Refine search
Actions for selected content:
48554 results in Computer Science
Matroid Duality from Topological Duality in Surfaces of Nonnegative Euler Characteristic
-
- Journal:
- Combinatorics, Probability and Computing / Volume 11 / Issue 5 / September 2002
- Published online by Cambridge University Press:
- 09 October 2002, pp. 515-528
-
- Article
- Export citation
Integer Partitions, Tilingsof 2D-gons and Lattices
-
- Journal:
- RAIRO - Theoretical Informatics and Applications / Volume 36 / Issue 4 / October 2002
- Published online by Cambridge University Press:
- 15 February 2003, pp. 389-399
- Print publication:
- October 2002
-
- Article
- Export citation
On Shuffle Ideals
-
- Journal:
- RAIRO - Theoretical Informatics and Applications / Volume 36 / Issue 4 / October 2002
- Published online by Cambridge University Press:
- 15 February 2003, pp. 359-384
- Print publication:
- October 2002
-
- Article
- Export citation
On a characteristic propertyof ARNOUX–RAUZY sequences
-
- Journal:
- RAIRO - Theoretical Informatics and Applications / Volume 36 / Issue 4 / October 2002
- Published online by Cambridge University Press:
- 15 February 2003, pp. 385-388
- Print publication:
- October 2002
-
- Article
- Export citation
About the decision of reachability for register machines
-
- Journal:
- RAIRO - Theoretical Informatics and Applications / Volume 36 / Issue 4 / October 2002
- Published online by Cambridge University Press:
- 15 February 2003, pp. 341-358
- Print publication:
- October 2002
-
- Article
- Export citation
-
We study the decidability of the following problem: given p affine functions ƒ1,...,ƒp over $\mathbb{N}^k$
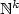 and two vectors $v_1, v_2\in\mathbb{N}^k$
and two vectors $v_1, v_2\in\mathbb{N}^k$ 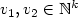 ,is v 2 reachable from v 1 by successive iterations of ƒ1,...,ƒp(in this given order)? We show that this question is decidable for p = 1, 2 and undecidable forsome fixed p.
,is v 2 reachable from v 1 by successive iterations of ƒ1,...,ƒp(in this given order)? We show that this question is decidable for p = 1, 2 and undecidable forsome fixed p.
An Upper Bound on the Space Complexityof Random Formulae in Resolution
-
- Journal:
- RAIRO - Theoretical Informatics and Applications / Volume 36 / Issue 4 / October 2002
- Published online by Cambridge University Press:
- 15 February 2003, pp. 329-339
- Print publication:
- October 2002
-
- Article
- Export citation
-
We prove that, with high probability, the space complexity of refutinga random unsatisfiable Boolean formula in k-CNF on nvariables and m = Δn clauses is $O\left(n \cdot \Delta^{-\frac{1}{k-2}}\right)$
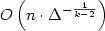 .
.
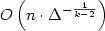
Preface
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp viii-xvi
-
- Chapter
- Export citation
Appendix A - Experimental protocols
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 177-184
-
- Chapter
- Export citation
Appendix D - CyberT: An online program for the statistical analysis of DNA array data
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 199-206
-
- Chapter
- Export citation
5 - Statistical analysis of array data: Inferring changes
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 53-72
-
- Chapter
- Export citation
Index
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 207-213
-
- Chapter
- Export citation
1 - A brief history of genomics
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 1-6
-
- Chapter
- Export citation
7 - The design, analysis, and interpretation of gene expression profiling experiments
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 97-134
-
- Chapter
- Export citation
3 - DNA array readout methods
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 17-28
-
- Chapter
- Export citation
8 - Systems biology
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 135-176
-
- Chapter
- Export citation
6 - Statistical analysis of array data: Dimensionality reduction, clustering, and regulatory regions
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 73-96
-
- Chapter
- Export citation
4 - Gene expression profiling experiments: Problems, pitfalls, and solutions
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 29-52
-
- Chapter
- Export citation
2 - DNA array formats
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 7-16
-
- Chapter
- Export citation
Appendix C - Internet resources
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 195-198
-
- Chapter
- Export citation
Appendix B - Mathematical complements
-
- Book:
- DNA Microarrays and Gene Expression
- Published online:
- 07 August 2009
- Print publication:
- 19 September 2002, pp 185-194
-
- Chapter
- Export citation
