with drugs used in affective and related disorders: AstraZeneca, Eli Lilly, Lundbeck, Sunovion, Servier, LivaNova, Janssen, Allegan, Bionomics, Sumitomo Dainippon Pharma. He is a consultant to LivaNova and Johnson & Johnson; has received honoraria for attending advisory boards and presenting talks at meetings organised by LivaNova; and is Principal Investigator on the following: the Restore-Life VNS registry study funded by LivaNova; ‘An open-label, long-term, safety and efficacy study of intranasal esketamine in treatment-resistant depression’ (ESKETINTRD3004); ‘The effects of psilocybin on cognitive function in healthy participants’ (IRAS ID 246135); and ‘The safety and efficacy of psilocybin in participants with treatment-resistant depression (P-TRD)’ (NCT03775200). He receives or has received grant funding from: NIMH (USA); CIHR (Canada); NARSAD (USA); Stanley Medical Research Institute (USA); MRC (UK); Wellcome Trust (UK); Royal College of Physicians (Edin); BMA (UK); UBC-VGH Foundation (Canada); WEDC (Canada); CCS Depression Research Fund (Canada); MSFHR (Canada); NIHR (UK); and Janssen (UK).
MCQs
Select the single best option for each question stem (Box 1 explores the MCQs in more detail)
1 Which of the following is true regarding brain neurotransmitter systems implicated in major depressive disorder (MDD)?
a Cell bodies of serotonergic neurons lie in the locus ceruleus of the brainstem
b Serotonin neurons project to wide areas of the neocortex
c Monoamines such as serotonin, noradrenaline and dopamine are likely to contribute equally to symptoms of MDD
d Most first-line antidepressants target dopaminergic neurotransmission
e Glutamatergic neurotransmission is second in prevalence only to GABAergic neurotransmission.
2 Which one of the following does not represent one of the actions of vortioxetine in serotonergic transmission?
a Inhibition of the serotonin reuptake transporter (SERT)
b Agonism at 5-HT2A receptors
c Antagonism at 5-HT1D receptors
d Antagonism at 5-HT3 receptors
e Agonism at 5-HT1A receptors.
3 Which statement about this psychedelics is true?
a Psychedelic drugs universally act as 5-HT1A agonists
b Psychedelic drugs seem to inhibit cortical plasticity
c Psychedelic drugs predominantly act on the cholinergic system
d The effect of unmasking in psychedelic drug trials may be mitigated using an active placebo
e Like SSRIs, the antidepressant effects of psychedelic drugs seem to build gradually over 1–4 weeks.
4 Which one of the following is true regarding ketamine's mechanism of action?
a S-ketamine and R-ketamine have approximately equal affinity for the NMDA receptor
b The sustained antidepressant effects of ketamine are unrelated to effects on neural plasticity
c The antidepressant effects of ketamine may depend on plastic changes in prefrontal–striatal circuitry
d The acute effect of ketamine seems to involve inhibition of glutamate surges in the prefrontal cortex
e Metabolites of ketamine do not seem to play a role in its antidepressant efficacy.
5 Which one of the following statements is true regarding the contribution of inflammation to major depressive disorder (MDD)?
a It is highly likely every case of MDD is contributed to by excess inflammation
b The association between inflammation and depression can be explained by the severity of physical symptoms alone
c Microglia release antibodies in response to inflammatory stimuli
d Cytokines can cross the blood–brain barrier via saturable transporter mechanisms
e It remains unclear whether there is a correlative link between inflammation and depression.
Exploration of the MCQs
MCQ1
Option b is correct – serotonergic cells in the raphe nucleus of the brainstem project to many cortical areas (and subcortical areas). Option a is incorrect – the locus ceruleus contains noradrenergic neurons. Option c is incorrect, as research suggests that different monoamines may contribute differently to depressive symptoms. Most first-line antidepressants target the serotonergic system, so d is incorrect. Finally, e is incorrect as glutamatergic transmission is the most common type of neurotransmission in the brain.
MCQ2
Option b is correct – vortioxetine is not a 5-HT2A agonist: this is the mechanism of action of psychedelic drugs and therefore this is the only incorrect option. Vortioxetine inhibits SERT (a), is an antagonist at 5-HT1D receptors (b) and 5-HT3 receptors (d) and acts as an agonist at 5-HT1A receptors (e).
MCQ3
Option d is correct – the effect of unmasking in psychedelic drug trials may be mitigated (but not completely avoided) by using active placebos. Psychedelic drugs act as 5-HT2A agonists, so a is incorrect (although some psychedelic drugs do have an additional action on 5-HT1A receptors), and through this mechanism they promote cortical plasticity, so b is incorrect. Psychedelic drugs predominantly act on the serotonergic system; therefore, c is incorrect. Option e is incorrect as psychedelic drugs seem to have rapid antidepressant effects and do not take 1–4 weeks to have their effects.
MCQ4
Option c is correct – studies such as Mkrtichian et al (2021) show that ketamine modulates prefrontal–striatal connectivity. Option a is incorrect because S-ketamine has a higher affinity for the NMDA receptor than R-ketamine. Option b is incorrect because neural plasticity seems to be a key mechanism underlying ketamine's relatively sustained antidepressant effects. Option d is incorrect because ketamine, acutely, seems to stimulate a glutamate surge through NMDA receptor inhibition on GABAergic interneurons. Finally, e is incorrect because (2R,6R)-HNK acts as an AMPA receptor agonist and seems to mediate, at least in part, ketamine's antidepressant effect.
MCQ5
Option d is correct – peripheral cytokines can cross the blood–brain barrier using transporters. Given that current evidence suggests that a subset of patients with depression show evidence of peripheral inflammation, a is incorrect. Option b is also incorrect because studies controlling for symptom severity still show an association between peripheral inflammation and depression. Microglia are phagocytes and do not release antibodies; therefore, c is incorrect. Option e is incorrect because a correlative link between peripheral inflammation and depression is well established.
MCQ answers
1 b 2 b 3 d 4 c 5 d






 Open access
Open access
eLetters
No eLetters have been published for this article.