



Butyrate as a friend
The extensive evidence for the importance of butyrate for the health of the colonic epithelium led to exploration of the hypothesis that failure of butyrate uptake or metabolism might possibly be a pathogenic mechanism in ulcerative colitis(Reference Roediger1). Butyrate is a short chain (short implies less than 6 carbon atoms, 4 in case of butyrate) fatty acid (SFA) generated in the colon mainly by anaerobic bacterial fermentation of dietary fibre. There is an extensive literature (> 120 000 entries for ‘butyrate’ on PubMed), the overwhelming majority of which emphasises its beneficial effects for health. It is the preferred carbon and energy source for the colonic epithelium, encourages differentiation rather than proliferation and induces apoptosis in colorectal cancer cell lines, so is credited with cancer-preventing properties(Reference Fung, Cosgrove and Lockett2,Reference Donohoe, Collins and Wali3) . Cellular effects may differ though, even being reversed, between cancerous and normal cells(Reference Mariadason, Velcich and Wilson4). Butyrate leads to increased synthesis of tight junction proteins and consequent enhanced mucosal barrier function(Reference Hodgkinson, El Abbar and Dobranowski5,Reference Sun, Chen and Zang6) . SFA including butyrate also have potentially beneficial immunological effects. Interaction with G protein-coupled receptors on the epithelial apical membrane induces activation of the NOD-, LRR-, and pyrin domain-containing protein 3 inflammasome with downstream induction of IL-18 relevant to defence against pathogens, induction of the antimicrobial and phagocytic function of macrophages, inhibition of the transcription of pro-inflammatory cytokines including IL-8, IL-12 and tumour necrosis factor, and an increased ratio of T helper 1 (TH1) to TH17 cells(Reference Gibson and Rosella7–Reference Mann, Lam and Uhlig10). Peroxisome proliferation-activated receptorγ activity is increased resulting in inhibition of NF-kappaB activity(Reference Hodgkinson, El Abbar and Dobranowski5). The immunological effects of SFA are probably considerably contributed to by propionate(Reference Smith, Howitt and Panikov11,Reference Arpaia, Campbell and Fan12) which is much less metabolised by the epithelium than butyrate. Portal vein blood sampled post-mortem showed a mean butyrate concentration of 88 μM with a molar ratio 4:1 of propionate to butyrate(Reference Cummings, Pomare and Branch13). Tissue concentrations of butyrate within the colonic mucosa have not been directly measured. Effects of butyrate on immunological cells, e.g. Treg cells, in vitro have been demonstrated at 64 μM and above(Reference Arpaia, Campbell and Fan12), so a direct effect on immune cells within the mucosa mediated by butyrate, and perhaps more particularly by propionate, is likely given their portal vein concentrations. Plasma concentrations of butyrate are very low, typically around 2–3 μM(Reference Gill, Muir and Gibson14), at least one thousand fold lower than the ≤ 2 mM most likely relevant to the colonic epithelium or the c20 mM typically present in the healthy colonic lumen(Reference Cummings, Pomare and Branch13,Reference Duncan, Belenguer and Holtrop15) .
