Refine search
Actions for selected content:
106129 results in Materials Science
High-temperature X-ray powder diffraction analysis of selected ceramic mixtures
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 38-43
-
- Article
- Export citation
Commercial Announcements
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 4 / December 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 245-246
-
- Article
-
- You have access
- Export citation
Regional Reports
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 4 / December 1996
- Published online by Cambridge University Press:
- 10 January 2013, p. 322
-
- Article
- Export citation
Anomalous peaks in grazing incidence thin film X-ray diffraction
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 44-49
-
- Article
- Export citation
Book Review - Mineral Reference Manual, E.H. Nickel and M.C. Nichols, Van Nostrand Reinhold, New York, 1991. $21.95 - Rentgenografie mehomogenních napětovych polí (X-Ray Diffraction Analysis of Inhomogeneous Stress Fields)Ivo Kraus, Academia Prague, Czechoslovakia, 1990, 157 pp. ISBN 80-200-0254-5 - Multivariate Calibration, Harald Martens and Tormod Naes, John Wiley and Sons, Chichester 1989, 419pp. ISBN 0 471 90979 3, Hard cover £ 75.00/$138.00 or paperback £ 29.50/$67.85
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 119-120
-
- Article
- Export citation
Application of the overlap integral in X-ray diffraction powder pattern recognition
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 2 / June 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 124-135
-
- Article
- Export citation
PDJ volume 13 issue 2 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 2 / June 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b3
-
- Article
-
- You have access
- Export citation
Intensity Round Robin Report
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 2 / June 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 74-100
-
- Article
- Export citation
RIR - Measurement and Use in Quantitative XRD
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 74-77
-
- Article
-
- You have access
- Export citation
X-ray powder diffraction data for BaPbO3
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 56-59
-
- Article
- Export citation
Milling effects upon quantitative determinations of chrysotile asbestos by the reference intensity ratio method
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 1 / March 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 26-29
-
- Article
- Export citation
Regional Reports
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 1 / March 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 60-64
-
- Article
- Export citation
X-Ray Diffractometry of Low-Mass Samples
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 2 / June 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 78-81
-
- Article
- Export citation
Use of X-Ray Powder Diffraction Rietveld Pattern-Fitting for Characterising Preferred Orientation in Gibbsites
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 2 / June 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 79-85
-
- Article
- Export citation
About the crystal structure of La1−xSrxCoO3−δ (0≤x≤0.6)
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 31-34
-
- Article
- Export citation
Rietveld refinement of the solid-solution series: (Cu,Mg)SO4·H2O
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 3 / September 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 189-194
-
- Article
- Export citation
-
The kieserite-type solid-solution series of synthetic (Cu,Mg)SO4·H2O was investigated by TG-analysis and X-ray powder diffraction using the Rietveld method. Representatives with Cu≥20 mol% are triclinic distorted (
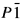 ) analogous to the poitevinite (Cu,Fe)SO4·H2O compounds. Cation site ordering with preference of Cu for the more distorted M1 site was additionally proven by the structure refinement.
) analogous to the poitevinite (Cu,Fe)SO4·H2O compounds. Cation site ordering with preference of Cu for the more distorted M1 site was additionally proven by the structure refinement.
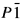 ) analogous to the poitevinite (Cu,Fe)SO
) analogous to the poitevinite (Cu,Fe)SOThe Value of X-ray Powder Diffraction Patterns, and the Structure of the Manganese-Aluminum Alloys with Apparent Icosahedral Symmetry
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 1 / March 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 14-15
-
- Article
- Export citation
ICDD Activities at the 1997 Denver Conference
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 2 / June 1997
- Published online by Cambridge University Press:
- 10 January 2013, p. 69
-
- Article
-
- You have access
- Export citation
X-ray powder diffraction data of anilinium trimolybdate tetrahydrate and anilinium trimolybdate dihydrate
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 4 / December 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 280-283
-
- Article
- Export citation
Determination of the Relative Amounts of α-carbamazepine and β-carbamazepine in a Mixture by Powder X-Ray Diffractometry
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 155-159
-
- Article
- Export citation
