Refine search
Actions for selected content:
106129 results in Materials Science
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 124-125
-
- Article
- Export citation
Cumulative Author Index
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 3 / September 1993
- Published online by Cambridge University Press:
- 10 January 2013, p. 200
-
- Article
- Export citation
Crystal structure of an unusual corrosion deposit, bis(ethylammonium) tetrachloroiron(II)
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 4 / December 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 261-266
-
- Article
- Export citation
Correspondent's Reports
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 1 / March 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 59-60
-
- Article
- Export citation
PDJ volume 9 issue 1 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b2
-
- Article
-
- You have access
- Export citation
Crystallite-size distributions and diffraction line profiles near the peak maximum
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 102-106
-
- Article
- Export citation
Crystalline structure of Ba2YCu0.25W0.75O6: A member of the new set of Ba2YzCuxW1−xO6 solid solutions
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 42-44
-
- Article
- Export citation
PDJ volume 9 issue 3 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 3 / September 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f8
-
- Article
-
- You have access
- Export citation
PDJ volume 6 issue 4 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 4 / December 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f19
-
- Article
-
- You have access
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 4 / December 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 269-270
-
- Article
- Export citation
A temperature-dependent powder diffraction study of chromium lanthanum nitrate, LaCr(NO3)6·12H2O
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 1 / March 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 23-25
-
- Article
- Export citation
Quantitative Determination by X-Ray Diffractometry of Calcium Sulfate and Calcium Carbonate in Airborne Dusts
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 86-90
-
- Article
- Export citation
Standard X-Ray Diffraction Powder Patterns of Fourteen Ceramic Phases
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 1 / March 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 40-54
-
- Article
- Export citation
Automation of a Filmscanner for Evaluation of Guinier Films and Applications to Determination of Diffraction Data of Pb2Sr2Ho0.625Ca0.375Cu3O8 and Measurement of Thermal Expansion of GeO2
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 82-84
-
- Article
- Export citation
An external standard method for angular calibration of powder diffractometers
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 3 / September 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 173-174
-
- Article
- Export citation
X-Ray Powder Data for Guanidiniuin Hexafluoro-chromate and -ferrate [C(NH2)3]3CrF6 and [C(NH2)3]3FeF6
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 3 / September 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 170-171
-
- Article
- Export citation
-
The complexes [C(NH2)3]3MF6, M = Cr, Fe have been isolated from aqueous solutions of the corresponding fluorides. The compounds are isostructural and crystallize in the cubic space group
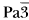 with a = 14.0667 (6) Å for M = Cr, a = 14.1246(8) Å for M = Fe and Z = 8. The MF63− anions are arranged as Na+ and Cl− in the NaCl lattice. Guanidinium cations are distributed on 24-fold general position.
with a = 14.0667 (6) Å for M = Cr, a = 14.1246(8) Å for M = Fe and Z = 8. The MF63− anions are arranged as Na+ and Cl− in the NaCl lattice. Guanidinium cations are distributed on 24-fold general position.
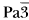 with a = 14.0667 (6) Å for M = Cr, a = 14.1246(8) Å for M = Fe and Z = 8. The MF
with a = 14.0667 (6) Å for M = Cr, a = 14.1246(8) Å for M = Fe and Z = 8. The MFEffects of sample transparency in powder diffraction
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 2 / June 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 107-113
-
- Article
- Export citation
PDJ volume 4 issue 2 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 2 / June 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f19
-
- Article
-
- You have access
- Export citation
An Application of Calculated X-Ray Diffraction Patterns in the Analysis of Reference Powder Data: Trivalent Metal Sulfates
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 215-218
-
- Article
- Export citation
Crystal and powder XRD data of Mg3(PO4)2-III: High-temperature and high-pressure form
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 4 / December 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 293-295
-
- Article
- Export citation