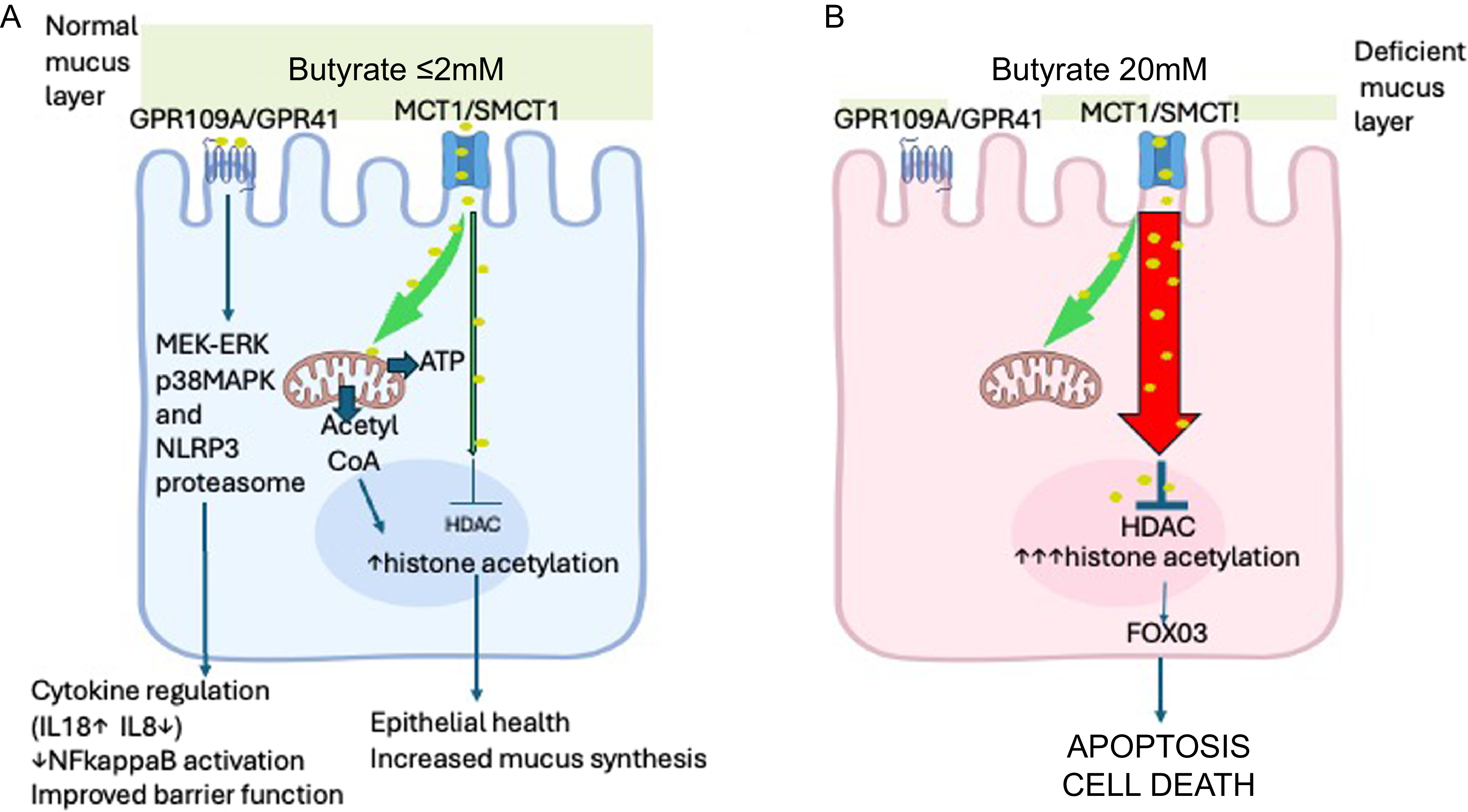
The effects of butyrate on epithelial cells are chiefly mediated either by activation of G protein-coupled receptors including GPR41 and GPR109A on the apical cell membrane or by uptake into the cell and subsequent inhibition of nuclear histone deacetylase which is thought to be central to its cancer-preventing properties(Reference Koh, De Vadder and Kovatcheva-Datchary9). The consequent increase in histone acetylation is also thought to be responsible for the apoptosis and increased cell death seen at higher concentrations of butyrate(Reference Donohoe, Collins and Wali3). Butyrate uptake into the cell is primarily via the proton-coupled monocarboxylate transporter 1 (MCT1) or via the sodium-coupled monocarboxylate transporter 1 (SMCT1). MCT1 is mainly expressed in the colon, with higher levels in the distal colon and in the upper part of crypts; SMCT1 also shows highest expression in the distal colon but is also expressed in the proximal colon and ileum(Reference Gonçalves and Martel16). Fermentation of fibre yields the SFA acetate (C2), propionate(C3) and butyrate(C4) at a ratio of about 60:25:15(Reference Stumpff17). All three SFA are internalised via the MCT1 but butyrate is then extensively metabolised by the epithelial cell whereas acetate and propionate pass through into the portal venous system largely unmetabolised and have much less impact on histone acetylation or cytotoxicity(Reference Sealy and Chalkley18–Reference Waldecker, Kautenburger and Daumann20). Maximal oxidation of butyrate has been reported for human colonocytes in culture at 1 mM butyrate(Reference Clausen and Mortensen21) and for colon cancer cells at 2 mM butyrate(Reference Andriamihaja, Chaumontet and Tome22), and this may account for much of the toxicity of higher extracellular concentrations since the intracellular concentration will rapidly rise once the metabolic capacity of the cell is exceeded(Reference Andriamihaja, Chaumontet and Tome22). More butyrate will then pass into the nucleus leading to greater inactivation of histone deacetylase and consequent histone hyperacetylation(Reference Donohoe, Collins and Wali3). At low butyrate concentrations (< 2 mM), its uptake and metabolism within mitochondria leads via the tricarboxylic acid cycle to the release of citrate which is converted in the cytosol by adenosine triphosphate citrate lyase to acetyl-CoA. This can then enter the nucleus and facilitate relatively low-level histone acetylation that is substantially independent of histone deacetylase inhibition. This results in activation of fewer genes than seen with higher butyrate concentrations, e.g. in one experiment performed on HCT116 colon cancer cells, 1416 genes were activated at 0·5 mM butyrate, 2827 at 2 mM and 4241 at 5 mM; moreover, not all genes activated at lower concentrations were also activated at higher concentrations(Reference Donohoe, Collins and Wali3). Histone hyperacetylation can be found in cell lines treated in vitro with butyrate at 3 mM without obvious short-term cytotoxicity(Reference Sealy and Chalkley18), but higher concentrations around 5–6 mM or greater are associated with apoptosis and reduced cell viability(Reference Hodgkinson, El Abbar and Dobranowski5,Reference Sealy and Chalkley18,Reference Candido, Reeves and Davie23) . Unsurprisingly, this is associated with impairment of barrier function(Reference Mariadason, Kilias and Catto-Smith24,Reference Huang, Li and Zhu25) . This toxicity of higher concentrations of butyrate for the colonic epithelium has often been overlooked probably because many studies have only evaluated the effects of butyrate at a single concentration. (Figure 1)
