Refine search
Actions for selected content:
1868 results in Physiology and biological physics

Origins of life: first came evolutionary dynamics
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 22 March 2023, e4
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
How sequence alterations enhance the stability and delay expansion of DNA triplet repeat domains
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 06 November 2023, e8
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Chasing collective variables using temporal data-driven strategies
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 06 January 2023, e2
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Mouth breathing, dry air, and low water permeation promote inflammation, and activate neural pathways, by osmotic stresses acting on airway lining mucus
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 14 February 2023, e3
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Energy landscapes and heat capacity signatures for peptides correlate with phase separation propensity
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 05 September 2023, e7
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Phase separation plays an important role in the formation of membraneless compartments within the cell and intrinsically disordered proteins with low-complexity sequences can drive this compartmentalisation. Various intermolecular forces, such as aromatic–aromatic and cation–aromatic interactions, promote phase separation. However, little is known about how the ability of proteins to phase separate under physiological conditions is encoded in their energy landscapes and this is the focus of the present investigation. Our results provide a first glimpse into how the energy landscapes of minimal peptides that contain
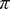 $ \pi $–
$ \pi $– $ \pi $ and cation–
$ \pi $ and cation– $ \pi $ interactions differ from the peptides that lack amino acids with such interactions. The peaks in the heat capacity (
$ \pi $ interactions differ from the peptides that lack amino acids with such interactions. The peaks in the heat capacity (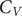 $ {C}_V $) as a function of temperature report on alternative low-lying conformations that differ significantly in terms of their enthalpic and entropic contributions. The
$ {C}_V $) as a function of temperature report on alternative low-lying conformations that differ significantly in terms of their enthalpic and entropic contributions. The 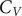 $ {C}_V $ analysis and subsequent quantification of frustration of the energy landscape suggest that the interactions that promote phase separation lead to features (peaks or inflection points) at low temperatures in
$ {C}_V $ analysis and subsequent quantification of frustration of the energy landscape suggest that the interactions that promote phase separation lead to features (peaks or inflection points) at low temperatures in 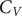 $ {C}_V $. More features may occur for peptides containing residues with better phase separation propensity and the energy landscape is more frustrated for such peptides. Overall, this work links the features in the underlying single-molecule potential energy landscapes to their collective phase separation behaviour and identifies quantities (
$ {C}_V $. More features may occur for peptides containing residues with better phase separation propensity and the energy landscape is more frustrated for such peptides. Overall, this work links the features in the underlying single-molecule potential energy landscapes to their collective phase separation behaviour and identifies quantities (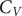 $ {C}_V $ and frustration metric) that can be utilised in soft material design.
$ {C}_V $ and frustration metric) that can be utilised in soft material design.