(A) Healthy epithelium. When the mucus layer is intact, butyrate concentration at the apical epithelial surface can be assumed to be non-toxic (≤ 2 mM). Butyrate effects are mediated either by interaction with G protein-coupled membrane receptors (GPR109A or GPR41) or by internalisation through MCT1 or SMCT1 transporters. Downstream signalling mechanisms by G protein-coupled receptors are incompletely known but end results include anti-inflammatory effects and improved barrier function. Butyrate uptake into the cell constitutes the major energy source for the cell. Entry of butyrate into the TCA cycle results in the generation of citrate which is converted in the cytosol into acetyl-CoA. This can pass into the nucleus and facilitate histone acetylation. This upregulates various genes to beneficial effects including mucus synthesis. (B) Inflamed epithelium with deficient mucus layer. Here the normal faecal butyrate concentration (c20 mM) will be in contact with the apical surface of the epithelium. Maximal butyrate oxidation occurs at 1–2 mM above which large amounts of butyrate will enter the nucleus causing a major inhibition of histone deacetylase and consequential marked increase in histone acetylation that leads to activation of many genes (> 2000) and apoptotic cell death. Its mediation via FOX03 has to date only been demonstrated in colon crypt stem cells. Some components of this figure were downloaded under licence from BioRender: https://BioRender.com/565kb82.

Butyrate as a foe – dose–response and epithelial toxicity
The dose–response of colon epithelial cells to butyrate is also not linear when other physiological endpoints are studied. We first became aware of this when studying the effect of butyrate on mucus synthesis by colon mucosal explants in short-term in vitro culture. Butyrate in the culture medium had an optimal (approximately four-fold) effect of stimulating mucus synthesis at a concentration of 0·1 mM, falling to insignificant by 10 mM and with still higher concentrations of butyrate causing obvious histological damage(Reference Finnie, Dwarakanath and Taylor26). Earlier studies of the effects of butyrate on colon cancer epithelial cells in culture had already shown that butyrate concentrations greater than 2 mM(Reference Herz, Schermer and Halwer27) or 3 mM(Reference Sealy and Chalkley18) markedly inhibited cell proliferation. This has been a consistent finding in subsequent in vitro studies. Moreover, butyrate at concentrations above 2 mM induces significant loss of viability in human colorectal cancer cell lines(Reference Morita, Tsao and Kim28,Reference Li, He and Chen29) and in rat colonocytes, an effect that is exacerbated in the presence of tumour necrosis factorα(Reference Vancamelbeke, Laeremans and Vanhove30,Reference Souders, Aristizabal-Henao and Patuel31) . It has been reported recently that human colon organoids are able to tolerate butyrate concentrations up to 20 mM without obvious cytotoxicity or increased lactate dehydrogenase release(Reference Parada-Venegas, De la Fuente López and Dubois-Camacho32). Arguably, this might be due to their ability to synthesise mucus(Reference Saxena, Mitchell and Bogdon33) which, if closely mimicking that of the healthy colon, should generate a closely adherent mucus layer(Reference Johansson, Larsson and Hansson34).
Colon crypt stem cells are particularly sensitive to butyrate, showing marked suppression of proliferation with butyrate at 1 mM which becomes irreversible at concentrations 3–10 mM in association with cleavage of caspase 3 as evidence of apoptosis(Reference Kaiko, Ryu and Koues35). These effects were not seen with propionate or acetate. It is thought that the stem cells, which reside at the base of the colon crypts, are protected from excess butyrate by the avid butyrate metabolism of epithelial cells further up the crypt towards the lumen. The butyrate effects were shown to be independent of stimulation of G protein-coupled receptors but dependent on increased histone acetylation which relaxes chromatin structure allowing increased promoter access and consequent expression of many genes. Although this was found to impact on transcription of more than 2400 genes, the effect on proliferation was shown to be primarily mediated by increased activation and promoter binding of the forkhead box O3 transcription factor. In support of this, treatment with a FoxO inhibitor enema of dextran sulphate-induced colitis in mice was found to reverse butyrate-induced changes in epithelial proliferation.
The potential harmful impact of luminal butyrate on the ileal mucosa is relevant in the context of the ileo-anal pouch that is commonly constructed following colectomy if medical therapy for ulcerative colitis has failed. The ileal mucosa has no continuous adherent mucus barrier, indeed if it did adequate absorption of nutrients would likely be impossible. It follows that bacterial contamination, inevitable in the pouch, yields substantial butyrate levels(Reference Nordgaard-Andersen, Clausen and Mortensen36) that are likely to be toxic to the epithelium. The puzzle then is not why the pouch of patients with ulcerative colitis is prone to pouchitis but why the pouch of patients undergoing colectomy for familial adenomatous polyposis is relatively resistant. There are some reports that the familial adenomatous polyposis epithelium responds differently to butyrate(Reference Tonelli, Dolara and Batignani37,Reference Bordonaro, Lazarova and Augenlicht38), and further investigation should be considered.
Butyrate and ulcerative colitis – metabolic studies
Once the importance of butyrate to colon epithelial health had been recognised, it seemed a plausible hypothesis that ulcerative colitis might be the result of defective butyrate metabolism. Initial experiments by Roediger showed that isolated colon epithelial cells from quiescent ulcerative colitis biopsies had reduced metabolism of 10 mM butyrate but normal oxygen consumption and cells isolated from active colitis samples showed markedly reduced butyrate metabolism and oxygen consumption(Reference Roediger1). This was supported by a study showing reduced metabolism of 5 mM butyrate by quiescent or mildly active ulcerative colitis mucosal biopsies(Reference Chapman, Grahn and Boyle39). Conversely, studies by our group performed on cultured colonic and ileal biopsies from patients with inactive colitis showed no difference from controls in the oxidation of butyrate 1 mM performed in the presence of 11 mM glucose(Reference Finnie, Taylor and Rhodes40). Other studies reported no defects in the activity of enzymes involved in butyrate oxidation(Reference Allan, Winter and Light41) and no difference between ulcerative colitis patients and controls in metabolism of 13C-labelled butyrate given by enema(Reference Simpson, Chapman and Dawson42). A later study showed a moderate (c50 %) impairment in oxidation of 1 mM butyrate in quiescent colitis but a much more severe defect (c90 %) in biopsies from severe colitis(Reference De Preter, Arijs and Windey43). This was partly accounted for by a reduction in butyrate uptake in keeping with the previously reported reduced expression of MCT1 in active colitis(Reference Thibault, De Coppet and Daly44,Reference Erdmann, Bruckmueller and Martin45) . A similar down-regulation of MCT1 is seen in early-stage colon cancer(Reference Lambert, Wood and Ellis46). Inhibition of MCT1 expression has been shown to block butyrate-induced cell cycle arrest(Reference Cuff, Dyer and Jones47), so it is likely to mitigate against mucosal damage from butyrate. A recent study has shown a normal response to transcriptional regulation by butyrate in ulcerative colitis-derived organoids but with impaired response in the presence of tumour necrosis factorα(Reference Ferrer-Picón, Dotti and Corraliza48). It seems reasonable to conclude that any defect in butyrate uptake or metabolism is modest when the disease is inactive but may become more marked with active disease(Reference Colgan, Wang and Hall49).