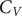

On the micelle formation of DNAJB6b
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 15 August 2023, e6
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Sulfur-mediated chalcogen versus hydrogen bonds in proteins: a see-saw effect in the conformational space
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 27 April 2023, e5
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
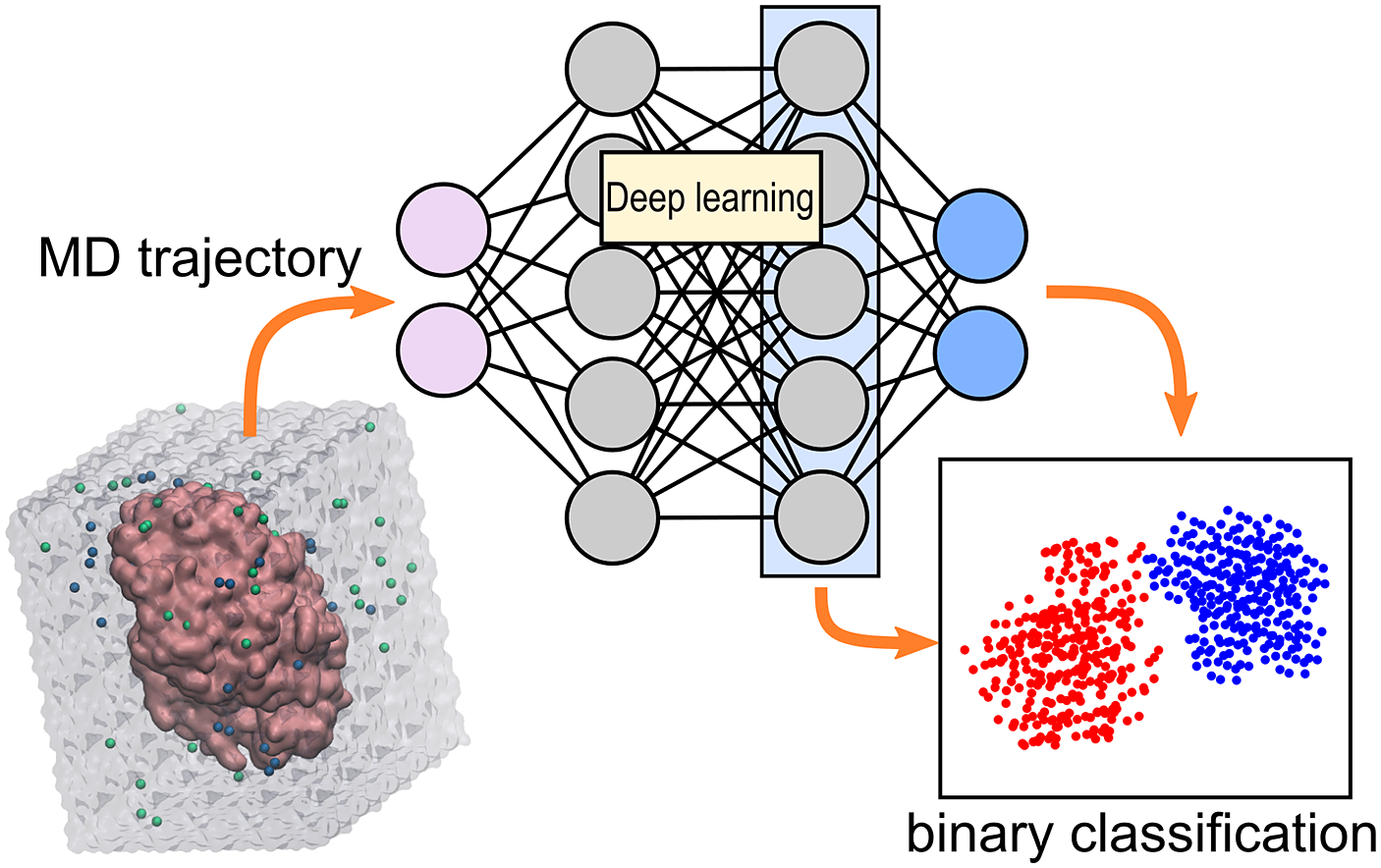
Computational prediction of ω-transaminase selectivity by deep learning analysis of molecular dynamics trajectories
-
- Journal:
- QRB Discovery / Volume 4 / 2023
- Published online by Cambridge University Press:
- 12 December 2022, e1
- Print publication:
- 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Preface
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp xix-xxii
-
- Chapter
- Export citation
Dedication
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp v-vi
-
- Chapter
- Export citation
Copyright page
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp iv-iv
-
- Chapter
- Export citation
Index
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 142-148
-
- Chapter
- Export citation
2 - One Molecule, Two Molecules, Red Molecules, Blue Molecules
- from Part I - Super-Resolution Microscopy and Molecular Imaging Techniques to Probe Biology
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 20-37
-
- Chapter
- Export citation
1 - Introduction on Single-Molecule Science
- from Part I - Super-Resolution Microscopy and Molecular Imaging Techniques to Probe Biology
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 3-19
-
- Chapter
- Export citation
5 - Single-Molecule Mechanics of Protein Nanomachines
- from Part II - Protein Folding, Structure, Confirmation, and Dynamics
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 67-79
-
- Chapter
- Export citation
8 - Atomic Force Microscopy and Detecting a DNA Biomarker of a Few Copies without Amplification
- from Part III - Mapping DNA Molecules at the Single-Molecule Level
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 111-124
-
- Chapter
- Export citation
Part III - Mapping DNA Molecules at the Single-Molecule Level
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 95-124
-
- Chapter
- Export citation
9 - Single-Molecule Detection in the Study of Gene Expression
- from Part IV - Single-Molecule Biology to Study Gene Expression
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 127-141
-
- Chapter
- Export citation
4 - Long-Read Single-Molecule Optical Maps
- from Part I - Super-Resolution Microscopy and Molecular Imaging Techniques to Probe Biology
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 49-64
-
- Chapter
- Export citation
3 - Multiscale Fluorescence Imaging
- from Part I - Super-Resolution Microscopy and Molecular Imaging Techniques to Probe Biology
-
-
- Book:
- Single-Molecule Science
- Published online:
- 05 May 2022
- Print publication:
- 26 May 2022, pp 38-48
-
- Chapter
- Export citation