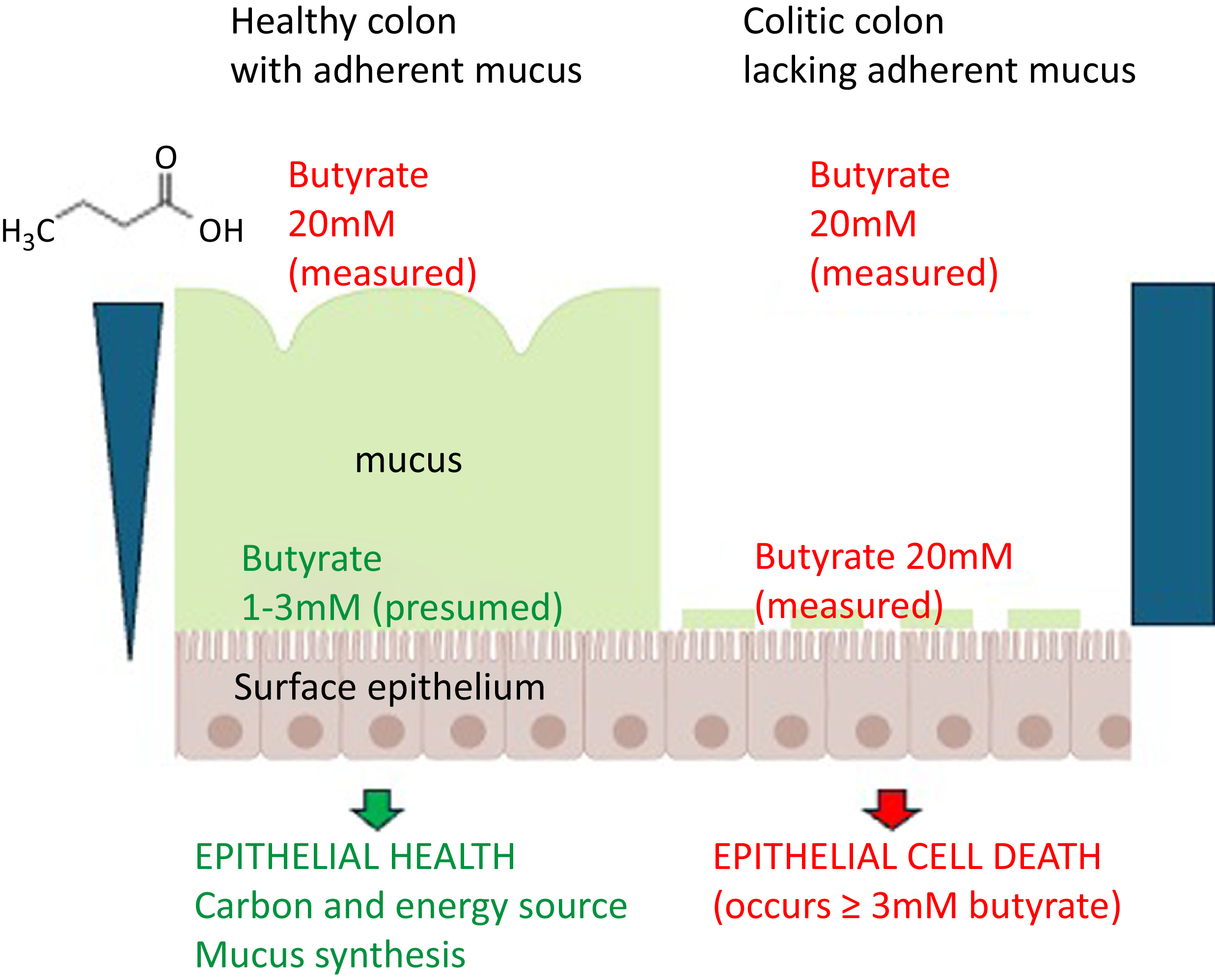
Faecal butyrate concentration has been reported to be increased in ulcerative colitis with levels up to 35 mM in active disease(Reference Roediger, Heyworth and Willoughby50) although other studies have reported reduced concentrations, e.g. 11 mM(Reference Hallert, Björck and Nyman51), but increased cytotoxicity of faecal water(Reference Poppe, Boesmans and Vieira-Silva52). The colon, unlike the small intestine, has a continuous adherent mucus layer but this is often depleted or completely absent in active ulcerative colitis(Reference Pullan, Thomas and Rhodes53,Reference van der Post, Jabbar and Birchenough54) . Although butyrate concentrations through the healthy mucus layer have not been directly measured, it is assumed that a concentration gradient must be present and presumably ensures that butyrate concentration at the mucosal surface approximates to the c2–3 mM or less that is the maximal concentration compatible with epithelial cell health in vitro (Reference Donohoe, Collins and Wali3,Reference Sealy and Chalkley18) . The damage to the mucus barrier found in severe ulcerative colitis allows penetration of bacteria through to the mucosal surface(Reference Johansson, Gustafsson and Holmén-Larsson55) which is itself likely to aggravate inflammation but will also allow the much higher luminal concentrations of butyrate to interact with the epithelium.
There is some experimental evidence to support a harmful effect of access to the mucosa by higher butyrate concentrations. In an IL-10 receptor antibody model of colitis, a diet supplemented with the soluble fibre inulin, which approximately tripled the faecal butyrate concentration, exacerbated the colitis whereas another soluble fibre, citrus pectin, which was only associated with a modest butyrate increase, was protective(Reference Singh, Yeoh and Walker56). Pectins, such as plantain (banana) pectin, can have marked anti-inflammatory effects on the colonic epithelium, including reduced IL-8 response to clostridial toxins(Reference Simpson, Roberts and Thompson57), and this might contribute to the very different response. Not all dietary fibres are equal(Reference Rhodes58).
Butyrate and ulcerative colitis – therapeutic studies
A recent systematic review that included eight randomised trials failed to find convincing evidence of a therapeutic effect of butyrate enemas in ulcerative colitis(Reference Jamka, Kokot and Kaczmarek59). This confirmed the conclusion of a previous narrative review that included five randomised trials of butyrate enemas in active ulcerative colitis, all negative for their primary endpoint, plus one negative trial of oral butyrate supplementation(Reference Gill, van Zelm and Muir60). It can be argued that these trials have not generally paid much attention to the likely pharmacokinetics of intraluminal butyrate. Oral butyrate supplementation is rapidly taken up by the small intestinal epithelium, and butyrate administered by enema also has a very short half-life as a consequence of rapid colonic mucosal uptake. Modelling suggests that sustained release formulations(Reference Korsten, Smits and Garssen61) or other forms of targeted delivery such as butyrate-functionalised polymethacrylates(Reference Fan, Zhang and Gao62) or other novel delivery technologies(Reference Zhang, Pan and Guo63) would be needed to ensure maintenance of therapeutic concentrations at the colonic mucosa. Such an approach has shown some efficacy in experimental colitis(Reference Fan, Zhang and Gao62,Reference Sun, Li and Li64) but has not been studied in human colitis. Dietary supplementation with prebiotics, usually intended to increase colonic SFA content, has been unconvincing. One small study of the fructooligosaccharide kestose showed possible benefit but meta-analysis showed no significant benefit from other prebiotics, including inulin, either in induction or maintenance of remission in ulcerative colitis(Reference Limketkai, Godoy-Brewer and Shah65). Similarly, a systematic review of trials of dietary fibre supplementation has been largely negative, with weak evidence for benefit from ispaghula in maintenance of remission and for germinated barley in active colitis, but in both cases based on single studies(Reference Wedlake, Slack and Andreyev66).
Potential therapies based on lowering or blocking the toxic effects of butyrate on the mucus-denuded epithelium
Although an early study of bowel rest and intravenous nutrition showed no benefit in severe ulcerative colitis(Reference Dickinson, Ashton and Axon67), a recent trial of total enteral nutrition has suggested possible benefit(Reference Sahu, Kedia and Vuyyuru68). This is despite, or perhaps because of, the inevitable fall in faecal butyrate as a consequence of the absence of dietary fibre. Total enteral nutrition in Crohn’s disease, whilst very effective, has been shown to be associated with an approximate 50 % fall in faecal butyrate concentration(Reference Kerbiriou, Dickson and Nichols69).
Hop beta acids instilled in drinking water have been shown to reduce generation of SFA, including butyrate, in mice(Reference Zou, Chassaing and Singh70) and were found to substantially abrogate the inulin-related worsening of colitis in IL-10-inhibited mice(Reference Singh, Yeoh and Walker56). Hop beta acids, also known as lupulones, are approved by the FDA as ‘generally recognised as safe’ and are used in food products such as processed meats, so could be potential therapeutics(Reference Li, Dalabasmaz and Gensberger-Reigl71). They have an antibacterial effect and are highly hydrophobic so likely to be minimally absorbed.
It should be noted that metronidazole which substantially blocks bacterial fermentation of fibre was also effective in the same experimental model(Reference Singh, Yeoh and Walker56), yet oral metronidazole as sole therapy was inferior to sulphasalazine in acute ulcerative colitis(Reference Gilat, Suissa and Leichtman72). At first sight, this looks like a major weakness in the hypothesis that butyrate is part of the problem in active colitis; however, studies in healthy adult humans have shown that metronidazole taken orally is not detectable in faeces, being very efficiently absorbed in the upper digestive tract with almost 100 % bioavailability(Reference Lamp, Freeman and Klutman73) and has no significant impact on faecal SFA concentration(Reference Høverstad, Carlstedt-Duke and Lingaas74). The situation is different in people with diarrhoea due to Clostridioides difficile when therapeutically effective faecal concentrations of metronidazole are achieved after oral dosing(Reference Bolton and Culshaw75), and faecal excretion of up to 15 % of dose has been reported after intravenous use(76).
Simply binding butyrate in the gut lumen to prevent its uptake via MCT1 could be another approach but there do not seem to be any currently available pharmaceuticals which do this. The anion-exchange resin, cholestyramine, perhaps unexpectedly, has been shown to increase faecal butyrate levels, particularly in the caecum in mice(Reference Nishida, Horinouchi and Higashimura77) and dogs(Reference Alexander, Guard and Suchodolski78), probably because unabsorbed fat can be metabolised to SFA.
An alternative therapeutic approach could be to blunt the deleterious effect of high butyrate concentrations on the colonic epithelium. There is evidence that corticosteroids and mesalazine, the mainstay of therapy for ulcerative colitis over many years, have such effects. Butyrate, propionate and acetate, all tested at 8 mM, were found to be toxic to the colorectal cancer cell line MCE301, probably causing death of this cell line by necrosis rather than apoptosis, and the toxicity of butyrate was substantially inhibited by therapeutically relevant concentrations of prednisolone and mesalazine(Reference Matsumoto, Hayasaki and Nishimura79). The mechanism for these effects is not known.
A plausible approach to treating active colitis might be to block the butyrate transporter MCT1. This is being trialled in cancer therapy, partly on the basis that MCT1 also facilitates export of lactate and thus encourages cancer cell growth(Reference Bola, Chadwick and Michopoulos80). Various inhibitors are being investigated including drugs currently used against other targets such as syrosingopine, used to treat hypertension, and natural products including the flavonoid quercetin(Reference Wang and Morris81,Reference Faghihkhorasani, Moosavi and Rasool Riyadh Abdulwahid82) . MCT1 activity, although reduced in early-stage colorectal cancer, can also be increased in metastatic disease, and MCT1 inhibition has been shown to increase apoptosis in cancer cells, so its effects in the postulated butyrate-toxicity colitis model could be unpredictable. Encouraging preliminary studies have however shown a possible therapeutic effect of quercetin in experimental colitis(Reference Sánchez de Medina, Gálvez and Romero83–Reference Jiang, Yan and Qin85). The reported impacts of quercetin on butyrate effects in vitro have been rather inconsistent, and other mechanisms have also been suggested for its possible therapeutic effect. Goncalves and colleagues have shown that quercetin modestly reduced butyrate uptake by Caco-2 colon epithelial cells without any impact on butyrate-induced mortality(Reference Gonçalves, Araújo and Pinho86) and also modestly reduced butyrate uptake by rat small intestinal epithelial cells-6 without any effect on MCT1 expression(Reference Gonçalves, Araújo and Martel87). An earlier study had shown a marked impact of quercetin and other flavonoids on reducing uptake of 14C benzoic acid by Caco-2 cells as a marker of MCT1 function, possibly by competitive inhibition of binding to MCT1(Reference Shim, Cheon and Kang88). More recent studies have tended to emphasise a contrary impact of quercetin on enhancing butyrate-induced cytotoxicity in cancer cells(Reference Betts, Deveci Ozkan and Yuksel89). However, more encouragingly, a study based on the UK Biobank has shown a reduced risk for ulcerative colitis (adjusted hazard ratio 0·69, P for trend 0·001) amongst people in the highest v. lowest quintile for quercetin intake(Reference Lu, Dan and Sun90). Quercetin is mainly obtained from fruit and vegetables including tomatoes, onions, broccoli and apples.
A more targeted approach could be to inhibit the forkhead box O3 transcription factor, shown by Kaiko et al. to mediate butyrate-induced apoptosis in cell lines(Reference Kaiko, Ryu and Koues35). This is supported by their demonstration of beneficial effects of a FoxO inhibitor enema in mice with dextran sulphate colitis. However, most of the literature suggests that activation of forkhead box O3 and its nuclear localisation has an anti-inflammatory effect(Reference Shimazu, Hirschey and Newman91) and that enhancement of this effect by saxagliptin(Reference Elmorsy, Youssef and Abdel-Hamed92) or linagliptin(Reference Alshahrani, Saber and Alruwaili93) ameliorates dextran sulphate colitis.
In conclusion, although the uptake and metabolism of butyrate generated by fermentation of dietary fibre is important for maintenance of colonic epithelial health, in situations such as severe ulcerative colitis when the normal adherent colonic mucus layer is lost or fragmented, faecal butyrate concentrations are likely to be highly toxic to the epithelium. (Figure 2). This also applies to the ileal pouch since ileal mucosa lacks an adherent mucus layer even in health. The current ‘best bet’ approaches to reducing the toxic effect of high-concentration butyrate in active ulcerative colitis are therapies already in use. These include corticosteroids and mesalazine, which reduce the cytotoxic effect of butyrate on the epithelium, and enteral nutrition which reduces the luminal butyrate concentration. Dietary supplementation with high quercetin foods or hop beta acids also deserves investigation. Research to gain a better understanding of how corticosteroids and mesalazine protect the epithelium against high-concentration butyrate could identify novel therapeutic targets. Meanwhile, investigators need to have a clear understanding of the dose–response of the colonic epithelium to butyrate and the implications of the denuded mucus barrier in active colitis. More butyrate is not always better.
Overview showing how the healthy adherent mucus layer, present in the colon but not in the small intestine, generates resistance to butyrate diffusion resulting in a concentration gradient. Butyrate at the relatively low concentration in contact with the epithelium, certainly < 3 mM but possibly substantially lower, has important beneficial effects on the mucosa. When the adherent mucus layer is damaged or missing, as in active ulcerative colitis, the luminal butyrate concentration (c20 mM) now in contact with the epithelium will inevitably be highly toxic. Some components of this figure were downloaded under licence from BioRender: https://BioRender.com/565kb82.

Acknowledgements
Grateful thanks to team members, collaborators and other friends in inflammatory bowel disease research, nutrition and glycobiology for stimulating conversations over the years.
No financial support.
J. R. is sole author. AI was not used in the writing of this article.
With the University of Liverpool and Provexis (IBD) Ltd, the author held patents for the treatment of inflammatory bowel disease and antibiotic-associated diarrhoea with soluble plantain fibre but these were allowed to lapse in 2022. No other conflicts.

 Open access
Open access